
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIridium-catalysed C–H borylation of β-aryl-aminopropionic acids†
Henry
Robinson
a,
Joe
Stillibrand‡
a,
Klemensas
Simelis
a,
Simon J. F.
Macdonald
b and
Andrew
Nortcliffe
 *a
*a
aGlaxoSmithKline Carbon Neutral Laboratories for Sustainable Chemistry, School of Chemistry, University of Nottingham, Triumph Road, Nottingham, NG7 2TU, UK. E-mail: andrew.nortcliffe@nottingham.ac.uk; Tel: +44(0)1158 466887
bGlaxoSmithKline, Medicines Research Centre, Gunnels Wood Road, Stevenage, SG1 2NY, UK
First published on 17th August 2020
Abstract
Iridium-catalysed catalytic, regioselective C–H borylation of β-aryl-aminopropionic acid derivatives gives access to 3,5-functionalised protected β-aryl-aminopropionic acid boronates. The synthetic versatility of these new boronates is demonstrated through sequential one-pot functionalisation reactions to give diverse building blocks for medicinal chemistry. The C–H borylation is also effective for dipeptide substrates. We have exemplified this methodology in the synthesis of a pan αv integrin antagonist.
Introduction
Natural and non-natural amino acids are widespread and have been extensively exploited in the synthesis of therapeutic molecules.1 As a result, there is a continued need to develop novel strategies for the synthesis of these fundamentally important building blocks. Of particular importance in the context of medicinal chemistry is the development of methods for the construction of structurally diverse β-aryl-aminopropionic acids. This motif is present in biologically active molecules with activity against Chagas disease,2 breast cancer,3 and in αv integrin antagonists as potential treatments for osteoporosis,4 melanoma5 and idiopathic pulmonary fibrosis.6 Additionally, β-phenyl-aminopropionic acid is an intermediate in the synthesis of Maraviroc,7 a CCR-5 receptor antagonist used for the treatment and prevention of HIV. Therefore, a unified strategy to the synthesis of functionalised β-aryl-aminopropionic acid would be of significant value.Three general strategies have been developed for the synthesis of β-aryl-aminopropionic acids to date (Scheme 1). The Rodionov reaction (Scheme 1A) represents the simplest option and involves the multicomponent coupling of an aldehyde, malonic acid and ammonium acetate to generate racemic β-aryl-aminopropionic acids.8–10 A second protocol involves diastereoselective Reformatsky reactions of sulfinimines (Scheme 1B).11 An alternative auxiliary-based approach is depicted in Scheme 1C and involves the addition of chiral lithium amides to cinnamic acid derivatives (Scheme 1C).12–14 These strategies for the synthesis of β-aryl-aminopropionic acids are dependent on the availability of highly functionalised starting aldehydes (Scheme 1A and B) or aryl bromides (Scheme 1C). While some of these are commercially available, they are available in limited supply and, since the diversity is introduced in the first step, a multistep synthesis is required for each different β-aryl-aminopropionic acid. This makes the exploration of structure–activity relationships around the aryl ring time and resource consuming.
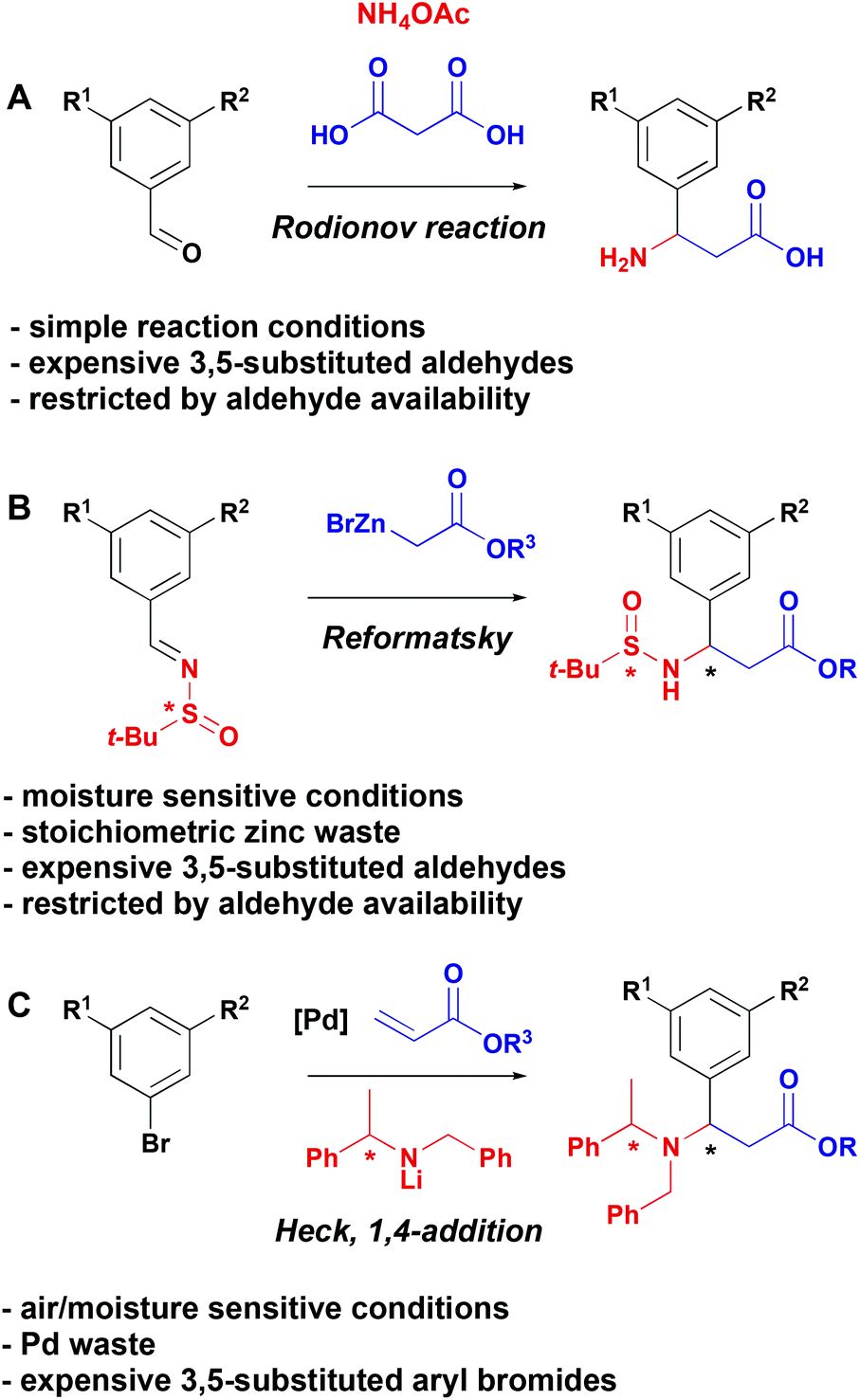 | ||
| Scheme 1 Common approaches to β-aryl-aminopropionic acids. | ||
As part of our research into novel biologically active peptidomimetics for therapeutic application we sought to incorporate β-aryl-aminopropionic acids with diverse substituents in the 3- and 5-positions. Given that existing methods, require a bespoke aldehyde or aryl bromide for every new compound and we designed an alternative method based on a late-stage functionalisation approach. Specifically, we envisioned that an appropriately pre-functionalised 3-substituted β-aryl-aminopropionic acid derivative, accessed via a Rodionov multicomponent reaction from more widely available 3-substituted aldehyde, could undergo a C–H functionalisation reactions (Scheme 2) providing a platform for further chemistry.
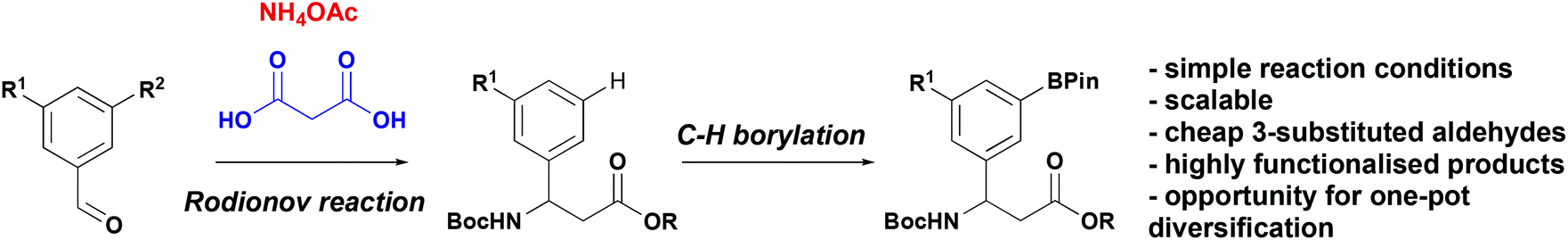 | ||
| Scheme 2 C–H functionalisation to diverse β-aryl-aminopropionic acids. | ||
In the past decade C–H activation chemistry emerged into the forefront of modern organic chemistry. The innovative work of Hartwig and Miyaura,15 and Smith and Maleczka16 in iridium-catalysed aromatic C–H borylation offers a potentially powerful method for functionalisation given that the synthetic versatility of boronate groups is ever increasing.17–19
Encouraged by previous examples of C–H borylation on simple phenylalanine20 and tryptophan21 derivatives we now report the substrate directed iridium borylation of β-3-aryl-aminopropionic acid precursors together with examples of “one-pot” transformations to diversely functionalised 3,5-substituted products and implementation of this methodology in the preparation of an integrin antagonist bearing a β-3,5-aryl-aminopropionic acid motif.
Results and discussion
A series of 3-substituted β-aryl-aminopropionic acid derivatives were prepared by Rodionov multicomponent reaction or addition of the bromozincacetate to the corresponding sulfinimine followed by suitable protection as the N-Boc esters. Using 1d as a model substrate we performed a brief screen of previously reported ligands and solvents to determine the optimum conditions.22 Methoxy(cyclooctadiene)iridium(I) dimer ([Ir(OCH3)(COD)]2) was chosen as the catalyst with bis(pinacolato)diboron (B2pin2) as the boron source. We evaluated three solvents (MTBE, THF and iPrNEt2) with two ligands – 4-4′-di-tert-butyl-2,2′-bipyridine (dtbpy) and 3,4,7,8-tetramethyl-1,10-phenanthroline (tmphen). The reactions were heated at 80 °C for 24 h and a qualitative assessment of conversion determined by LCMS. The reaction did not reach completion when iPrNEt2 was used a solvent. Conversion was comparable for both ligands and when ether solvents were used. Under standard heating conditions, shortening the reaction time from 24 h reduced the conversion, but complete conversion could be achieved using microwave irradiation in three hours. Based on this screen we selected MTBE and dtbpy to pair with ([Ir(OCH3)(COD)]2) and B2Pin2. MTBE was selected over THF. Under these conditions a range of β-aryl-aminopropionic acid derivatives were converted into the corresponding borylated derivatives (Scheme 3). As predicted, the reaction predominantly or exclusively yielded the 3,5-substituted products, in accordance with the reported directing regiochemistry of iridium-catalysed borylation reactions.23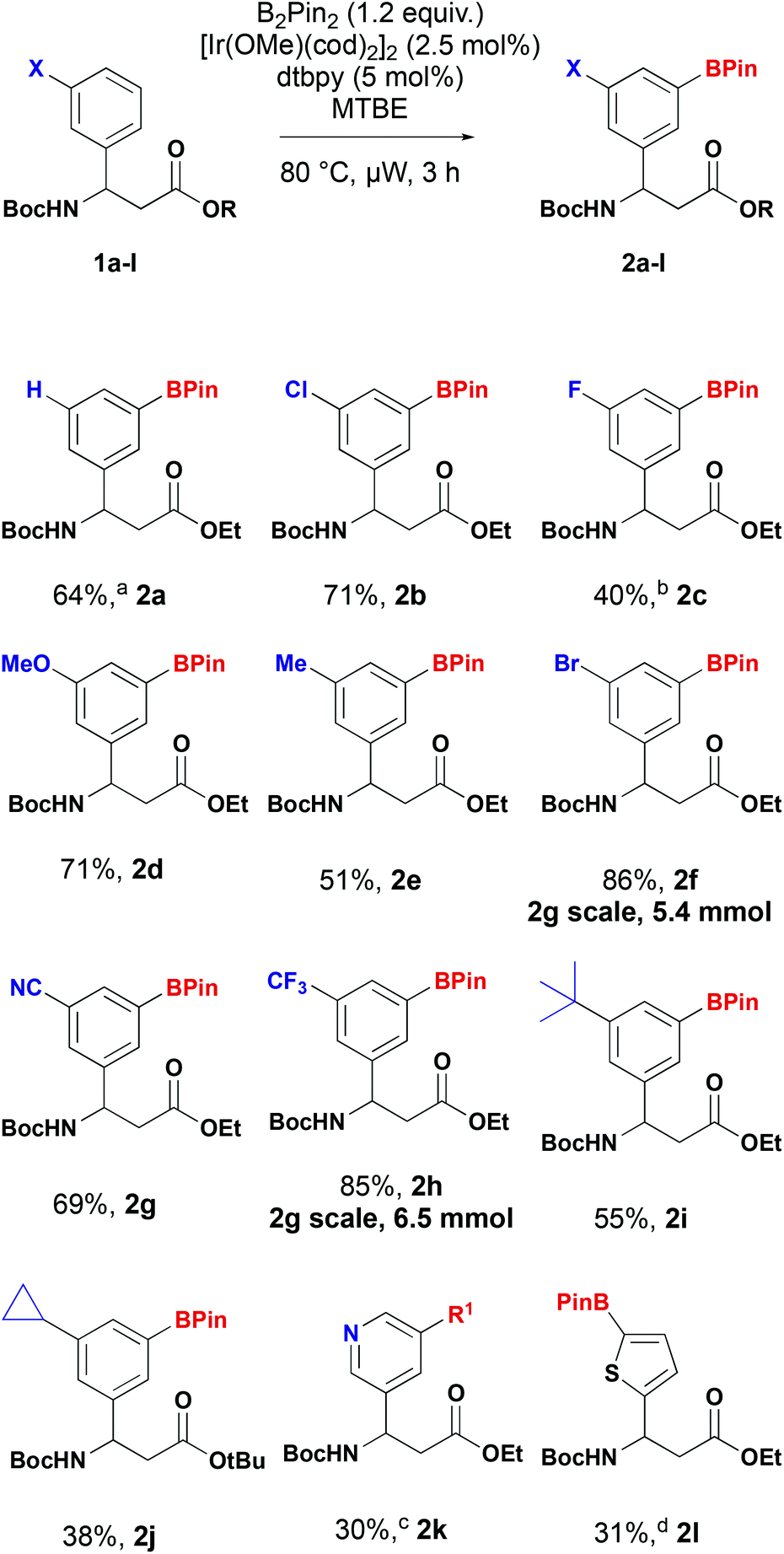 | ||
Scheme 3 Substrate scope of iridium-catalysed C–H borylation of β-aryl-aminopropionic acids. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1:1:3 mixture of regioisomers (2a: 4-substitued:3,5-substituted), b2:1 mixture of regioisomers (2c: 3,4-substituted), cborylated product (R1 = Bpin) unstable to column chromatography; yield over two steps following Suzuki coupling with p-nitroiodobenzene to yield product 2k (R1 = p-nitrophenyl), dreaction conducted at room temperature. 1:1:3 mixture of regioisomers (2a: 4-substitued:3,5-substituted), b2:1 mixture of regioisomers (2c: 3,4-substituted), cborylated product (R1 = Bpin) unstable to column chromatography; yield over two steps following Suzuki coupling with p-nitroiodobenzene to yield product 2k (R1 = p-nitrophenyl), dreaction conducted at room temperature. | ||
The reaction of unsubstituted β-aryl-aminopropionic acid derivative (X = H) yielded a mixture of 3- and 4-isomers along with and 3,5-diborylated product in a ratio of 1:1:3, as determined by 1H NMR. 3-Substituted substrates were readily tolerated in the reaction delivering the corresponding 3,5-substituted products in 38–86% yield (Scheme 3). 3-Fluorosubstituted substrate 1c yielded a 2:1 mixture of 3,5 (2c) and 3,4-isomers that were inseparable by column chromatography. This is consistent with observations by other workers.20,24 The borylated products were stable and could be readily purified by column chromatography to isolate them. Furthermore, the 3-trifluoromethyl 2f and 3-bromo 2h derivatives could be prepared on multigram scale with improved isolated yields. Under the standard borylation conditions, 2-thienyl derivative 1l yielded exclusively the 3,5-diborylated product. The 5-borylated product 2l was produced exclusively by conducting the reaction at room temperature. C–H borylation of the 3-pyridyl derivative 1k yielded the desired product, however, attempts to purify the compound by chromatography resulted in protodeborylation and recovery of starting material 1k. The yield reported is for the two-step procedure, involving Suzuki–Miyaura coupling of the unstable boronate ester with 4-nitroiodobenzene (R1 = 4-nitrophenyl). When the enantioenriched substrate (S)-1f was subjected to C–H borylation followed by oxidation, phenol (S)-3 was obtained with an 97% ee indicating no racemisation occurs in the borylation process (Scheme 4).
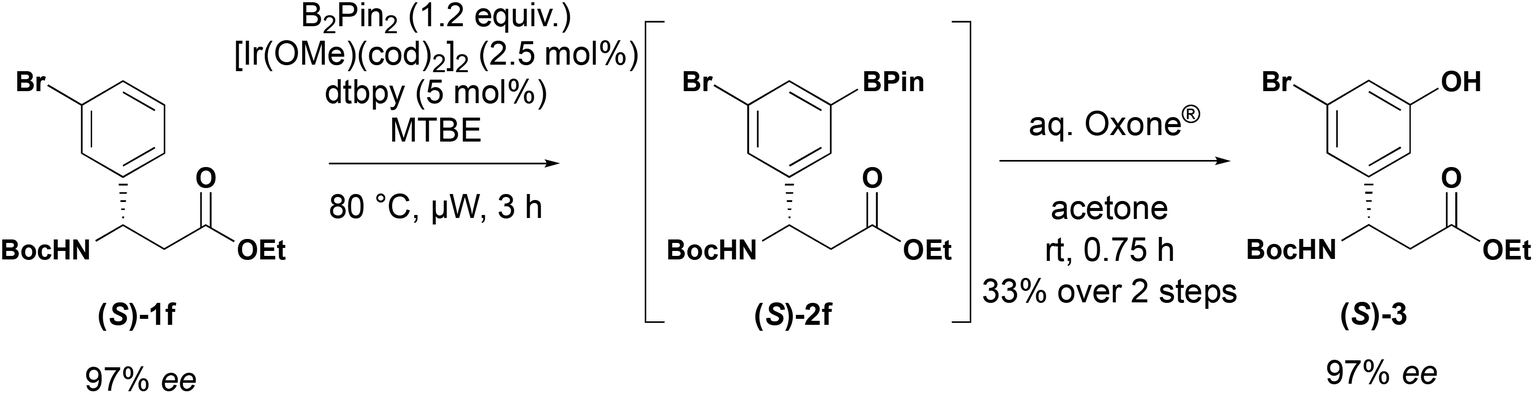 | ||
| Scheme 4 Iridium-catalysed C–H borylation of enantiopure β-aryl-aminopropionic acids. | ||
To demonstrate the versatility of these β-aryl-aminopropionic acid boronate derivatives in synthesis we have exemplified their applicability in typical reactions used in the synthesis of biologically active molecules (Scheme 5). In each example, the 3-substituted β-aryl-aminopropionic acid derivative was borylated under the previously described conditions and then telescoped into functionalisation without purification of the boronate. Oxidation of the boronate with aqueous Oxone® provided the phenols 3–6.16 3-Trifluoromethyl derivative 1h was borylated and transformed into biaryl ether 7 under standard Suzuki–Miyaura conditions. Attempts to alkylate the phenols under standard conditions (RBr, Cs2CO3) were unsuccessful, resulting in ester hydrolysis and transesterification only. To circumvent this and access ether containing products we utilised the Chan–Lam coupling to provide phenyl ethers 8 and 9.25 The Chan–Lam coupling was also an effective methodology to access N-linked heteraraomatic β-aryl-aminopropionic acid derivatives, the 1,3-dimethylpyrazole derivates 10 and 11 were accessed in 74 and 68% yield respectively over two steps.26 These transformations are further examples of the Chan–Lam coupling as a viable sustainable alternative to Pd-catalysed etherification and amination and are compatible with tandem iridium borylation. Bromination of 2i with CuBr2 provided the 3-bromo-5-tert-butyl derivative 12, this motif is featured in recently reported selective integrin antagonists.27,28 3-Bromo-5-fluoroderivative 13 was prepared via metallation of the intermediate boronate to the organosilver reagent and reaction with Selectfluor®.29
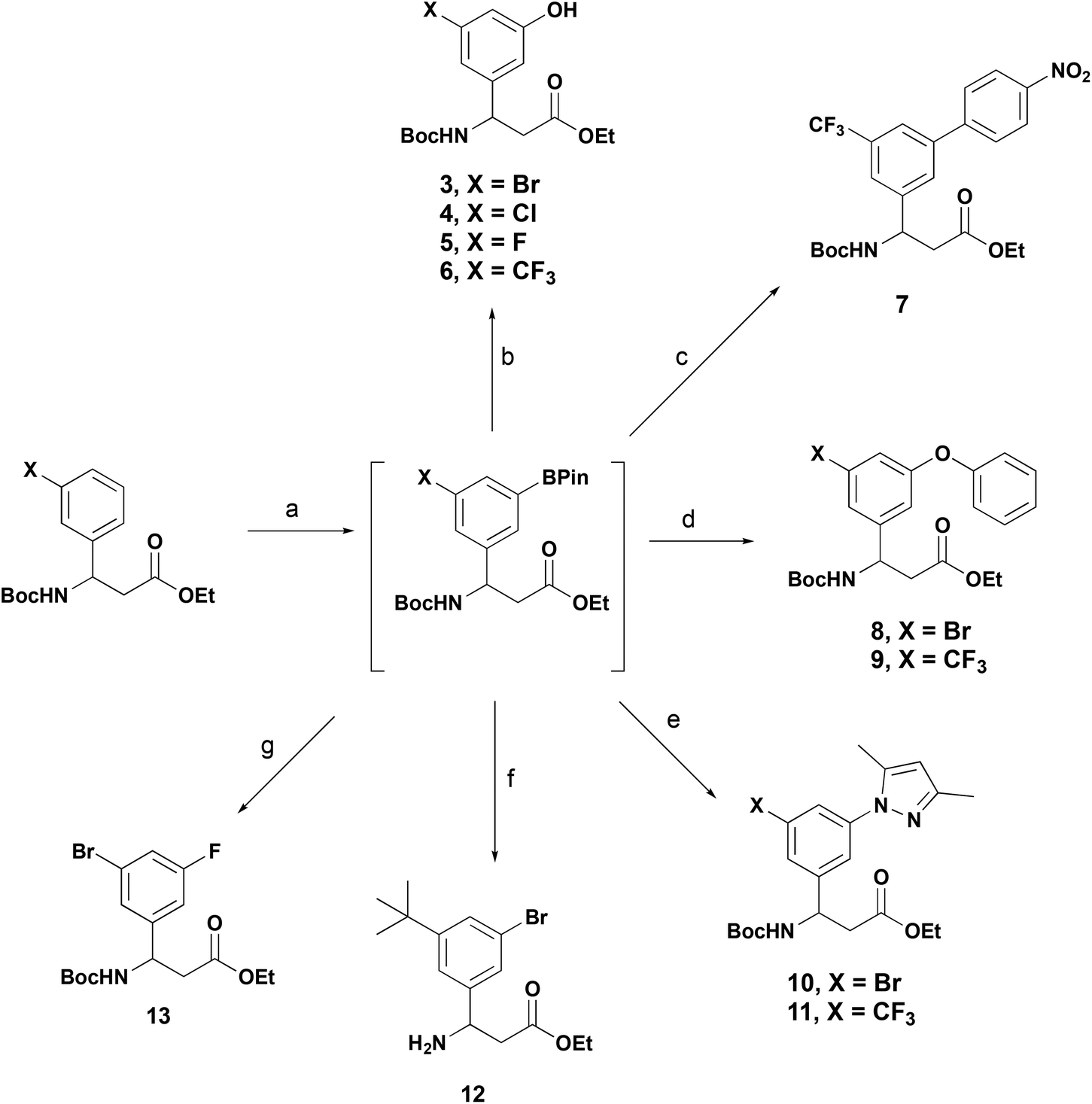 | ||
| Scheme 5 One-pot functionalisation of BPin-β-aryl-aminopropionic acids. Reagents and conditions: (a) Standard conditions Scheme 2; (b) Oxone®, aq. acetone, r.t., 0.75 h, 3 = 52%, 4 = 77%, 5 = 33%, 6 = 92% over two steps; (c) 1-iodo-4-nitrobenzene, Pd(dppf)·CH2Cl2, K3PO4, 1,2-DME:H2O, 70 °C, 4.5 h, 93% over two steps; (d) phenol, CuOAc2, B(OH)3, 4 Å molecular sieves, 70 °C, 16 h, 8 = 36%, 9 = 34% over two steps; (e) 1,4-dimethylpyrazole, Cu(OAc)2, B(OH)3, 4 Å molecular sieves, 80 °C, 16 h, 10 = 74%, 11 = 68% over two steps; (f) CuBr2, MeOH:H2O (2:1), 70 °C, 16 h, 39%; (g) i. AgOTf, NaOH, MeOH, 0 °C, 0.5 h; ii. Selectfluor®, 3 Å MS, acetone, r.t., 3 h, 59%. | ||
While tert-butoxycarbonyl groups present a robust, stable protecting group for the substrates, further N-functionalisation requires deprotection before further reaction. To demonstrate the utility of the C–H borylation in peptidomimetics, we replaced the tert-butoxycarbonyl group with an α-amino acid (Scheme 6). Under the established C–H borylation conditions the amino acid derived substrates 14a–c were converted to the corresponding dipeptide boronates 15a–c in good yield showing the tolerance of the conditions to more complex conditions with additional hydrogen bond donors.
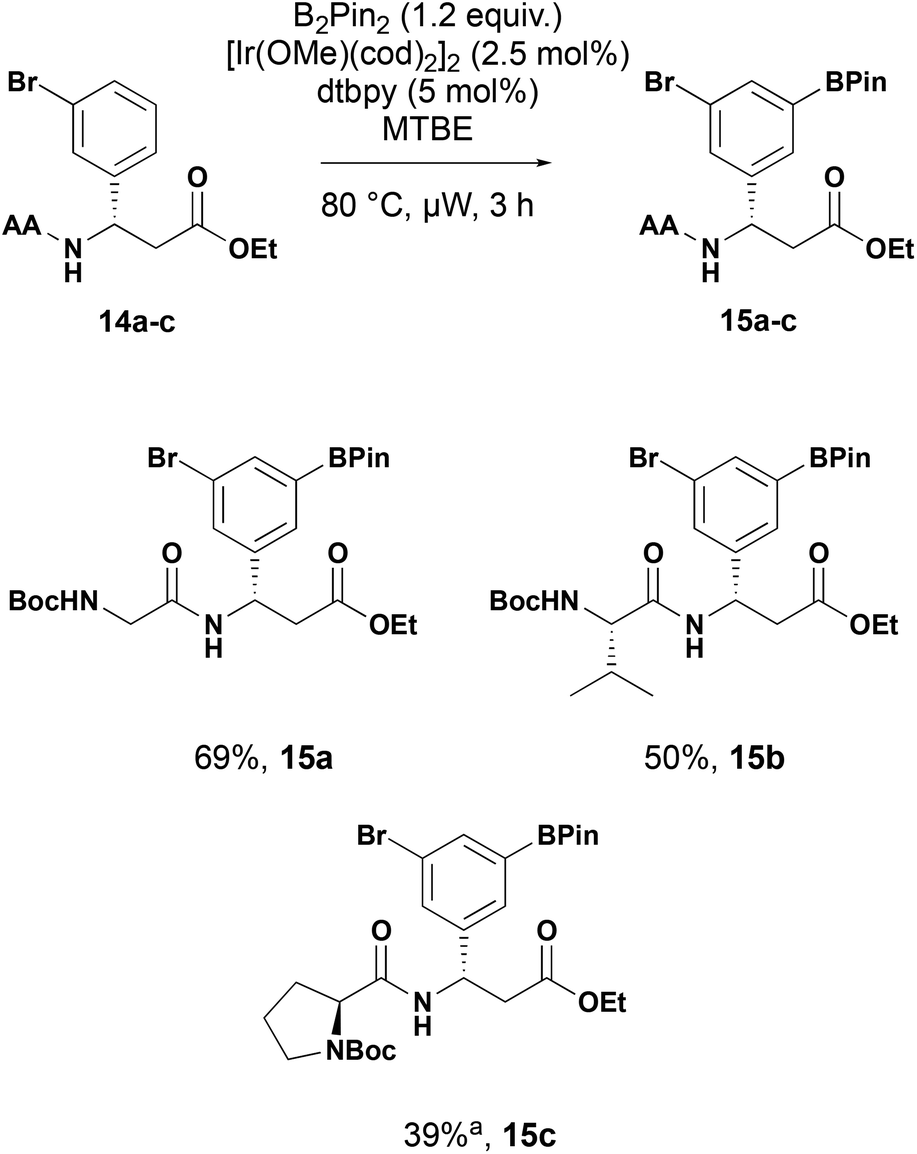 | ||
| Scheme 6 Iridium-catalysed C–H borylation of β-aryl-aminopropionic dipeptides. AA = amino acid. aExtended reaction time of 6 h. | ||
Finally, we used this C–H borylation strategy to prepare a reported selective integrin antagonist.27,28 Utilising the C–H borylation strategy to prepare 3-bromo-5-tert-butyl derivative 12 (Scheme 5) we converted this through to dipeptide 16 by amide coupling with Boc-glycine in 50% yield (Scheme 7). Further acidic deprotection and coupling with 3-guanidinobenzoic acid and final ester hydrolysis provided the integrin antagonist 17. While comparable in step count to the reported, this approach allows installation of the stereocenter by Reformatsky addition of ethyl bromozincacetate to the chiral sulfinamide, obviating a chiral HPLC separation as previously reported. The reported synthetic procedures for similar compounds require a bespoke aldehyde synthesis for each target compound, whereas a C–H borylation strategy allows a more sustainable synthesis of similar analogs by functionalising a common intermediate.
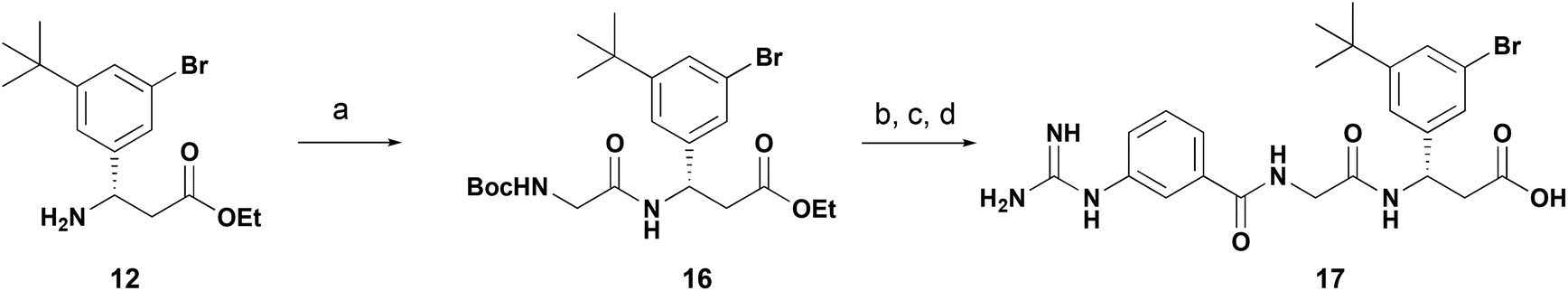 | ||
| Scheme 7 Application of C–H borylation chemistry in the synthesis of an integrin antagonist. Reagents and conditions: (a) Boc-glycine, HATU, iPr2EtN, MeCN, 0 °C to r.t., 16 h, 50%; (b) 4 M HCl in dioxane, 5 h; (c) 3-guanidinobenzoic acid, diisopropylcarbodiimide, HOBt, CH2Cl2:DMF (1:1), r.t., 64 h; (c) 1 M aq. LiOH, THF, r.t., 64 h, 52% over three steps. | ||
In conclusion, we have developed an efficient iridium-catalysed C–H borylation procedure of β-aryl- and heteroaryl-aminopropionic acids and explored the scope of the procedure with respect to functional group tolerance and retention of enantiointegrity. We have further explored the C–H borylation procedure in dipeptide substrates further demonstrating its synthetic utility. By applying telescoped “one-pot” reaction conditions we have shown that the C–H borylation approach allows an effective route to diversely functionalised β-aryl-aminopropionic acids across chemical space relevant to medicinal chemistry.
Funding
We thank GlaxoSmithKline and the University of Nottingham for providing funds for consumables associated with this work, including a part-studentship for H. Robinson.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank Thomas McInally and Prof. Christopher J. Moody for helpful discussions. Determination of chiral purity was undertaken by Reach Separations, Bio City, Nottingham, UK.References
- M. A. T. Blaskovich, J. Med. Chem., 2016, 59, 10807–10836 CrossRef CAS PubMed.
- M. Kashif, K. F. Chacón-Vargas, J. C. López-Cedillo, B. Nogueda-Torres, A. D. Paz-González, E. Ramírez-Moreno, R. Agusti, M. L. Uhrig, A. Reyes-Arellano, J. Peralta-Cruz, M. Ashfaq and G. Rivera, Eur. J. Med. Chem., 2018, 156, 252–268 CrossRef CAS PubMed.
- Y. Guo, Y. Zhao, G. Wang, Y. Chen, Y. Jiang, L. Ouyang and B. Liu, Eur. J. Med. Chem., 2018, 143, 402–418 CrossRef CAS PubMed.
- J. H. Hutchinson, W. Halczenko, K. M. Brashear, M. J. Breslin, P. J. Coleman, L. T. Duong, C. Fernandez-Metzler, M. A. Gentile, J. E. Fisher, G. D. Hartman, J. R. Huff, D. B. Kimmel, C.-T. Leu, R. S. Meissner, K. Merkle, R. Nagy, B. Pennypacker, J. J. Perkins, T. Prueksaritanont, G. A. Rodan, S. L. Varga, G. A. Wesolowski, A. E. Zartman, S. B. Rodan and M. E. Duggan, J. Med. Chem., 2003, 46, 4790–4798 CrossRef CAS PubMed.
- A. Trabocchi, G. Menchi, N. Cini, F. Bianchini, S. Raspanti, A. Bottoncetti, A. Pupi, L. Calorini and A. Guarna, J. Med. Chem., 2010, 53, 7119–7128 CrossRef CAS PubMed.
- J. Adams, E. C. Anderson, E. E. Blackham, Y. W. R. Chiu, T. Clarke, N. Eccles, L. A. Gill, J. J. Haye, H. T. Haywood, C. R. Hoenig, M. Kausas, J. Le, H. L. Russell, C. Smedley, W. J. Tipping, T. Tongue, C. C. Wood, J. Yeung, J. E. Rowedder, M. J. Fray, T. McInally and S. J. F. Macdonald, ACS Med. Chem. Lett., 2014, 5, 1207–1212 CrossRef CAS PubMed.
- S. J. Haycock-Lewandowski, A. Wilder and J. Åhman, Org. Process Res. Dev., 2008, 12, 1094–1103 CrossRef CAS.
- W. M. Rodionow, J. Am. Chem. Soc., 1929, 51, 847–852 CrossRef.
- W. M. Rodionow and E. A. Postovskaja, J. Am. Chem. Soc., 1929, 51, 841–847 CrossRef.
- C. Y. K. Tan and D. F. Weaver, Tetrahedron, 2002, 58, 7449–7461 CrossRef CAS.
- K. Brinner, B. Doughan and D. J. Poon, Synlett, 2009, 991–993 CrossRef CAS.
- S. G. Davies, A. D. Smith and P. D. Price, Tetrahedron: Asymmetry, 2005, 16, 2833–2891 CrossRef CAS.
- S. G. Davies, A. M. Fletcher, P. M. Roberts and J. E. Thomson, Tetrahedron: Asymmetry, 2012, 23, 1111–1153 CrossRef CAS.
- S. G. Davies, A. M. Fletcher, P. M. Roberts and J. E. Thomson, Tetrahedron: Asymmetry, 2017, 28, 1842–1868 CrossRef CAS.
- T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390–391 CrossRef CAS PubMed.
- R. E. Maleczka, F. Shi, D. Holmes and M. R. Smith, J. Am. Chem. Soc., 2003, 125, 7792–7793 CrossRef CAS PubMed.
- C. Sandford and V. K. Aggarwal, Chem. Commun., 2017, 53, 5481–5494 RSC.
- J. W. B. Fyfe and A. J. B. Watson, Chem, 2017, 3, 31–55 CAS.
- S. M. Nicolle, A. Nortcliffe, H. E. Bartrum, W. Lewis, C. J. Hayes and C. J. Moody, Chem. – Eur. J., 2017, 13623–13627 CrossRef CAS PubMed.
- F.-M. Meyer, S. Liras, A. Guzman-Perez, C. Perreault, J. Bian and K. James, Org. Lett., 2010, 12, 3870–3873 CrossRef CAS PubMed.
- V. A. Kallepalli, F. Shi, S. Paul, E. N. Onyeozili, R. E. Maleczka and M. R. Smith, J. Org. Chem., 2009, 74, 9199–9201 CrossRef CAS PubMed.
- S. M. Preshlock, B. Ghaffari, P. E. Maligres, S. W. Krska, R. E. Maleczka and M. R. Smith, J. Am. Chem. Soc., 2013, 135, 7572–7582 CrossRef CAS PubMed.
- T. Ishiyama, Y. Nobuta, J. F. Hartwig and N. Miyaura, Chem. Commun., 2003, 2924–2925 RSC.
- T. E. Hurst, T. K. Macklin, M. Becker, E. Hartmann, W. Kügel, J.-C. Parisienne-La Salle, A. S. Batsanov, T. B. Marder and V. Snieckus, Chem. – Eur. J., 2010, 16, 8155–8161 CrossRef CAS PubMed.
- J. C. Vantourout, H. N. Miras, A. Isidro-Llobet, S. Sproules and A. J. B. Watson, J. Am. Chem. Soc., 2017, 139, 4769–4779 CrossRef CAS PubMed.
- H. Robinson, S. A. Oatley, J. E. Rowedder, P. Slade, S. J. F. Macdonald, S. P. Argent, J. D. Hirst, T. McInally and C. J. Moody, Chem. – Eur. J., 2020, 26, 7678–7684 CrossRef CAS PubMed.
- P. G. Ruminski and D. W. Griggs, WO2017117538, 2009.
- P. G. Ruminski and D. W. Griggs, WO2014015054, 2014.
- T. Furuya and T. Ritter, Org. Lett., 2009, 11, 2860–2863 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra and chiral HPLC chromatograms. See DOI: 10.1039/d0ob01495h |
| ‡ J. S. Deceased December 2018. |
| This journal is © The Royal Society of Chemistry 2020 |