
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDNA-organized artificial LHCs – testing the limits of chromophore segmentation†
Nutcha
Bürki
a,
Elena
Grossenbacher
a,
Andrea
Cannizzo
 b,
Thomas
Feurer
b,
Simon M.
Langenegger
a and
Robert
Häner
*a
b,
Thomas
Feurer
b,
Simon M.
Langenegger
a and
Robert
Häner
*a
aDepartment of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, CH-3012 Bern, Switzerland. E-mail: robert.haener@dcb.unibe.ch
bInstitute of Applied Physics, University of Bern, Sidlerstrasse 5, CH-3012 Bern, Switzerland
First published on 26th August 2020
Abstract
DNA-organized multi-chromophoric systems containing phenanthrene and pyrene derivatives exhibit a highly efficient excitation energy transfer from phenanthrene (donor) to pyrene (acceptor). The energy transfer also occurs if the phenanthrene antenna is interrupted by intervening DNA base pairs. Artificial light-harvesting complexes composed of up to five phenanthrene-DNA alternations with fluorescence quantum yields as high as 68% are described.
The use of DNA to arrange fluorophores in precisely defined distances and orientations allows the construction of nanoarchitectures with a wide variety of different geometries.1–12 The defined positioning of chromophores in a framework is an important aspect in the design of artificial light-harvesting complexes (LHCs).13–22 In such systems, light is collected by precisely arranged donor chromophores and the absorbed energy is transferred to a suitable acceptor.23–30 The light-harvesting properties resulting from the supramolecular assembly of phenanthrene and pyrene molecules within the scaffold of the DNA duplexes was demonstrated before.17 In this type of LHC, the phenanthrene units serve as donor chromophores.31 In an efficient process, the absorbed light is transferred to the pyrene acceptor molecule. Transient absorption spectroscopy indicates that the energy is transferred over a delocalized excited state of the phenanthrenes.13 Subsequent studies showed that excitation energy transfer also takes place if the aromatic stack of phenanthrenes is divided into two segments by intervening base pairs. Excitation energy is still efficiently transmitted along the phenanthrene antenna if the latter is interrupted by up to three DNA base pairs. In fact, the fluorescence quantum yield even slightly increased by going from a continuous chromophore stack to a bi-segmental arrangement.13 The present study shows that the efficiency of energy transfer in multi-segmental phenanthrene arrangements, in which phenanthrenes and DNA base pairs are arranged in an alternating fashion (see Fig. 1 for an illustration), is not only maintained but even grows until it reaches a maximum after three to four segments.
 | ||
| Fig. 1 Illustration of differently arranged phenanthrene antennas in DNA-organized artificial LHCs. Top: Single, contiguous phenanthrene stack; bottom: alternating arrangement of base pairs and phenanthrene units. | ||
Table 1 gives an overview on the different pyrene- and/or phenanthrene-modified oligodeoxynucleotides that were used in the present study. Their preparation was accomplished by automated oligonucleotide synthesis using the corresponding phenanthrene and pyrene building blocks (see ESI†).14 A total of nine different hybrids containing 3,6-dibutynylphenanthrene (α) building blocks as donor chromophores and 1,8-dibutynylpyrene (Y) as the acceptor molecule were assembled and investigated. Duplex 1 contains a single α donor besides the pyrene acceptor. Duplexes 2 to 5 possess an increasing number of additional α pairs that are always separated by an AT base pair. Duplexes 6 to 8 have the same composition as duplex 4 but they contain one or two base pair mismatches. Finally, Ref1, in which the stack of ten phenanthrenes is not interrupted by base pairs, serves as the reference duplex.
| Duplex | Sequence | T m [°C] | ϕ F [%] (SD) |
|---|---|---|---|
| 1 | 5′ GGC TAA YTA AAT TTA AAT CGC 3′ | 60.7 | 47.2 (±2.2) |
| 3′ CCG ATT αAT TTA AAT TTA GCG 5′ | |||
| 2 | 5′ GGC TAA YTα AAT TTA AAT CGC 3′ | 57.4 | 51.0 (±2.7) |
| 3′ CCG ATT αAα TTA AAT TTA GCG 5′ | |||
| 3 | 5′ GGC TAA YTα AαT TTA AAT CGC 3′ | 52.5 | 68.1 (±3.2) |
| 3′ CCG ATT αAα TαA AAT TTA GCG 5′ | |||
| 4 | 5′ GGC TAA YTα AαT αTA AAT CGC 3′ | 49.8 | 64.0 (±2.1) |
| 3′ CCG ATT αAα TαA αAT TTA GCG 5′ | |||
| 5 | 5′ GGC TAA YTα AαT αTα AAT CGC 3′ | 42.6 | 46.3 (±2.3) |
| 3′ CCG ATT αAα TαA αAα TTA GCG 5′ | |||
| 6 | 5′ GGC TAA YTα AαT αTA AAT CGC 3′ | 50.7 | 65.1 (±0.6) |
3′ CCG ATT α![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) α TαA αAT TTA GCG 5′ α TαA αAT TTA GCG 5′ |
|||
| 7 | 5′ GGC TAA YTα AαT αTA AAT CGC 3′ | 48.3 | 54.6 (±0.6) |
| 3′ CCG ATT αAα Tα αAT TTA GCG 5′ |
|||
| 8 | 5′ GGC TAA YTα AαT αTA AAT CGC 3′ | 51.3 | 69.6 (±1.2) |
| 3′ CCG ATT αα Tα αAT TTA GCG 5′ |
|||
| Ref1 | 5′ GGC TAA Yαα ααT TTA AAT CGC 3′ | 64.0 | 70.0 (±3.0) |
| 3′ CCG ATT ααα ααA AAT TTA GCG 5′ |
The melting curves of the different hybrids were recorded by monitoring (260 nm) the change in absorption during cooling and heating cycles (80 °C to 20 °C and reversed) at a rate of 0.5 °C min−1 (see Fig. S37–S44†). The melting temperatures (Tm) correspond to the maximum of the first derivative of the melting curve and are given in Table 1. In hybrids 1–5, the Tm decreases with an increasing number of modifications. Each replacement of an AT base pair with a pair of α results in a considerable destabilisation of 3–7 °C. Duplex Ref1, which contains the same number of modifications as duplex 5 but arranged in an uninterrupted way, has a much higher duplex stability than duplex 5, 64 °C compared to 43 °C, which is comparable to the melting temperature of duplex 1.
The addition of a base pair mismatch (T-T) in duplexes 6 or 7 resulted in nearly unchanged thermal stability (+0.9, −1.5 °C) compared to the fully complementary duplex 4. A bit surprising, however, is the fact that duplex 8 with two mismatches is slightly more stable than the matched duplex 4. We assume that this finding is explained by a duplex structure, in which the mismatched base pairs are flipped out of the DNA duplex, resulting in π–π interactions between the phenanthrene segments to compensate for the loss of stability caused by the two T-T mismatches.32 Along the same argument, this may also be the case for hybrids 6 and 7 and serve as an explanation for the absences of a destabilization in spite of the presence of a mismatch.
Fig. 2 shows the absorption spectra of duplexes 1–8 and Ref1. Phenanthrene absorption exhibits vibronic bands with maxima at 333 nm and 318 nm, while pyrene absorption results in the maxima observed at 370 nm and 391 nm. The absorption spectra of duplexes 1–8 show significant differences in the intensities of the phenanthrene absorption bands (333 nm and 318 nm), which is due to the different number of phenanthrene units present in the hybrids (one to nine units). At 370 nm and 391 nm, the absorbance remains approximately the same for all duplexes because they all contain only one pyrene molecule. Little information can be gained from the spectral range below 300 nm, since this area is the result of a combined absorption by pyrene, phenanthrene and the nucleobases.
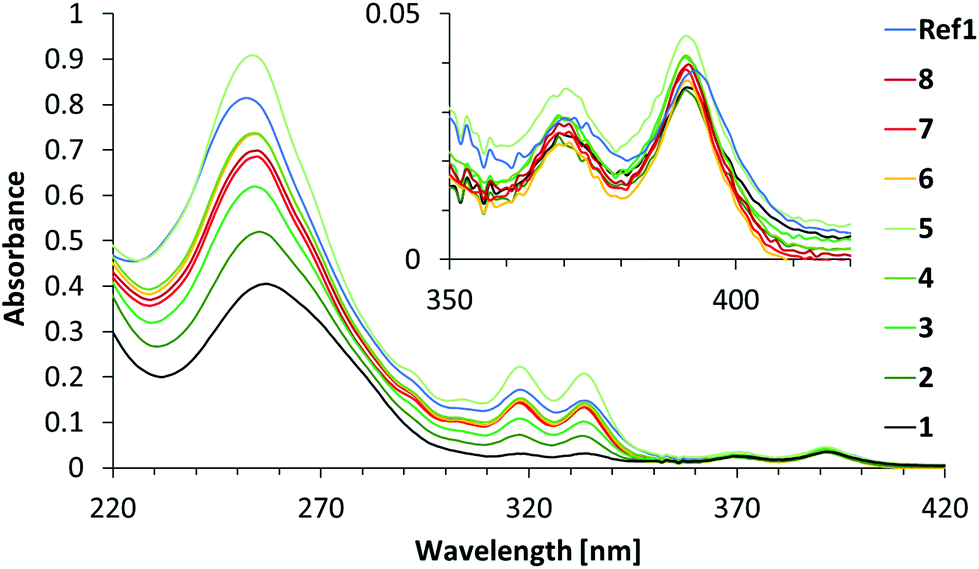 | ||
| Fig. 2 Absorption spectra of duplexes 1–8 and Ref1. Conditions: see Table 1; 20 °C. | ||
Duplex Ref1 contains nine α building blocks and one Y acceptor like duplex 5, but without intervening A-T base pairs. The absorption spectrum of duplex Ref1 shows, however, significant differences from the one of duplex 5. Thus, a considerable hypochromic effect is observed at 318 nm and, more pronounced, at 333 nm. No wavelength shifts are observed at these peaks. These findings are in agreement with a model of the Ref1 duplex structure, in which all phenanthrenes are organized in an uninterrupted stacking arrangement that leads to a strong electronic coupling. In hybrid 5, on the other hand, stacking among all nine phenanthrenes is not possible due to the intervening AT base pairs.
Fluorescence spectra were recorded by phenanthrene excitation at 316 nm. The resulting spectra are depicted in Fig. 3. Duplexes 1–5 all show significant pyrene emission with a maximum at 402 nm and a second band that is slightly shifted in a hypsochromic fashion (426 to 422 nm) by going from hybrid 1 to 5. Fluorescence spectra of duplexes 6–8 possess a similar shape as that of duplex 4 but with a somewhat increased intensity. While duplexes 1, 2 and 3 show almost no phenanthrene emission (365–380 nm range), a slight signal appears in this region for duplex 4 and grows even a bit stronger for duplex 5. This means that some of the excitation energy is emitted by α itself, rather than being transferred to the acceptor. The intensity of pyrene fluorescence increases steadily in the order of duplexes 1–4; no further increase is observed for duplex 5. In addition, all emission spectra exhibit an extended tailing with a shoulder around 450 nm, which is attributed to phenanthrene excimer and/or phenanthrene-pyrene exciplex formation.
 | ||
| Fig. 3 Emission spectra of duplexes 1–8 and Ref1. Conditions: see Table 1; λexc 316 nm, 20 °C; excitation slit: 2.5 nm, emission slit: 5 nm. | ||
The emission profile of duplex Ref1 differs significantly from those of duplexes 1–8, in which the phenanthrene stacks are interrupted by DNA base pairs. It has a maximum at 425 nm with a shoulder around 410 nm and a pronounced tailing up to 580 nm. This difference is a direct manifestation of the stacking arrangement and the resulting electronic coupling of the phenanthrenes in duplex Ref1, which is in contrast to the segmented arrangement obtained by interposed DNA base pairs in the other hybrids.
The fluorescence signals of the emission spectra were integrated over the range from 350 to 600 nm and the data are displayed in Fig. 4. The fluorescence intensity increases steadily for duplexes 1 to 4. Accordingly, each α unit in the antenna contributes approx. 2000 intensity units. From duplex 4 to 5 (i.e. from seven to nine α units) no further increase in intensity is observed. In contrast, by comparing duplex 4 to the duplex Ref1, which involves also a change from seven to nine α units, an increase of 4000 units in the fluorescence signal is observed, which corresponds exactly to the previously established contribution by two phenanthrenes. This shows, that the energy absorbed by an increasing number of phenanthrenes, also when separated by AT base pairs, leads to increasing pyrene fluorescence by up to four such segments (duplex 4). After that, further addition of phenanthrenes (duplex 5) has no effect on the energy transferred to pyrene. In the duplexes containing T-T mismatches, the fluorescence intensities are comparable to duplexes 4 and 5, with the exception of duplex 7, which shows a reduced pyrene fluorescence.
 | ||
| Fig. 4 Integrated fluorescence (350–600 nm) and fluorescence quantum yields (relative to quinine sulfate) of duplexes 1–8 and Ref1, schematically illustrated on the left (green: pyrene, blue: phenanthrene, grey: nucleobases, red: mismatches). Conditions: see Table 1; λexc 316 nm, 20 °C; excitation slit: 2.5 nm, emission slit: 5 nm. | ||
Fluorescence quantum yields (Fig. 4 and Table 1) of all assembled artificial LHCs were determined using quinine sulfate as a standard (see ESI†). Duplex Ref1 has the highest quantum yield (approx. 70%) in the entire series. The quantum yields increase for duplexes 1 to 3 before they level off (duplex 4) and then decrease by about 20% (duplex 5). This is expected on the basis of the fluorescence spectra discussed previously. Thus, duplex 4 is the maximum length in this type of ‘interrupted’ phenanthrene antenna that contributes to energy harvesting by light absorption and energy transfer to the pyrene acceptor. Surprisingly again, duplexes 6 and 8 containing one and two mismatches have a similar or slightly higher quantum yield than the fully matched duplex 4. Again, the possibility that the mismatched DNA base pairs adopt a ‘flipped out’ conformation, see above, may serve as an explanation for this unexpected observation with regard to the quantum yields.
The observed decrease in energy transfer efficiency from duplex 4 to duplex 5 may be the result of several factors. The first reason might reside in a decreased duplex stability as a result of adding more and more phenanthrene segments. In fact, a broadening of the melting curve was observed for duplex 5 (see Fig. S41†). Lower duplex stability is a result of reduced molecular interactions between the two strands, including reduced stacking interactions between phenanthrenes, hence less efficient energy transfer. The second reason for the observed drop in fluorescence quantum yield may simply reflect the increase in the distance between the last donor α in duplex 5 compared to duplex 4, in which this separation is shorter (see Fig. 1 for illustration). Assuming a distance of 3.4 Å per aromatic unit, this distance would be 34 Å in duplex 4 compared to 44 Å in duplex 5. More in-depth studies are required to further elucidate the relevant parameters for this behaviour.
Conclusions
In summary, DNA-organized artificial LHCs have been described which are composed of multi-segmental duplexes with alternating DNA and phenanthrene sections. The collective of phenanthrenes assembled in the DNA duplex in this way serves as a light harvesting antenna. Absorbed light is efficiently transferred to a pyrene acceptor chromophore, despite the discontinuity of the phenanthrene stack due to intervening DNA base pairs. All segmented constructs proved to be efficient light-harvesting systems with fluorescence quantum yields ranging from 47% to 68%. The efficiency of the LHCs first increases with the number of phenanthrenes until it reaches a maximum after three to four chromophore segments. Experiments with duplexes containing mismatched bases showed that the DNA base pairs placed between the phenanthrene chromophores are not obeying the selectivity rules of the Watson–Crick base pairing. The results demonstrate that the construction of artificial light-harvesting systems tolerates the integration of functionally non-related components, such as DNA base pairs, into the multi-chromophore array.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Swiss National Foundation (NCCR MUST and Grant 200020-188468) is gratefully acknowledged.Notes and references
- N. C. Seeman, Annu. Rev. Biochem., 2010, 79, 65–87 CrossRef CAS.
- E. Stulz and G. H. Clever, DNA in Supramolecular Chemistry and Nanotechnology, 2015 Search PubMed.
- E. Stulz, Acc. Chem. Res., 2017, 50, 823–831 CrossRef CAS.
- R. Varghese and H. A. Wagenknecht, Chem. Commun., 2009, 2615–2624 RSC.
- V. L. Malinovskii, D. Wenger and R. Häner, Chem. Soc. Rev., 2010, 39, 410–422 RSC.
- W. Pfeifer and B. Saccà, ChemBioChem, 2016, 17, 1063–1080 CrossRef CAS.
- Y. N. Teo and E. T. Kool, Chem. Rev., 2012, 112, 4221–4245 CrossRef CAS.
- A. V. Pinheiro, D. Han, W. M. Shih and H. Yan, Nat. Nanotechnol., 2011, 6, 763–772 CrossRef CAS.
- M. Numata, K. Sugiyasu, T. Kishida, S. Haraguchi, N. Fujita, S. M. Park, Y. J. Yun, B. H. Kim and S. Shinkai, Org. Biomol. Chem., 2008, 6, 712–718 RSC.
- E.-C. Wamhoff, J. L. Banal, W. P. Bricker, T. R. Shepherd, M. F. Parsons, R. Veneziano, M. B. Stone, H. Jun, X. Wang and M. Bathe, Annu. Rev. Biophys., 2019, 48, 395–419 CrossRef CAS.
- F. A. Aldaye, A. L. Palmer and H. F. Sleiman, Science, 2008, 321, 1795–1799 CrossRef CAS.
- H. Bui, S. A. Díaz, J. Fontana, M. Chiriboga, R. Veneziano and I. L. Medintz, Adv. Opt. Mater., 2019, 7, 1900562 CrossRef.
- C. D. Bösch, E. Abay, S. M. Langenegger, M. Nazari, A. Cannizzo, T. Feurer and R. Häner, Helv. Chim. Acta, 2019, 102, e1900148 CrossRef.
- C. B. Winiger, S. Li, G. R. Kumar, S. M. Langenegger and R. Häner, Angew. Chem., Int. Ed., 2014, 53, 13609–13613 CrossRef CAS.
- M. Probst, S. M. Langenegger and R. Häner, Chem. Commun., 2014, 50, 159–161 RSC.
- I. H. Stein, C. Steinhauer and P. Tinnefeld, J. Am. Chem. Soc., 2011, 133, 4193–4195 CrossRef CAS.
- F. Garo and R. Häner, Angew. Chem., Int. Ed., 2012, 51, 916–919 CrossRef CAS.
- J. G. Woller, J. K. Hannestad and B. Albinsson, J. Am. Chem. Soc., 2013, 135, 2759–2768 CrossRef CAS.
- P. Ensslen and H. A. Wagenknecht, Acc. Chem. Res., 2015, 48, 2724–2733 CrossRef CAS.
- J. S. Melinger, A. Khachatrian, M. G. Ancona, S. Buckhout-White, E. R. Goldman, C. M. Spillmann, I. L. Medintz and P. D. Cunningham, ACS Photonics, 2016, 3, 659–669 CrossRef CAS.
- E. A. Hemmig, C. Creatore, B. Wünsch, L. Hecker, P. Mair, M. A. Parker, S. Emmott, P. Tinnefeld, U. F. Keyser and A. W. Chin, Nano Lett., 2016, 16, 2369–2374 CrossRef CAS.
- F. Nicoli, A. Barth, W. Bae, F. Neukirchinger, A. H. Crevenna, D. C. Lamb and T. Liedl, ACS Nano, 2017, 11, 11264–11272 CrossRef CAS.
- S. D. Jo, J. S. Kim, I. Kim, J. S. Yun, J. C. Park, B. I. Koo, E. Lee and Y. S. Nam, Adv. Funct. Mater., 2017, 27, 1700212 CrossRef.
- E. Boulais, N. P. D. Sawaya, R. Veneziano, A. Andreoni, J. L. Banal, T. Kondo, S. Mandal, S. Lin, G. S. Schlau-Cohen, N. W. Woodbury, H. Yan, A. Aspuru-Guzik and M. Bathe, Nat. Mater., 2018, 17, 159–166 CrossRef CAS.
- W. P. Klein, S. A. Díaz, S. Buckhout-White, J. S. Melinger, P. D. Cunningham, E. R. Goldman, M. G. Ancona, W. Kuang and I. L. Medintz, Adv. Opt. Mater., 2018, 6, 1700679 CrossRef.
- M. Kownacki, S. M. Langenegger, S. X. Liu and R. Häner, Angew. Chem., 2019, 131, 761–765 CrossRef.
- H. Kashida, H. Kawai, R. Maruyama, Y. Kokubo, Y. Araki, T. Wada and H. Asanuma, Commun. Chem., 2018, 1, 1–8 CrossRef CAS.
- Y. Choi, L. Kotthoff, L. Olejko, U. Resch-Genger and I. Bald, ACS Appl. Mater. Interfaces, 2018, 10, 23295–23302 CrossRef CAS.
- P. K. Dutta, R. Varghese, J. Nangreave, S. Lin, H. Yan and Y. Liu, J. Am. Chem. Soc., 2011, 133, 11985–11993 CrossRef CAS.
- X. Zhou, S. Mandal, S. Jiang, S. Lin, J. Yang, Y. Liu, D. G. Whitten, N. W. Woodbury and H. Yan, J. Am. Chem. Soc., 2019, 141, 8473–8481 CrossRef CAS.
- M. Nazari, C. D. Bösch, A. Rondi, A. Francés-Monerris, M. Marazzi, E. Lognon, M. Gazzetto, S. M. Langenegger, R. Häner, T. Feurer, A. Monari and A. Cannizzo, Phys. Chem. Chem. Phys., 2019, 21, 16981–16988 RSC.
- T. A. Zeidan, M. Hariharan, K. Siegmund and F. D. Lewis, Photochem. Photobiol. Sci., 2010, 9, 916–922 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob01531h |
| This journal is © The Royal Society of Chemistry 2020 |