
Absolute handedness control of oligoamide double helices by chiral oxazolylaniline induction†
Brice
Kauffmann,  b
Quan
Gan
*a
b
Quan
Gan
*a
b
Quan
Gan
*a
Abstract
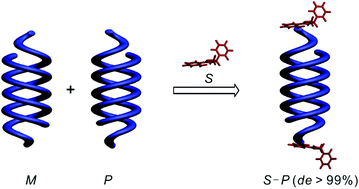
Aromatic oligoamide double helices bearing a chiral oxazolylaniline moiety at the C-terminus were synthesized and their helix handedness was completely controlled (de > 99%). The absolute helix sense and the de values were evaluated by using 1H NMR, X-ray crystallography, and circular dichroism (CD). Using crystal structure analysis, the high efficiency of helix handedness induction was attributed to the close location of the asymmetric carbon center to the helix orbits via intramolecular hydrogen bonding. The CD experiments also showed that there is no loss of chiral induction either in the interconversion of single and double helices or by elongation of the sequences.
- This article is part of the themed collections: Editor’s Collection and Supramolecular chemistry in OBC
 Please wait while we load your content...
Please wait while we load your content...

