
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
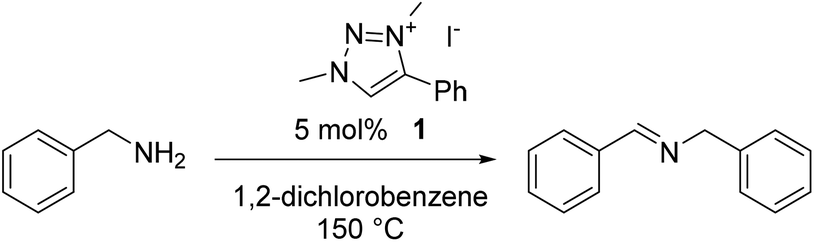
Anion–cation synergistic metal-free catalytic oxidative homocoupling of benzylamines by triazolium iodide salts†
Joseph P.
Byrne‡
 and
Martin
Albrecht
*
and
Martin
Albrecht
*
Department of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland. E-mail: martin.albrecht@dcb.unibe.ch
First published on 4th September 2020
Abstract
Triazolium iodide salts are excellent catalysts for the selective oxidative coupling of benzylamines to yield imines. This metal-free reaction proceeds in quantitative spectroscopic yields when run in refluxing 1,2-dichlorobenzene and open to the air. No catalytic activity was observed with related triazolium tetrafluoroborate salts. Variation of catalyst and reaction atmosphere provides mechanistic insights, and revealed dioxygen as the terminal oxidant and the iodine/iodide couple as key redox component in the catalytic dehydrogenation pathway. While molecular iodine is competent as a catalyst in its own right, the triazolium cation triples the reaction rate and reaches turnover frequencies up to 30 h−1, presumably through beneficial interactions of the electron-poor azolium π system and I2, which facilitate the electron transfer from the substrate to iodine and concomitant formation of I−. This acceleration is specific for triazolium cations and represents a hybrid anion/cation catalytic process as a simple and straightforward route towards imine products, with economic advantages over previously reported metal-based catalytic systems.
Introduction
Imines are an important class of compounds that serve as building blocks in fine chemicals synthesis, pharmaceuticals and biologically active molecules.1 Traditionally, imines, or Schiff bases, have been prepared by condensation of aldehydes and amines, often employing dehydrating agents to prevent the backward reaction, as well as Lewis acid catalysts. The instability of many aldehydes and the inherent impossibility of intermolecular homocoupling of a single reagent are limitations of this approach and significant research into alternative methods to access this class of molecules have been reported.1,2Recently, increasing interest has focussed on oxidative coupling of amines and alcohols to form imine intermediates, with particular interest directed towards the oxidation and coupling of amines.3 This process typically relies on the use of metal catalysts. Noble metals have been employed to catalyze these reactions, including bulk Au, supported Au and HAuCl4·3H2O/CeO2 nanoparticles,4,5 Pd coordination compounds,6 and Ru(OH)x/TiO2 systems.7 Research from our lab and others has shown that Ru and Ir complexes of triazolylidene N-heterocyclic carbene (NHC) complexes to catalyze dehydrogenative amine homocoupling, when reacted at 150 °C in toluene or 1,2-dichlorobenzene.8–10 In an effort to lower costs and increase sustainability of the process, recent work has focussed on the use of non-noble metals such as Cu,11–14 Mn,15,16 and Fe,17,18 with impressive results.
Strategies for conversion of amines to imines without involving metal-based catalysts offer benefits in terms of cost and purification.1 To this end, diverse purely organic systems have been reported for this transformation, such as bleach,19 catalytic AIBN,20 graphene oxide,21 bio-inspired ortho-quinone organocatalysts,22 and salicylic acid derivatives,23 the latter of which were also used to construct benzimidazoles. Photocatalytic reactions have been reported with WS2 nanosheets24 and truxene-based polymers.25 Molten ionic liquids like (NBu4)Br have also recently been reported to mediate this transformation, when used as a solvent.26
Based on our interest in triazolium salts as ligand precursors to 1,2,3-triazolylidene complexes,27–31 and inspired by the activity of some of the ionic liquids for amine homocoupling,32 we decided to investigate the possibility for these organic salts to act as molecular catalysts for oxidative amine coupling. An attractive feature of triazolium salts is their versatility for incorporating (functionalized) substituents, since triazole precursors are conveniently prepared through Cu(I)-catalyzed azide–alkyne cycloaddition chemistry.33–38 Here we report an efficient amine oxidation protocol based on a dual catalysis system consisting of triazolium cations and iodide as redox-active counteranion, with synergistic efficiencies that surpass by far the most active organic catalyst for this oxidative transformation,23 even when carried out under air, and allowing for a shorter timescale for quantitative yields.
Results and discussion
Model reaction and controls
The catalytic oxidative dehydrogenation of benzyl amine was chosen as a model reaction for this study (Table 1). 1,2,3-Triazolium iodide salt 1, with methyl and phenyl substituents, was first explored as a catalyst for this reaction, in refluxing 1,2-dichlorobenzene. These conditions have previously been used for this transformation when catalyzed by precious metal complexes.8–10 With a loading of 5 mol% of 1, selective formation of the imine product in quantitative spectroscopic yields was observed within 3 h (entry 1), with no other side-products detected by 1H NMR analysis of the reaction mixture (Fig. S13†). No reaction was observed over the same timescale in the absence of triazolium salt (entry 2). Lowering the temperature to 115 °C or refluxing in lower-boiling solvents (toluene, 1,2-dichloroethane) resulted in no discernible reaction progress (Table S1†).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Additive | Yieldb (%) | ||||
| mol% | mol% | 1 h | 3 h | 4 h | |||
| a General conditions: Benzyl amine (0.25 mmol), triazolium salt 1 (5 mol%) and hexamethylbenzene (internal standard), 1,2-dichlorobenzene (2 mL), 150 °C. b Spectroscopic yield determined by 1H NMR analysis with respect to hexamethylbenzene. c 99% after 6 h. d 6% benzaldehyde formed in addition. | |||||||
| 1 | 1 | 5 | — | — | 60 | 100 | 99 |
| 2 | — | 0 | — | — | 0 | <2 | <2 |
| 3 | 1 | 2.5 | — | — | 33 | 79 | 94 |
| 4 | 1 | 0.5 | — | — | 24 | 60 | 75c |
| 5 | 1 | 5 | H2O | 1000 | 51 | 93 | 88d |
| 6 | 1 | 5 | H2O | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
0 | 0 | 0 |
| 7 | 1 | 5 | KOtBu | 5 | 47 | 93 | 98 |
| 8 | 1 | 5 | TsOH | 5 | 47 | 91 | 96 |
Decreasing catalyst loading to 2.5 mol% gave 94% yield after 4 h, with the rate of reaction approximately halved in line with the decrease in substrate/catalyst ratio. A low catalyst loading of 0.5 mol% also gave quantitative yields, albeit after 6 h (entries 3 and 4; Fig. S1†). The robustness of the catalytic species derived from 1 was tested by performing the reaction with only 0.05 mol% triazolium salt. After 16 h a maximum yield of 36% was reached, indicating a catalyst turnover number of approximately 740 under these conditions.
No special precautions were taken to dry 1,2-dichlorobenzene for these reactions in order to achieve selective imine formation. Indeed, deliberate addition of 10 equivalents of H2O did not significantly slow the transformation (entry 5), although, notably, a drop in yield occurred towards the end of the reaction (at 4 h) and a corresponding amount of benzaldehyde was observed by 1H NMR analysis, indicating that the presence of water leads to hydrolysis of the imine product. Increasing the water content tenfold completely inhibited catalysis (entry 6). Neither base nor acid additives were beneficial, as neither stoichiometric KtOBu nor TsOH accelerated conversion (entries 7 and 8). Notably, with either of the additives, quantitative yields of the imine product were still achieved after 4 h.
Different benzylamines with electronically active para-substituents were successfully oxidized with triazolium salt 1 (Table 2, entries 1–4). No specific trend correlating the Hammett parameter σp and catalytic activity was noted. Aliphatic n-butylamine was not oxidized to an imine under these conditions, demonstrating a rather limitated substrate scope for dehydrogenation. Nonetheless the possibility to convert benzylic amines selectively offers attractive synthetic opportunities.
a
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | σ para | Yieldb (%) | TOF25 (h−1) | |||
| R1 | R2 | 1 h | 3 h | 4 h | |||
| a General conditions: Benzyl amine (0.25 mmol), triazolium salt 1 (5 mol%), hexamethylbenzene (internal standard), 1,2-dichlorobenzene (2 mL), 150 °C. b Spectroscopic yield determined by 1H NMR analysis with respect to hexamethylbenzene. c 8% aldehyde formed in addition. d Acetophenone formed in addition (9% after 1 h, 20% after 3 h, 26% after 4 h; total conversion after 4 h is 59%). | |||||||
| 1 | OMe | H | −0.27 | 32 | 97 | 92c | 6 |
| 2 | H | H | +0.00 | 60 | 100 | 99 | 16 |
| 3 | F | H | +0.06 | 19 | 71 | 91 | 4 |
| 4 | CF3 | H | +0.54 | 65 | 98 | 100 | 21 |
| 5 | H | CH3 | +0.00 | 26d | 35d | 33d | 5 |

Catalysts for oxidative coupling of amines are often applied to alcohol oxidation,1,10,39 however 1 was specific for amines as benzyl alcohol was not converted at all. Reaction of a 1:1 mixture of benzyl alcohol and benzylamine led to exclusive formation of the same imine product as is formed in the presence of benzylamine only (>98% yield after 4 h), while benzyl alcohol remained unreacted and was still present in the final reaction mixture after 6 h. Finally, branched primary amine α-methylbenzylamine was also employed as a substrate for the homocoupling reaction (entry 5), but this transformation was less selective. While complete conversion occurred, only 33% yield of imine was seen within 4 h in addition to acetophenone (26%) and other unidentified products.
Catalyst variation
In order to better understand this new catalytic system and identify its relevant components, a number of related species were tested for their catalytic activity. Variation of the wingtip groups of triazolium iodide catalyst 1 was investigated through use of salts 2–4 (Table 3, entries 2–4), which have been reported previously for applications including as ionic liquids32 and precursors to 1,2,3-triazolylidene organometallic ligands.27–29 All these triazolium iodide salts efficiently catalyze the oxidative homocoupling. Salts 2 and 4 displayed activities comparable to 1 (TOF50 = 9, 9 and 13 h−1, respectively) and give quantitative spectroscopic yields within 3 h, while 3 required 6 h to reach completion (TOF50 = 5 h−1). Bearing in mind the similarities between imidazolium and triazolium salts, 1,3-dimethylimidazolium iodide 5 was also assessed as a catalyst (entry 5) and did demonstrate activity. However, the imidazolium salt was a poorer catalyst than any of the triazolium salts, reaching only 19% spectroscopic yield after 3 h (TOF50 < 2). Considerably lower activity of 4 and, in particular, 5 strongly suggests that the azolium cation plays a relevant role in tuning the activity of catalysts for this dehydrogenative coupling.|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Heterocycle | R | R′ | X | Yieldb (%) | TOF50 (h−1) | |
| 1 h | 3 h | |||||||
| a General conditions as described in Table 1. b Spectroscopic yield determined by 1H NMR analysis with respect to hexamethylbenzene. c 96% after 6 h. d 48% after 6 h. | ||||||||
| 1 | 1 | Triazolium | Me | Ph | I | 60 | 100 | 13 |
| 2 | 2 | Triazolium | n Bu | n Bu | I | 45 | 99 | 9 |
| 3 | 3 | Triazolium | n Bu | Ph | I | 16 | 75c | 5 |
| 4 | 4 | Triazolium | n Bu | Mes | I | 47 | 100 | 9 |
| 5 | 5 | Imidazolium | Me | Me | I | 5 | 19d | <2 |
| 6 | 1′ | Triazolium | Me | Ph | BF4 | 0 | 0 | — |
| 7 | 2′ | Triazolium | n Bu | n Bu | BF4 | 0 | 0 | — |
| 8 | 3′ | Triazolium | n Bu | Ph | BF4 | 0 | 0 | — |
| 9 | 5′ | Imidazolium | Me | Me | BF4 | 0 | 0 | — |
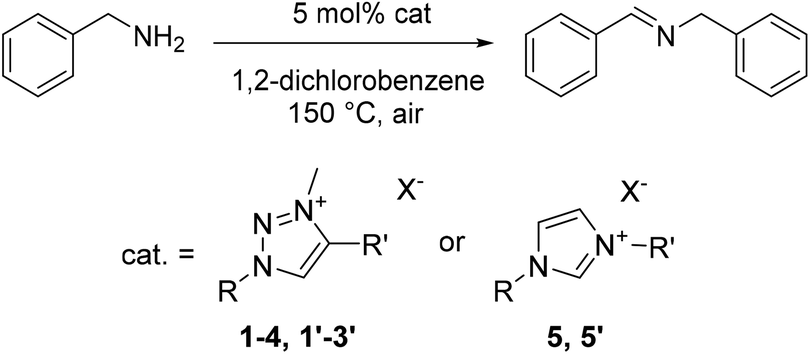
To evaluate potential interaction between the triazolium cation and the anion, triazolium tetrafluoroborate 1′ was synthesized by treating 1 with AgBF4. 1H NMR analysis (300 MHz, CDCl3) of 1′ showed only minor changes in the spectrum compared to 1, most diagnostically a significantly upfield shifted resonance at 8.65 ppm attributed to the triazolium CH (cf. δH 9.36 for 1).27 Such a resonance shift in the spectra of these azolium salts is indicative of less hydrogen-bonding counterions in solution.40
Both 1 and 1′ were further characterised by single crystal X-ray diffraction analysis (Fig. 1 and Table S4†). The molecular structure of iodide salt 1 displays weak non-classical interaction between the triazolium CH and iodide counterion (Fig. 1a), with a H⋯I distance of 2.857 Å. Similar interactions exist in other triazolium iodide structures,41,42 and molecular modelling of iodide anion binding by tetra-triazolium receptors by Beer and co-workers predicted binding distances on the same scale.43 There was also a potential anion–π interaction with a centroid⋯I distance of 3.710 Å. The structure of the tetrafluoroborate salt 1′ revealed the BF4− counterion disordered over two sites without a clear hydrogen bonding pattern (Fig. 1b), in agreement with solution analysis.
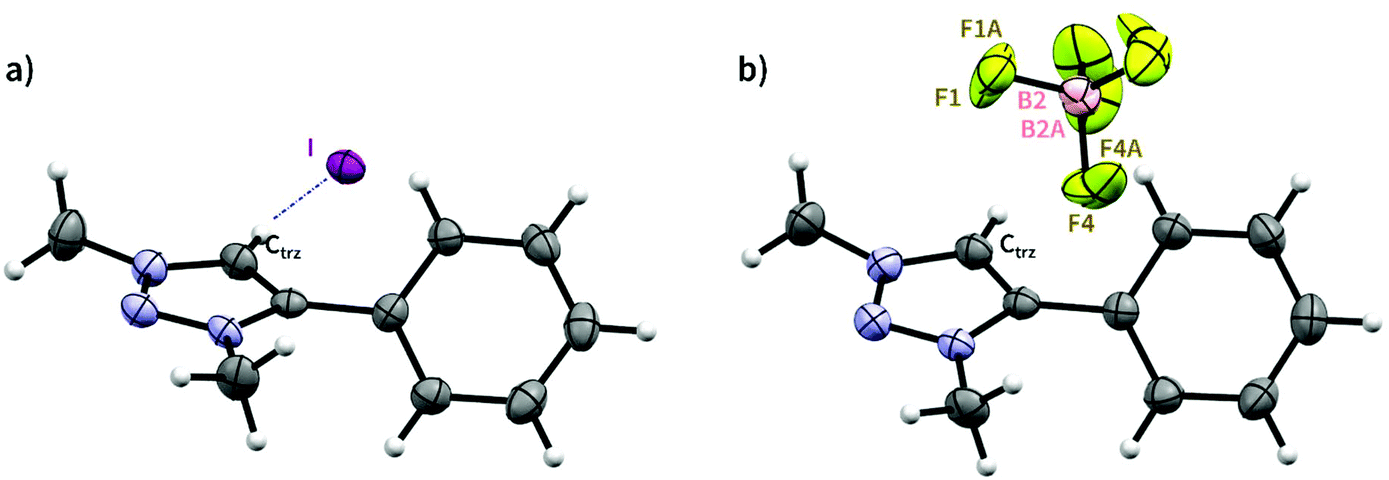 | ||
| Fig. 1 (a) Molecular structure of triazolium salt 1, determined by single crystal X-ray diffraction analysis. Selected bond lengths (Å) and angles (°): |Ctrz⋯I| = 3.783(3), |Htrz⋯I| = 2.857, |∠Ctrz–Htrz⋯I| = 173.69. (b) Molecular structure of triazolium salt 1′, determined by single crystal X-ray diffraction analysis. BF4 anion is disordered over two sites B2 (65.2%) and B2A (34.8%). | ||
This tetrafluoroborate analogue allowed the role of the anion in the catalysis to be probed. In contrast to the triazolium iodide 1, the tetrafluoroborate analogue 1′ was catalytically completely inactive under the same conditions (Table 3, entry 6). This inactivity was also observed with tetrafluoroborate analogues 2′, 3′30 and 5′ (entries 7–9), pointing towards a critical role of the iodide anion in catalysis.
To further explore the role of cations and anions for catalysing this transformation, various other salts were evaluated (Table S2†). Ionic liquid (Bu4N)Br, previously reported to catalyze amine homocoupling of benzylamines when used as a solvent,26 gave negligible conversion when added in catalytic amounts. Similarly (H4N)Cl was inactive, while (Et4N)I gave modest yields and reached nearly 40% after 4 h, a level of activity similarly to imidazolium salt 5 and indicative for an essential role of iodide for inducing catalytic activity. In agreement with this model, KI gave comparable yields, which implies a notable 8 turnover numbers (TONs) for this simple salt. While active, these yields are significantly lower than those of the triazolium iodides, which suggests a prominent role of the cation for inducing high conversion. In line with this notion, experiments with KI in the presence of 5 mol% of 18-crown-6 to passivate the cation quenched activity completely.
Mechanistic insight
From the catalytic reactions described above, it is possible to gain some insight into mechanistic aspects of this process, and rule out certain pathways. For instance, involvement of an N-heterocylic carbene as catalytically active species is unlikely, since addition of stoichiometric KtOBu to triazolium salt 1 induced slower conversion (cf. Table 1, entry 7), even though this base promotes the formation of free carbenes.44 Although NHCs generated in situ have been reported as organocatalysts for various transformations,45–48 a similar reaction pathway is evidently not occurring here.Instead, iodide plays an essential role, which may be related to its unique redox properties and the low potential to form molecular iodine, I2.49 To probe this hypothesis, reactions were performed under deliberately oxidizing or reducing conditions. Thus, when the reaction was performed in deaerated solvent and under inert atmosphere, significantly slower conversion rates were observed (Table 4, entry 1 vs. 2), while saturation with O2 accelerates amine oxidation when compared to standard conditions in air (entry 3, TOF50 ∼ 30 h−1vs. 13 h−1). Dependence on dioxygen as an oxidant for amine homocoupling has previously been observed in ionic liquid solvents,26 as well as with photocatalytic systems.24,25 Carrying out the reaction in the absence of light had no notable impact on catalyst activity (entry 4), discounting the possibility of photocatalysis occurring under these conditions.
| Entry | Catalyst | Atmosphereb | Yieldc (%) | TOF50 (h−1) | ||
|---|---|---|---|---|---|---|
| 1 h | 3 h | 4 h | ||||
| a General conditions: Benzyl amine (0.25 mmol), catalyst (5 mol%), hexamethylbenzene (internal standard), 1,2-dichlorobenzene (2 mL), 150 °C. b Solution saturated with the corresponding gas (N2 or O2) for 10 min before substrate addition. c Spectroscopic yield determined by 1H NMR analysis with respect to hexamethylbenzene. d Yield at 2 h. | ||||||
| 1 | 1 | Air | 60 | 100 | 99 | 13 |
| 2 | 1 | N2 | 0 | 19 | 32 | — |
| 3 | 1 | O2 | 96 | 97d | — | 30 |
| 4 | 1 | Air (darkness) | 52 | 97 | — | 11 |
| 5 | I2 | Air | 42 | 91 | 98 | 7 |
| 6 | I2 | N2 | 10 | 15 | 28 | — |
| 7 | 6 | Air | 57 | 100 | — | 11 |
| 8 | 6 | N2 | 13 | 22 | 36 | — |
| 9 | 1′ + I2 | Air | 87 | 100d | — | 17 |
| 10 | 1′ + I2 | N2 | 11 | 20 | 33 | — |
These results point to a potential catalytic activity of molecular iodine for this transformation. I2-Catalyzed and -mediated reactions are of increasing interest for organic chemistry, including e.g. oxidative couplings of 2-aminopyridines and 1,3-diones, and for synthesis of other heterocycles.13,50–52 Of relevance to this work, iodine was reported to produce imines as either proposed intermediates or low-yielding by-product upon oxidation of benzylamines to nitriles, indicating that high loadings of I2 (0.25–3 equivalents) induce transformations of these substrates.53,54 However, no efficient and selective intermolecular imine synthesis by related catalytic methods has been reported.
Indeed, when 5 mol% molecular iodine was reacted with benzyl amine under the conditions described above for triazolium iodide 1 (Table 4, entry 5), quantitative spectroscopic yields of the corresponding imine were obtained within 4 h. This activity is only slightly lower than with the triazolium iodide (7 vs. 13 h−1, cf. entry 1, though the mol% in iodide is doubled). Upon addition of the substrate benzylamine, the initially purple I2 solution in 1,2-dichlorobenzene underwent a characteristic color change to yellow, which may be attributed to an amine-induced reduction of iodine to iodide.55 The activity of I2 as a catalyst, as well as the key role of dioxygen described above, suggests that molecular iodine may indeed be on the catalytic cycle of this reaction. Interestingly, when catalysis is carried out with I2 (5 mol%) under inert atmosphere (entry 6), 10% of imine was observed after 1 h, indicating approximately one turnover achieved in the absence of O2. Notably, the triazolium iodide 1 is inactive under identical inert conditions.
We therefore propose the reaction mechanism shown in Scheme 1, consisting of a cascade redox process with as key process the presumably rate-limiting iodine-mediated oxidation of primary amine to the aldimine. Aldimine condensation with another equivalent of amine generates the final secondary imine, while the iodide by-product is regenerated under aerobic conditions back to iodine. The H2O2 by-product was not directly observed in the reaction mixture, but it is proposed that the well-known ability of iodide to catalyze H2O2 decomposition56 makes detection unlikely. Moreover, iodide-mediated peroxide detection strategies are based on the development of a diagnostic yellow color of the solution,57 which was observed to evolve when using 1 as catalyst (vide supra). The accelerating role of the triazolium salt has been tentatively attributed to potential interactions between the electron-poor azolium ring and molecular iodine, which induces polarisation of the I–I bond and facilitates the reduction to I−.52,58 The concomitant benzylamine C–H activation is postulated to be the rate-determining step and accordingly, activation of I2 through polarisation increases the observed reaction rate. A further beneficial role of the azolium cation may also be imparted by the stabilization of the iodide through hydrogen bonding or anion⋯π interactions.35,59,60 This amine oxidation may therefore be considered as an anion–cation dual catalytic process, where the cation assists the redox process of the anion, which in turn induces substrate oxidation.
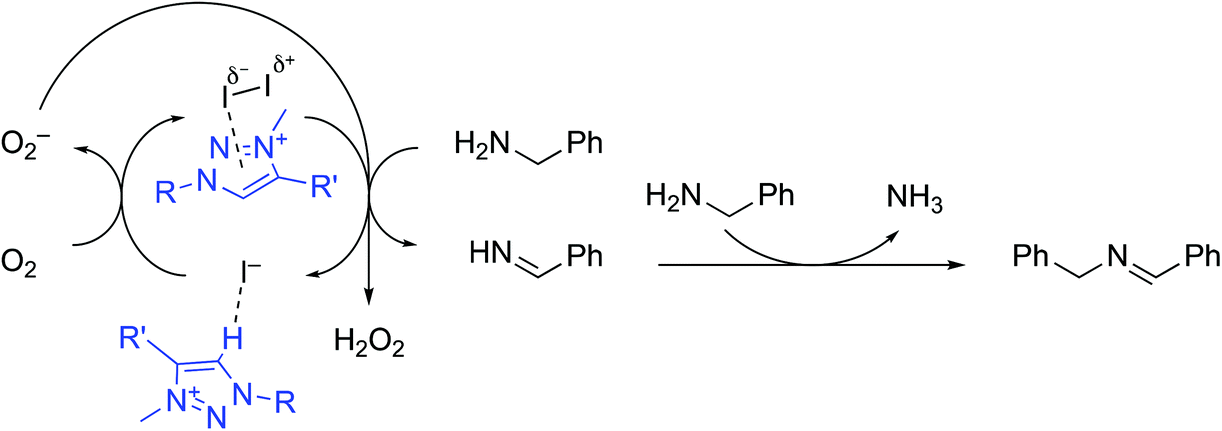 | ||
| Scheme 1 Proposed reaction pathway and role of the azolium salt and iodine in the amine oxidation. | ||
To evaluate the possible involvement of a triiodide anion due to the presence of both iodide and iodine,49,61 the triazolium triiodide salt 6 was prepared by mixing an equimolar ratio of 1 and I2 in solution (Scheme 2). In CDCl3, this reaction led to an immediate color change from yellow to orange and the triazolyl proton resonance shifted about 1 ppm upfield to δH = 8.55 in the 1H NMR spectrum, indicative of weaker C–H⋯X hydrogen bonding upon transforming I− to I3− (Fig. S8†). Compound 6 crystallized upon slow evaporation and the single crystals were analyzed by X-ray diffraction (Fig. S11†). The use of this triiodide salt 6 as precatalyst gave identical results, under air as the iodide salt 1 (Table 4, entry 7, TOF50 11 vs. 13 h−1). Under inert conditions compound 6 achieved 13% yield after 1 h (entry 8), again suggesting one turnover in the absence of dioxygen. This behavior is reminiscent of that of I2. Finally, when the inactive triazolium tetrafluoroborate 1′ was used in combination with I2, excellent catalytic activity was observed that was far superior to I2 alone (entry 9, TOF50 17 vs. 7 h−1 in air), and in fact even more active than 1 or 6 under aerobic conditions. Again, under inert atmosphere (entry 10), only a single turnover was observed in the first hour of reaction. These data support the mechanistic model with I2 as the catalytically active species for oxidation of the amine, and the triazolium cation playing a crucial role in accelerating turnover frequency.
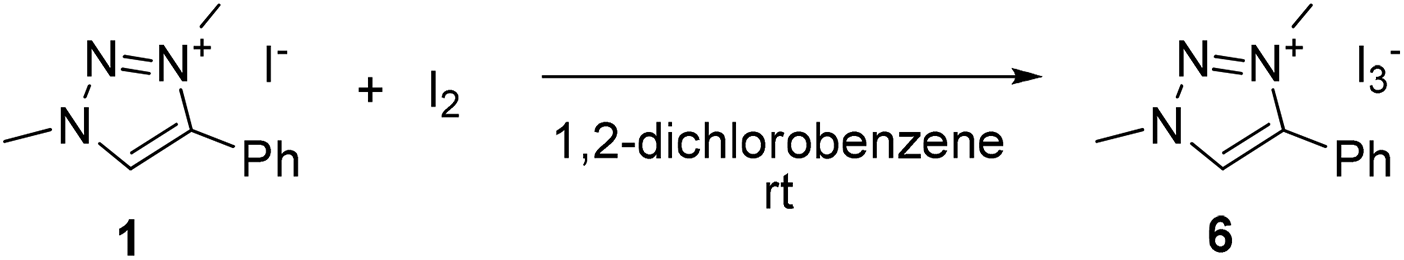 | ||
| Scheme 2 Synthesis of triiodide salt 6. | ||
Conclusions
We report a novel metal-free approach to the oxidative homocoupling of benzylic amines catalyzed by triazolium iodide salts, selectively giving imine products in quantitative yields. Turnover frequencies of up to 13 h−1 were achieved with 1 under aerobic conditions and up to 30 h−1 under O2 atmosphere. Preliminary mechanistic investigations indicate critical roles of both the cation and the anion of the precatalyst, which has been attributed to an iodide/iodine couple for the actual oxidation, and a triazolium⋯I2 interaction for facile iodine reduction and concomitant amine oxidation. Moreover, the activity of the triazolium iodide catalysts is dependent on dioxygen as benign terminal oxidant. While the substrate scope is limited to benzylic amines, the simplicity of the system is appealing. This triazolium iodide/iodine catalysis constitutes a more cost-effective approach to amine oxidation than the more widely reported transition-metal based strategies. Notably, the synergistic effects of triazolium cations and iodide/iodine-mediated redox cataylsis may be of broader relevance and an advantageous new concept to other organocatalytic redox reactions such as the azapyrrole synthesis or Bayer–Villiger oxidation.Experimental section
General considerations
All compounds, unless otherwise stated, were obtained from commercial sources and used as provided. Triazolium salts 1,272,322′,283,293′,30 and 428 were prepared according to literature procedures, as were imidazolium salts 562 and 5′.63 NMR spectra were recorded on a Bruker spectrometer operating at room temperature. Chemical shifts (δ in ppm, J in Hz) were referenced to residual solvent resonances and are reported downfield from SiMe4. High resolution mass spectrometry was performed by the Analytical Research Services at University of Bern.
General catalytic procedure
Benzyl amine (27 μL, 0.25 mmol) and triazolium salt 1 (3.6 mg, 0.012 mmol, 5 mol%) were dissolved in 1,2-dichlorobenzene (2 mL) and heated at 150 °C in round-bottomed flasks fitted with water-cooled condensers. Hexamethylbenzene was added as internal standard and reaction progress monitored by 1H NMR analysis (Fig. S1–S5 and S13–S18†). For reactions under N2 atmosphere or O2 atmosphere, reaction was carried out in a sealed Schlenk tube, and the reaction mixture was saturated with the relevant gas for 10 minutes before addition of benzyl amine, and the mixture was kept under a balloon of gas.Synthesis of 1′
Triazolium salt 1 (0.100 g, 0.33 mmol) was dissolved in CH2Cl2 (20 mL). AgBF4 (0.070 g, 0.34 mmol) was added and the reaction mixture stirred at room temperature for 2 h. This was filtered through Celite to remove insoluble silver salts and concentrated under reduced pressure to yield 1′ as an off-white solid (0.080 g, 0.31 mmol, 92%). HRMS (ESI+): Calculated for C10H12N3m/z = 174.1026 [M − BF4]+. Found m/z = 174.1022; 1H NMR (300 MHz, CDCl3): δ = 8.65 (s, 1H, CtrzH), 7.51–7.65 (m, 5H, CPhH), 4.43 (s, 3H, N–CH3), 4.26 (s, 3H, C–CH3); 13C NMR (75 MHz, CDCl3): δ = 132.0, 129.9 129.8, 129.5 (4 × CPh), 128.9, 125.8 (2 × Ctrz), 40.5, 38.3 (2 × NCH3).Synthesis of 6in situ
Triazolium salt 1 (0.006 g, 0.02 mmol) and I2 (0.005 g, 0.02 mmol) were dissolved in CDCl3 (or 1,2-dichlorobenzene). 1H NMR (300 MHz, CDCl3): δ = 8.55 (s, 1H, triazolium CtrzH), 7.69–7.75 (m, 5H, CPhH), 4.57 (s, 3H, N–CH3), 4.37 (s, 3H, C–CH3). Single crystals suitable for X-ray diffraction analysis grew upon slow evaporation of this sample.Crystal-structure determination
Triazolium iodide 1 crystallized upon slow diffusion of diethyl ether into a saturated methanol solution of the compound. Triazolium tetrafluoroborate 1′ and triazolium triiodide 6 crystallized by slow evaporation of CDCl3 solutions. All crystals were mounted in air at ambient conditions. The measurements for 1 were made on an Oxford Diffraction SuperNova area-detector diffractometer64 using mirror optics monochromated Mo Kα radiation (λ = 0.71073 Å) and Al filtered.65 The unit cell constants and an orientation matrix for data collection were obtained from a least-squares refinement of the setting angles of reflections in the range 2.6° < θ < 27.7°. A total of 1402 frames were collected using ω scans, with 1.0 + 1.0 seconds exposure time, a rotation angle of 1.0° per frame, a crystal-detector distance of 65.0 mm, at T = 173(2) K. All measurements for 1′ and 6 were made on a RIGAKU Synergy S area-detector diffractometer64 using mirror optics monochromated Cu Kα radiation (λ = 1.54184 Å). The unit cell constants and an orientation matrix for data collection were obtained from a least-squares refinement of the setting angles of reflections in the range 5.6° < θ < 76.9° (for 1′) or 4.4° < θ < 76.4° (for 6). A total of 2578 frames (1′) or 3102 frames (6) were collected using ω scans, with exposure times of 0.05 and 0.18 s (1′) or 5 and 20 s (6), a rotation angle of 0.5° per frame, a crystal-detector distance of 34.0 mm, at T = 173(2) K. Data reduction for all three data sets was performed using the CrysAlisPro64 program. The intensities were corrected for Lorentz and polarization effects. For 1, a numerical absorption correction based on Gaussian integration over a multifaceted crystal model was applied. For 1′ and 6, the intensities were corrected for Lorentz and polarization effects, and an absorption correction based on the multi-scan method using SCALE3 ABSPACK in CrysAlisPro1 was applied. Data collection and refinement parameters are given in Table S3.† The structure was solved by direct methods using SHELXT,66 which revealed the positions of all non-hydrogen atoms, which were refined anisotropically. All H-atoms were placed in geometrically calculated positions and refined using a riding model where each H-atom was assigned a fixed isotropic displacement parameter; for 1 and 1′, this value was equal to 1.2Ueq of its parent atom (1.5Ueq for the methyl groups), while for 6 the value was equal to 1.5Ueq of its parent atom. In the structure of 1′, the BF4 anion is disordered about two orientations. The geometries of the two moieties were restrained to be similar. Its ADP's were restrained by the SHELX SIMU and RIGU instructions. Refinement of the structure was carried out on F2 using full-matrix least-squares procedures, which minimized the function ∑w(Fo2 − Fc2)2. The weighting scheme was based on counting statistics and included a factor to downweight the intense reflections. All calculations were performed using the SHELXL-2014/767 program in OLEX2.68 The crystal of 6 used for structure determination was twinned, and thus the integration of just the major domain was used. The quality of this single crystal structure determination is lowered by this fact. The results are sufficient for a discussion of the crystal packing but do not allow a discussion of bonding geometry. Further details are given in Table S4.† Crystallographic data for all structures have been deposited with the Cambridge Crystallographic Data Centre (CCDC) as supplementary publication numbers 2016764 (1), 2016765 (1′), and 2016766 (6).†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge generous support from the European Commission for a Marie Skłodowska-Curie Individual Fellowship to JPB (Grant 749549) and from the European Research Council (CoG 615653). We thank G. M. Ó Máille and D. Verhoeven for useful discussion and Pamela Nylund for technical assistance.References
- B. Chen, L. Wang and S. Gao, Recent Advances in Aerobic Oxidation of Alcohols and Amines to Imines, ACS Catal., 2015, 5, 5851–5876 CrossRef CAS.
- K. O. Marichev, S. A. Patil and A. Bugarin, Recent advances in the synthesis, structural diversity, and applications of mesoionic 1,2,3-triazol-5-ylidene metal complexes, Tetrahedron, 2018, 74, 2523–2546 CrossRef CAS.
- R. D. Patil and S. Adimurthy, Catalytic Methods for Imine Synthesis, Asian J. Org. Chem., 2013, 2, 726–744 CrossRef CAS.
- B. Zhu, M. Lazar, B. G. Trewyn and R. J. Angelici, Aerobic oxidation of amines to imines catalyzed by bulk gold powder and by alumina-supported gold, J. Catal., 2008, 260, 1–6 CrossRef CAS.
- L. Aschwanden, T. Mallat, M. Maciejewski, F. Krumeich and A. Baiker, Development of a New Generation of Gold Catalysts for Amine Oxidation, ChemCatChem, 2010, 2, 666–673 CrossRef CAS.
- R. E. Rodríguez-Lugo, M. A. Chacón-Terán, S. De León, M. Vogt, A. J. Rosenthal and V. R. Landaeta, Synthesis, characterization and Pd(II)-coordination chemistry of the ligand tris(quinolin-8-yl)phosphite. Application in the catalytic aerobic oxidation of amines, Dalton Trans., 2018, 47, 2061–2072 RSC.
- J. W. Kim, J. He, K. Yamaguchi and N. Mizuno, Heterogeneously catalyzed one-pot synthesis of aldimines from primary alcohols and amines by supported ruthenium hydroxides, Chem. Lett., 2009, 38, 920–921 CrossRef CAS.
- A. Prades, E. Peris and M. Albrecht, Oxidations and Oxidative Couplings Catalyzed by Triazolylidene Ruthenium Complexes, Organometallics, 2011, 30, 1162–1167 CrossRef CAS.
- B. Bagh, A. M. McKinty, A. J. Lough and D. W. Stephan, 1,2,3-Triazolylidene ruthenium(II)(η6-arene) complexes: synthesis, metallation and reactivity, Dalton Trans., 2014, 43, 12842–12850 RSC.
- R. Pretorius, J. Olguín and M. Albrecht, Carbohydrate-Functionalized 1,2,3-Triazolylidene Complexes for Application in Base-Free Alcohol and Amine Oxidation, Inorg. Chem., 2017, 56, 12410–12420 CrossRef CAS.
- R. D. Patil and S. Adimurthy, Copper(0)-catalyzed aerobic oxidative synthesis of imines from amines under solvent-free conditions, RSC Adv., 2012, 2, 5119 RSC.
- Z. Hu and F. M. Kerton, Simple copper/TEMPO catalyzed aerobic dehydrogenation of benzylic amines and anilines, Org. Biomol. Chem., 2012, 10, 1618 RSC.
- B. Jiang, Y. Ning, W. Fan, S.-J. Tu and G. Li, Oxidative Dehydrogenative Couplings of Pyrazol-5-amines Selectively Forming Azopyrroles, J. Org. Chem., 2014, 79, 4018–4024 CrossRef CAS.
- T. Sonobe, K. Oisaki and M. Kanai, Catalytic aerobic production of imines en route to mild, green, and concise derivatizations of amines, Chem. Sci., 2012, 3, 3249–3255 RSC.
- A. Mukherjee, A. Nerush, G. Leitus, L. J. W. Shimon, Y. Ben David, N. A. Espinosa Jalapa and D. Milstein, Manganese-Catalyzed Environmentally Benign Dehydrogenative Coupling of Alcohols and Amines to Form Aldimines and H2: A Catalytic and Mechanistic Study, J. Am. Chem. Soc., 2016, 138, 4298–4301 CrossRef CAS.
- A. M. Meireles, V. S. da Silva, J. S. Rebouças and D. C. da Silva Martins, Unexpected Products of Benzylamine Oxidation Catalyzed by Manganese Porphyrins: Some Factors that Play a Critical Role for Imine Formation, ChemistrySelect, 2019, 4, 3275–3280 CrossRef CAS.
- R. I. Khusnutdinov, A. R. Baygusina and R. I. Aminov, Synthesis of N-benzylidenebenzylamine from benzylamine under the action of iron-containing catalysts in CCl4, Russ. J. Org. Chem., 2012, 48, 1059–1061 CrossRef CAS.
- G. Jaiswal, V. G. Landge, D. Jagadeesan and E. Balaraman, Sustainable iron-catalyzed direct imine formation by acceptorless dehydrogenative coupling of alcohols with amines, Green Chem., 2016, 18, 3232–3238 RSC.
- G. F. P. De Souza, T. W. Von Zuben and A. G. Salles, On Water Metal-Catalyst-Free Oxidative Coupling-Amidation of Amines to Access Imines and Amides, ACS Sustainable Chem. Eng., 2017, 5, 8439–8446 CrossRef CAS.
- L. Liu, Z. Wang, X. Fu and C. H. Yan, Azobisisobutyronitrile initiated aerobic oxidative transformation of amines: Coupling of primary amines and cyanation of tertiary amines, Org. Lett., 2012, 14, 5692–5695 CrossRef CAS.
- H. He, Z. Li, K. Li, G. Lei, X. Guan, G. Zhang, F. Zhang, X. Fan, W. Peng and Y. Li, Bifunctional Graphene-Based Metal-Free Catalysts for Oxidative Coupling of Amines, ACS Appl. Mater. Interfaces, 2019, 11, 31844–31850 CrossRef CAS.
- Y. Qin, L. Zhang, J. Lv, S. Luo and J.-P. Cheng, Bioinspired Organocatalytic Aerobic C–H Oxidation of Amines with an ortho -Quinone Catalyst, Org. Lett., 2015, 17, 1469–1472 CrossRef CAS.
- C. Dong, Y. Higashiura, K. Marui, S. Kumazawa, A. Nomoto, M. Ueshima and A. Ogawa, Metal-Free Oxidative Coupling of Benzylamines to Imines under an Oxygen Atmosphere Promoted Using Salicylic Acid Derivatives as Organocatalysts, ACS Omega, 2016, 1, 799–807 CrossRef CAS.
- F. Raza, J. H. Park, H. R. Lee, H. I. Kim, S. J. Jeon and J. H. Kim, Visible-Light-Driven Oxidative Coupling Reactions of Amines by Photoactive WS2 Nanosheets, ACS Catal., 2016, 6, 2754–2759 CrossRef CAS.
- V. R. Battula, H. Singh, S. Kumar, I. Bala, S. K. Pal and K. Kailasam, Natural Sunlight Driven Oxidative Homocoupling of Amines by a Truxene-Based Conjugated Microporous Polymer, ACS Catal., 2018, 8, 6751–6759 CrossRef CAS.
- A. Monopoli, P. Cotugno, F. Iannone, F. Ciminale, M. M. Dell'Anna, P. Mastrorilli and A. Nacci, Ionic-liquid-assisted metal-free oxidative coupling of amines to give imines, Eur. J. Org. Chem., 2014, 2014, 5925–5931 CrossRef CAS.
- D. Canseco-Gonzalez, A. Gniewek, M. Szulmanowicz, H. Müller-Bunz, A. M. Trzeciak and M. Albrecht, PEPPSI-Type Palladium Complexes Containing Basic 1,2,3-Triazolylidene Ligands and Their Role in Suzuki-Miyaura Catalysis, Chem. – Eur. J., 2012, 18, 6055–6062 CrossRef CAS.
- D. Canseco-Gonzalez and M. Albrecht, Wingtip substituents tailor the catalytic activity of ruthenium triazolylidene complexes in base-free alcohol oxidation, Dalton Trans., 2013, 42, 7424 RSC.
- A. Poulain, D. Canseco-Gonzalez, R. Hynes-Roche, H. Müller-Bunz, O. Schuster, H. Stoeckli-Evans, A. Neels and M. Albrecht, Synthesis and Tunability of Abnormal 1,2,3-Triazolylidene Palladium and Rhodium Complexes, Organometallics, 2011, 30, 1021–1029 CrossRef CAS.
- Y. Wei, A. Petronilho, H. Mueller-Bunz and M. Albrecht, Mesoionic Triazolylidene Nickel Complexes: Synthesis, Ligand Lability, and Catalytic C–C Bond Formation Activity, Organometallics, 2014, 33, 5834–5844 CrossRef CAS.
- J. P. Byrne, P. Musembi and M. Albrecht, Carbohydrate-functionalized N-heterocyclic carbene Ru(ii) complexes: synthesis, characterization and catalytic transfer hydrogenation activity, Dalton Trans., 2019, 48, 11838–11847 RSC.
- S. S. Khan, S. Hanelt and J. Liebscher, Versatile synthesis of 1, 2, 3-triazolium-based ionic liquids, ARKIVOC, 2009, 2009, 193 Search PubMed.
- H. C. Kolb and K. B. Sharpless, The growing impact of click chemistry on drug discovery, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS.
- V. D. Bock, H. Hiemstra and J. H. Van Maarseveen, Cu I-catalyzed alkyne-azide ‘click’ cycloadditions from a mechanistic and synthetic perspective, Eur. J. Org. Chem., 2006, 2006, 51–68 CrossRef.
- B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522–2571 RSC.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- E. Haldón, M. C. Nicasio and P. J. Pérez, Copper-catalysed azide-alkyne cycloadditions (CuAAC): An update, Org. Biomol. Chem., 2015, 13, 9528–9550 RSC.
- K. F. Donnelly, A. Petronilho and M. Albrecht, Application of 1,2,3-triazolylidenes as versatile NHC-type ligands: synthesis, properties, and application in catalysis and beyond, Chem. Commun., 2013, 49, 1145–1159 RSC.
- E. Zhang, H. Tian, S. Xu, X. Yu and Q. Xu, Iron-catalyzed direct synthesis of imines from amines or alcohols and amines via aerobic oxidative reactions under air, Org. Lett., 2013, 15, 2704–2707 CrossRef CAS.
- H. V. Huynh, T. T. Lam and H. T. T. Luong, Anion influences on reactivity and NMR spectroscopic features of NHC precursors, RSC Adv., 2018, 8, 34960–34966 RSC.
- D. Mendoza-Espinosa, R. González-Olvera, G. E. Negrón-Silva, D. Angeles-Beltrán, O. R. Suárez-Castillo, A. Álvarez-Hernández and R. Santillan, Phenoxy-Linked Mesoionic Triazol-5-ylidenes as Platforms for Multinuclear Transition Metal Complexes, Organometallics, 2015, 34, 4529–4542 CrossRef CAS.
- G. Dhimba, N. S. Lawal and M. D. Bala, New phenoxymethyl substituted mesoionic triazolium salts: Synthesis and structural characterisation, J. Mol. Struct., 2019, 1179, 100–107 CrossRef CAS.
- N. G. White, S. Carvalho, V. Félix and P. D. Beer, Anion binding in aqueous media by a tetra-triazolium macrocycle, Org. Biomol. Chem., 2012, 10, 6951 RSC.
- G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Crystalline 1H–1,2,3−Triazol–5−ylidenes: New Stable Mesoionic Carbenes (MICs), Angew. Chem., Int. Ed., 2010, 49, 4759–4762 CrossRef CAS.
- N. Marion, S. Díez-González and S. P. Nolan, N-Heterocyclic Carbenes as Organocatalysts, Angew. Chem., Int. Ed., 2007, 46, 2988–3000 CrossRef CAS.
- A. T. Biju, N. Kuhl and F. Glorius, Extending NHC-Catalysis: Coupling Aldehydes with Unconventional Reaction Partners, Acc. Chem. Res., 2011, 44, 1182–1195 CrossRef CAS.
- X. Bugaut and F. Glorius, Organocatalytic umpolung: N-heterocyclic carbenes and beyond, Chem. Soc. Rev., 2012, 41, 3511 RSC.
- S. Yetra, A. Patra and A. Biju, Recent Advances in the N-Heterocyclic Carbene (NHC)-Organocatalyzed Stetter Reaction and Related Chemistry, Synthesis, 2015, 47, 1357–1378 CrossRef CAS.
- F. Marken, in Encyclopedia of Electrochemistry, Wiley, 2007, pp. 273–302 Search PubMed.
- M. S. Yusubov and V. V. Zhdankin, Iodine catalysis: A green alternative to transition metals in organic chemistry and technology, Resour.-Effic. Technol., 2015, 1, 49–67 Search PubMed.
- J. Xie, H. Jiang, Y. Cheng and C. Zhu, Metal-free, organocatalytic cascade formation of C–N and C–O bonds through dual sp 3 C–H activation: oxidative synthesis of oxazole derivatives, Chem. Commun., 2012, 48, 979–981 RSC.
- M. Breugst and D. von der Heiden, Mechanisms in Iodine Catalysis, Chem. – Eur. J., 2018, 24, 9187–9199 CrossRef CAS.
- A. Goosen, C. W. McCleland and A. M. Sipamla, Reactions of Benzylamine and Selected Ring- and N-Substituted Derivatives with Iodine, Including a Synthesis for Benzonitriles, J. Chem. Res., 1995, 1, 18–19 Search PubMed.
- S. Iida and H. Togo, Direct and Facile Oxidative Conversion of Primary, Secondary, and Tertiary Amines to Their Corresponding Nitriles, Synlett, 2006, 2633–2635 CAS.
- J. C. Schug and M. J. Kogan, The nature of iodine-amine solutions, J. Magn. Reson., 1973, 11, 406–415 CAS.
- J. C. Hansen, The iodide-catalyzed decomposition of hydrogen peroxide: A simple computer-interfaced kinetics experiment for general chemistry, J. Chem. Educ., 1996, 73, 728–732 CrossRef CAS.
- A. Mátai and É. Hideg, A comparison of colorimetric assays detecting hydrogen peroxide in leaf extracts, Anal. Methods, 2017, 9, 2357–2360 RSC.
- C. Laurence, J. Graton, M. Berthelot and M. J. El Ghomari, The Diiodine Basicity Scale : Toward a General Halogen-Bond Basicity Scale, Chem. – Eur. J., 2011, 17, 10431–10444 CrossRef CAS.
- Z. Yacob and J. Liebscher, in Ionic Liquids - Classes and Properties, InTech, 2011 Search PubMed.
- A. Caballero, F. Zapata, L. González, P. Molina, I. Alkorta and J. Elguero, Discovery of anion-π interactions in the recognition mechanism of inorganic anions by 1,2,3-triazolium rings, Chem. Commun., 2014, 50, 4680–4682 RSC.
- R. L. Benoit, M. F. Wilson and S.-Y. Lam, Studies on the iodide–triiodide equilibrium, Can. J. Chem., 1977, 55, 792–797 CrossRef CAS.
- I. Dinarès, C. Garcia de Miguel, A. Ibáñez, N. Mesquida and E. Alcalde, Imidazolium ionic liquids: A simple anion exchange protocol, Green Chem., 2009, 11, 1507 RSC.
- M. Egashira, Y. Yamamoto, T. Fukutake, N. Yoshimoto and M. Morita, A novel method for preparation of imidazolium tetrafluoroborate ionic liquids, J. Fluor. Chem., 2006, 127, 1261–1264 CrossRef CAS.
- Oxford Diffraction. CrysAlisPro (Version 1.171.39.46), Oxford Diffraction Ltd., Yarnton, Oxfordshire, UK, 2018 Search PubMed.
- P. Macchi, H.-B. Bürgi, A. S. Chimpri, J. Hauser, Z. Gál and IUCr, Low-energy contamination of Mo microsource X-ray radiation: analysis and solution of the problem, J. Appl. Crystallogr., 2011, 44, 763–771 CrossRef CAS.
- G. M. Sheldrick, SHELXT - Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick and IUCr, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: A complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Catalytic and NMR spectroscopic data, crystallographic details. CCDC 2016764–2016766. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ob01472a |
| ‡ Present address: School of Chemistry, National University of Ireland Galway, University Road, Galway, Ireland. |
| This journal is © The Royal Society of Chemistry 2020 |