
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorinations of unsymmetrical diaryliodonium salts containing ortho-sidearms; influence of sidearm on selectivity†
Ahmed M. H.
Abudken
 ab,
Eric G.
Hope
a,
Kuldip
Singh
a and
Alison M.
Stuart
*a
ab,
Eric G.
Hope
a,
Kuldip
Singh
a and
Alison M.
Stuart
*a
aSchool of Chemistry, University of Leicester, Leicester, LE1 7RH, UK. E-mail: Alison.Stuart@le.ac.uk
bCollege of Pharmacy, Al-Qadisiyah University, Al-Qadisiyah, Iraq
First published on 24th July 2020
Abstract
Activated aromatics were reacted with two different fluoroidoane reagents 1 and 2 in the presence of triflic acid to prepare only the para-substituted diaryliodonium salts. With fluoroiodane 1 the unsymmetrical diaryliodonium salts contained an ortho-propan-2-ol sidearm, whereas the alcohol sidearm was eliminated to form an ortho-styrene sidearm in the reaction with fluoroiodane 2. Only the diaryliodonium salts containing a styrene sidearm were fluorinated successfully to deliver para-fluorinated aromatics in good yields.
Introduction
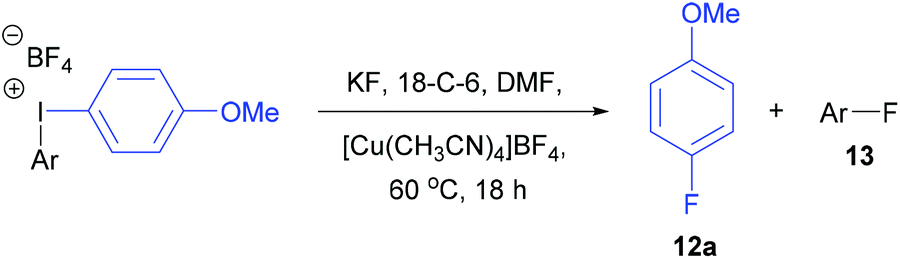
Fluoroaromatics constitute an important class of organic compounds because of their commercial applications in pharmaceuticals, agrochemicals, liquid crystals and positron emission tomography (PET).1 Fluorine-18 (18F) is the most commonly used radionuclide for PET, mainly due to its half-life of 110 minutes, and this has created a demand for new methods for the late-stage radiofluorination of aromatics. Consequently, the number of synthetic methods available for the preparation of aromatic fluorides has increased dramatically over the last eight years,2 and the synthesis of aryl carbon–fluorine bonds from fluoride normally requires pre-functionalisation of the aromatic with either boron,3 tin,4N-arylsydnone5 or hypervalent iodine.6–13The fluorination of unsymmetrical diaryliodonium salts was first reported in 1995 by Pike6 and it has now become a well-established method for the introduction of [18F]fluoride into aromatics.7 Since fluoride normally attacks the most electron-deficient aromatic ring, an effective strategy for directing fluoride into the desired aryl ring is to use an unsymmetrical aryl(spectator)iodonium salt where the spectator group is an electron-rich aromatic such as 4-methoxyphenyl or 2-thienyl.6–9 There is also an “ortho effect” in these fluorinations which overcomes the electronic effect. If one of the aromatic rings contains an ortho-substituent such as methyl, it will direct the fluorination to that ring. Sanford has shown that ortho-selectivity can be reversed by using a copper catalysed fluorination of aryl(mesityl)iodonium salts and the reaction is highly selective for fluorination to occur at the less sterically hindered arene, even with highly electron-rich aromatics.10,11 Recently, she described a two-step procedure involving an SEAr reaction between activated aromatics, MesI(OH)(OTs) and TMSOTf to generate aryl(mesityl)iodonium triflates which were used in situ in a copper-mediated radiofluorination with [18F]KF.10c Despite the presence of two ortho-methoxy groups, 2,4,6-trimethoxyphenyl was introduced by Chun in 2019 as an excellent new spectator group in the high-temperature radiofluorinations of aryl(2,4,6-trimethoxyphenyl)iodonium triflates where the aryl group was fluorinated with extremely high chemoselectivity.12 Iodonium ylides, (diacetoxyiodo)arenes and iodoarenes oxidised by m-CPBA have also been investigated as precursors to [18F]fluoroarenes.13
In 2013 we reported the preparation of the hypervalent iodine(III) reagent 1 from cheap sources of nucleophilic fluoride (Scheme 1) and its application as a new fluorinating reagent for the fluorination of 1,3-diketones, 1,3-ketoesters and 1,3-ketoamides.14a Since then, the reaction scope of fluoroiodane 1 has extended significantly from the difluorinations of styrenes and cyclopropanes, to intramolecular fluorocyclisations of unsaturated alcohols, amines, amides and carboxylic acids, as well as to radiofluorinations.14–16 Most of these reactions, however, required a transition metal to activate the fluoroiodane reagent 1 by coordinating to the fluorine atom.17 We have now established that 1 can be activated by hydrogen bonding to hexafluoroisopropanol and crucially, it removed the need for transition metals.14d
 | ||
| Scheme 1 Preparation of fluoroiodane reagents and unsymmetrical diaryliodonium salts. | ||
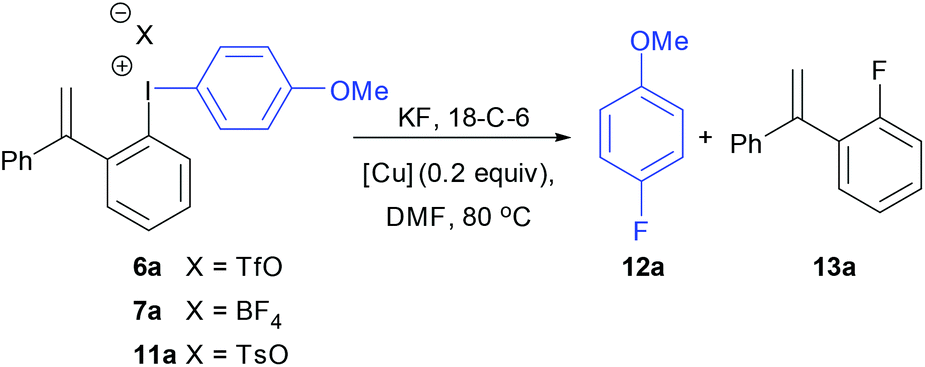
We were interested in preparing fluorinated aromatics using fluoroiodane 1 and here, we report the preparation of two new classes of unsymmetrical diaryliodonium salts containing ortho-sidearms, 5 and 7, by a site selective SEAr reaction between activated aromatics and the fluoroiodane reagents 1 and 2 with triflic acid (Scheme 1). We will also discuss the fluorinations of these unsymmetrical diaryliodonium salts using Sanford's copper catalysed conditions to reverse the “ortho effect” and fluorinate selectively the activated aromatics.
Results and discussion
Initially, anisole was reacted with fluoroiodane 1 and 1.5 equivalents of triflic acid in acetonitrile at room temperature to form exclusively the para-substituted diaryliodonium triflate 4a in 86% yield in just 1 hour at room temperature (Table 1). 1,3-Dimethoxybenzene and mesitylene were reacted with fluoroiodane 1 and triflic acid to form the diaryliodonium salts 4b and 4c in 91–92% yield. When the same reaction conditions were applied to the less activated aromatic, 2-bromoanisole, the diaryliodonium salt 4d was obtained in a moderate 58% yield. After further optimisation (see Table S3 in ESI†), the yield increased to 86% when 2-bromoanisole was reacted with fluoroiodane 1 and 3 equivalents of triflic acid in acetonitrile at 0 °C for 2 hours. The optimised protocol was applied to other less activated aromatics such as diphenyl ether, methyl-2-methoxybenzoate and m-xylene to form the unsymmetrical diaryliodonium salts 4e, 4f and 4g in moderate to good yields (22–69%). All the diaryliodonium triflates were obtained as stable, white powders which were purified simply by washing with hexane and diethyl ether.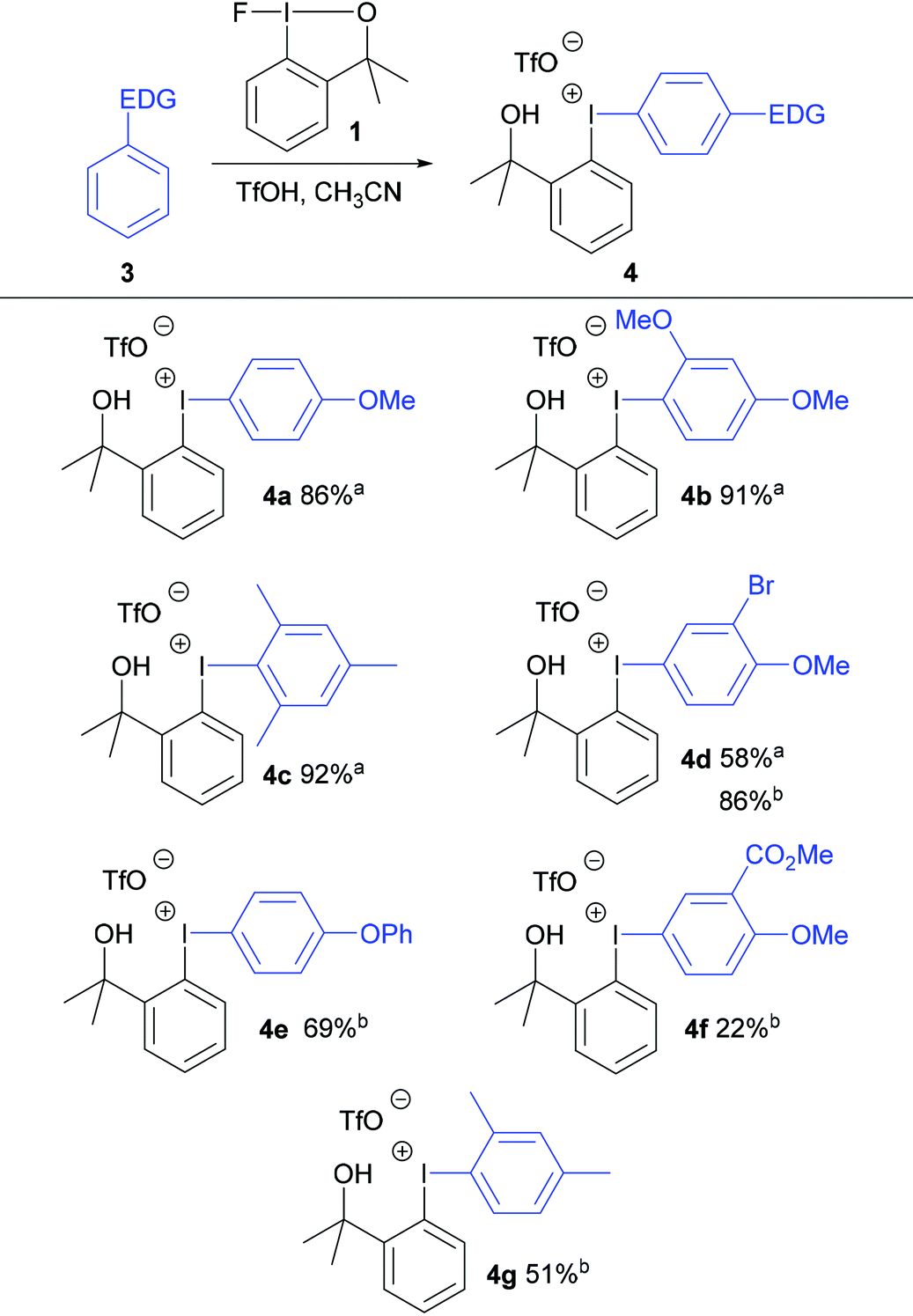
The triflate counteranions in the unsymmetrical diaryliodonium salts 4 were exchanged easily by stirring the salt in dichloromethane with aqueous sodium tetrafluoroborate at room temperature for 18 hours (Scheme 2). This simple procedure delivered the diaryliodonium tetrafluoroborates 5 in good yields (79–91%).
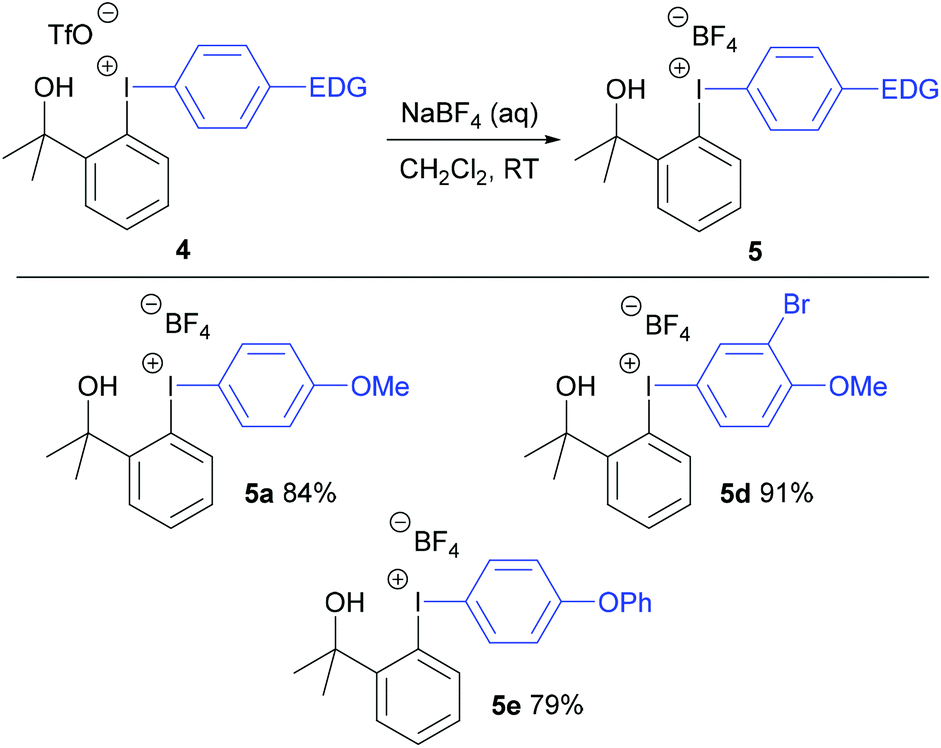 | ||
| Scheme 2 Metathesis reactions using NaBF4 (aq). | ||
A new analogue of the fluoroiodane reagent 1, where one of the methyl groups was replaced by a phenyl group, was then prepared by a five-step synthesis (see ESI†). The phenyl group was introduced to encourage the alcohol sidearm in the diaryliodonium salts to eliminate and form an alkene in conjugation with two aromatic rings under the strong acidic conditions used in their synthesis. Hence, a new class of unsymmetrical diaryliodonium salts containing an ortho-styrene sidearm was prepared by reacting a series of activated aromatics with fluoroiodane 2 and triflic acid in acetonitrile at 0 °C for 2 hours. After stirring the diaryliodonium triflates 6 in dichloromethane with an aqueous solution of sodium tetrafluoroborate, the diaryliodonium tetrafluoroborates 7a to 7h were produced in good yields (74–89%) over the two steps (Scheme 3). The reaction worked particularly well with less activated aromatics such as diphenyl ether, methyl-2-methoxybenzoate and m-xylene delivering the diaryliodonium triflates 6b, 6f and 6g in better yields (85%, 90% and 89% respectively) than the same reactions with fluoroiodane 1 where the salts, 4e, 4f and 4g, were obtained in 69%, 22% and 51% yields respectively. In the reaction between benzyl phenyl ether and fluoroiodane 2, the benzyl protecting group was removed under the strong acidic conditions producing the p-phenol salt 6c and subsequently 7c in 59% overall yield after anion exchange. Interestingly, the first general procedure for preparing para-hydroxy substituted diaryliodonium salts was only reported in 2018 by Zhdankin using Koser's reagent, triisopropylsiloxyarenes and triflic acid to deprotect the aryl silyl ethers.18 Some arenes, however, were incompatible with the reaction (Scheme 3) including 1,3-dimethoxybenzene, 1,2-dimethoxybenzene, 1-methoxy-2-methylbenzene, acetanilide and N-methylindole.
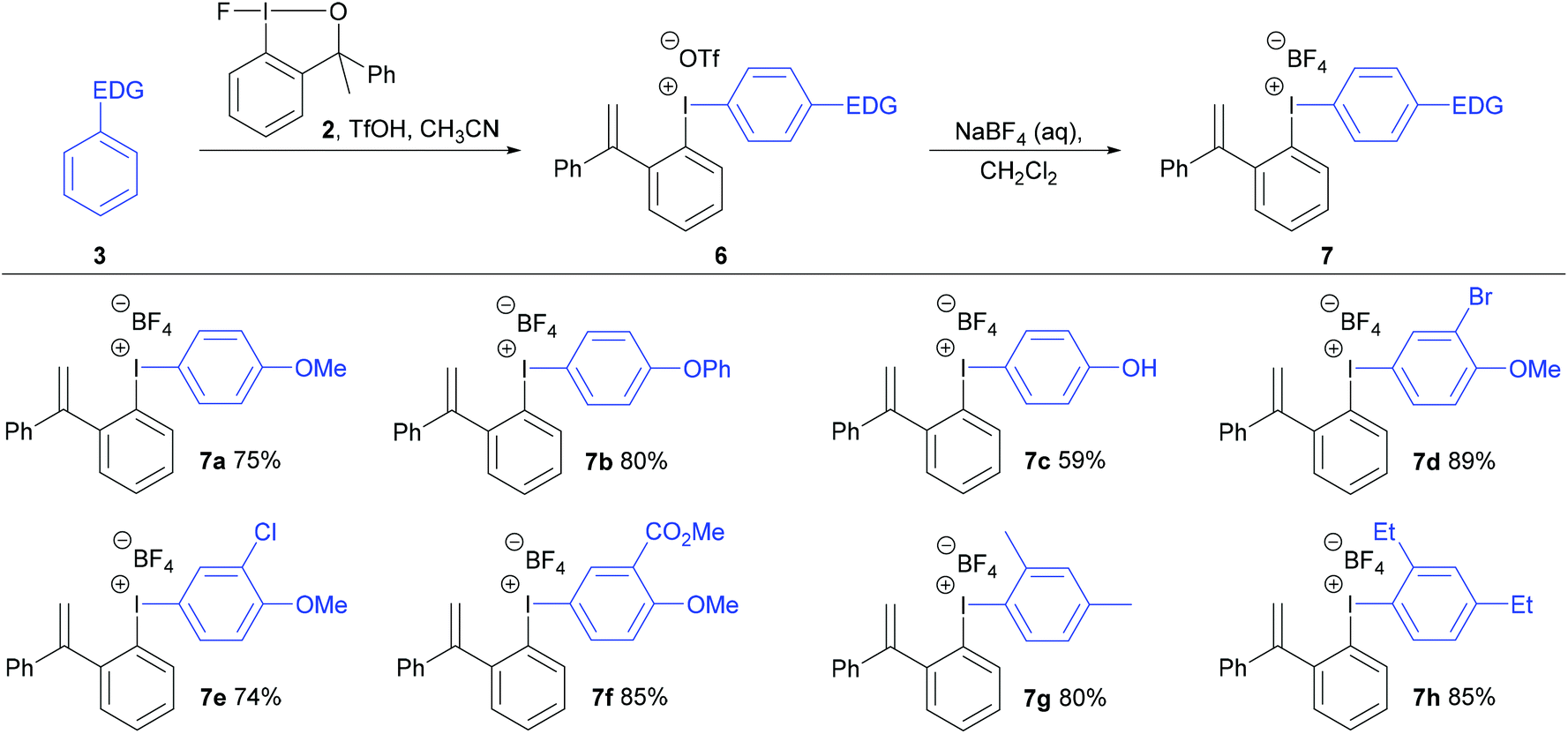 | ||
| Scheme 3 Preparation of diaryliodonium salts 7 from fluoroiodane reagent 2 and the overall yields for the two steps are reported. | ||
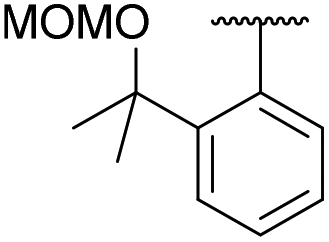
The diaryliodonium tetrafluoroborate 5a was also prepared in one-step by the direct reaction between anisole, fluoroiodane reagent 1 and the Lewis acid, boron trifluoride (Scheme 4). Unfortunately, this reaction did not work with less activated aromatics such as 2-bromoanisole. When anisole was reacted with fluoroiodane reagent 2 and boron trifluoride, the alcohol sidearm did not undergo elimination and the diaryliodonium tetrafluoroborate 8a containing a 1-phenylethan-1-ol sidearm was formed in 90% yield. Before attempting to fluorinate these unsymmetrical diaryliodonium salts, the free hydroxy groups in 5a and 8a were protected with MOMCl (Scheme 4). An excess of both MOMCl and N,N-diisopropylethylamine (Hunig's base) were used to ensure complete conversion to products 9a and 10a which were isolated in excellent yields after stirring the crude products with an aqueous solution of sodium tetrafluoroborate. The last step was essential because without it, some of the salt was obtained with a chloride counteranion which affected the subsequent fluorinations.
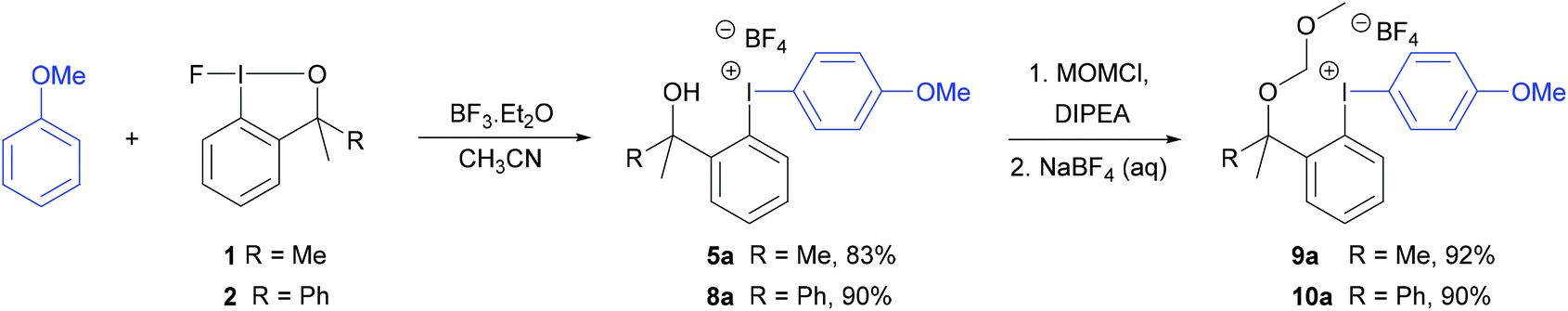 | ||
| Scheme 4 Preparation of diaryliodonium tetrafluoroborates, 5a and 8a, and protection of alcohol sidearm. | ||
Solid-state structures
The solid-state structures of 5a containing a propan-2-ol sidearm and 8a containing a 1-phenylethan-1-ol sidearm are shown in Fig. 1 and 2. The I(1)–C(1) (2.132(3) and 2.134(3) Å) and I(1)–C(10)/C(15) (2.087(3) and 2.088(3) Å) bond lengths are practically identical for the two diaryliodonium salts. The distinct C(15)/C(10)–I(1)–C(1) bond angles are typical for the pseudo T-shape of diaryliodonium salts and range from 97.1(1) to 98.4(1)°. In both solid-state structures there is a strong intramolecular interaction between I(1) and O(1) (2.576(2) and 2.589(2) Å) with an O(1)⋯I(1)–C(10) bond angle of 169°.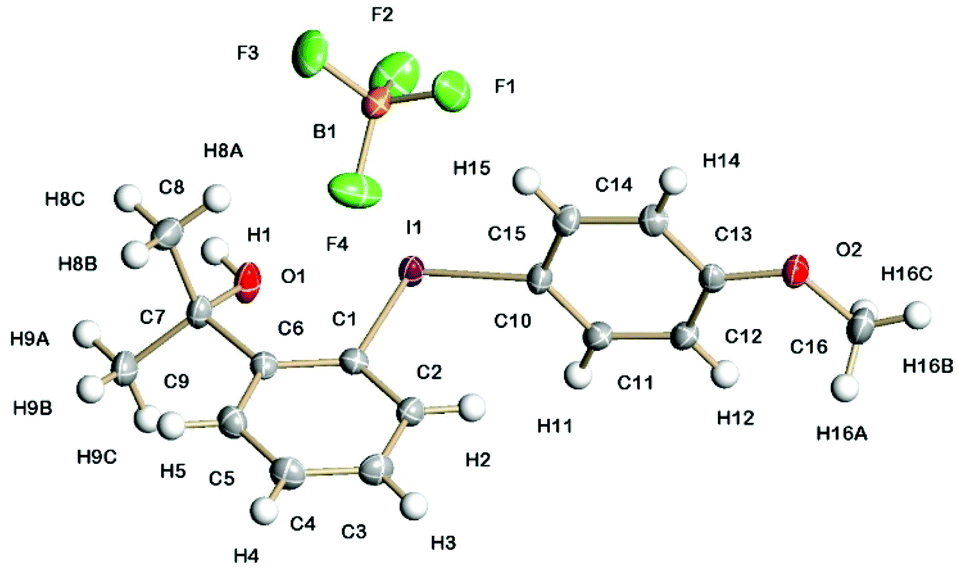 | ||
| Fig. 1 Molecular structure of 5a containing a propan-2-ol sidearm and showing 50% displacement ellipsoids. Selected bond lengths and angles: I(1)–C(1) 2.132(3) Å; I(1)–C(10) 2.087(3) Å; I(1)⋯O(1) 2.576(2) Å; C(10)–I(1)–C(1) 98.35(10)°; O(1)⋯I(1)–C(10) 169.24(8)°. | ||
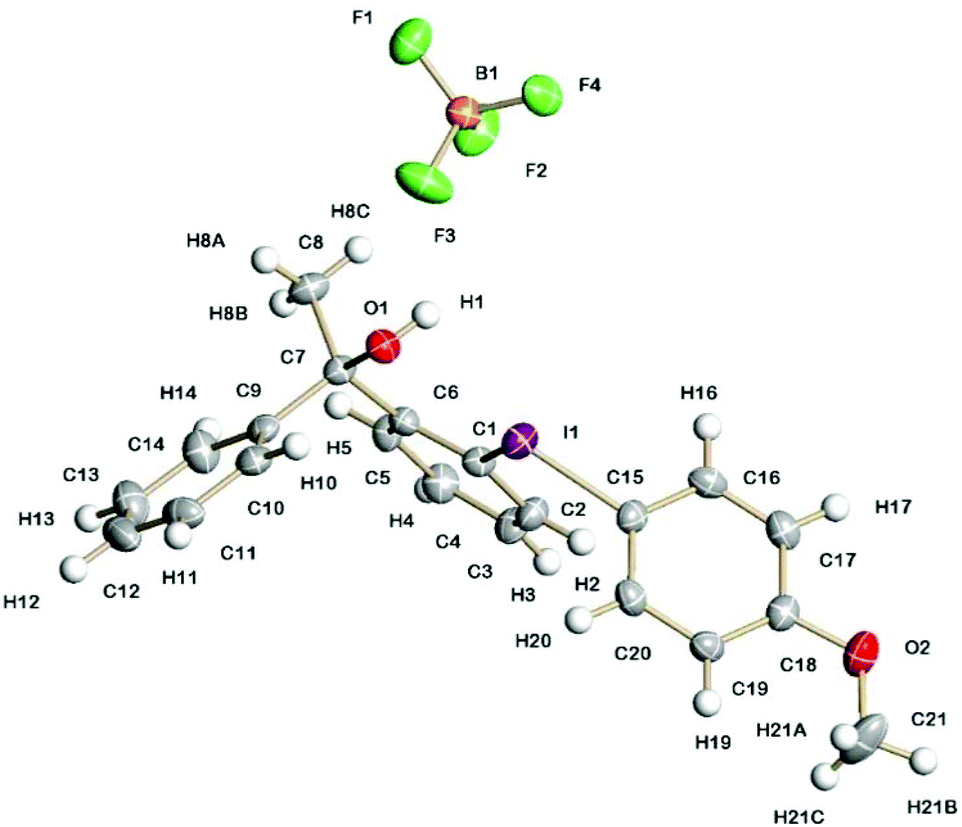 | ||
| Fig. 2 Molecular structure of 8a containing a 1-phenylethan-1-ol sidearm and showing 50% displacement ellipsoids. Selected bond lengths and angles: I(1)–C(1) 2.134(3) Å; I(1)–C(15) 2.088(3) Å; I(1)⋯O(1) 2.589(2) Å; C(15)–I(1)–C(1) 97.12(11)°; O(1)⋯I(1)–C(15) 168.84(8)°. | ||
Fig. 3 shows the solid-state structure of 7a containing an o-styrene sidearm and the X-ray crystal structures of the related triflate 6a and tosylate 11a that both contain the same o-styrene sidearm are presented in the ESI.† In this series of diaryliodonium salts, the I(1)–C(1) bond lengths (2.102(3)–2.118(4) Å) are very similar to the I(1)–C(15) bond lengths (2.094(3)–2.100(4) Å) and the C(15)–I(1)–C(1) bond angles range from 92.8(1) to 98.0(1)°. The C(7)–C(8) bond lengths are 1.314(5)–1.331(5) Å which is typical for an alkene C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond length (1.322 Å)19 and the bond angles around C(7) are normal for an sp2 hybridised carbon atom. There is secondary bonding between the iodine and the oxygen atoms of the tosylate and triflate counteranions in 11a and 6a respectively. There are two intramolecular interactions in 11a, I(1)⋯O(4) = 2.721(3) Å and I(1A)⋯O(2) = 2.850(3) Å, as well as two intermolecular interactions, I(1)⋯O(2A) = 2.805(3) Å and I(1A)⋯O(3A) = 2.859(3) Å. The intramolecular and intermolecular interactions in 6a are slightly weaker with the triflate anion, I(1)⋯O(3) = 2.930(2) Å and I(1)⋯O(4) = 3.094(3) Å respectively. An intramolecular interaction (3.023(3) Å) between F(3) of the tetrafluoroborate counteranion and I(1) is also present in 7a.
C bond length (1.322 Å)19 and the bond angles around C(7) are normal for an sp2 hybridised carbon atom. There is secondary bonding between the iodine and the oxygen atoms of the tosylate and triflate counteranions in 11a and 6a respectively. There are two intramolecular interactions in 11a, I(1)⋯O(4) = 2.721(3) Å and I(1A)⋯O(2) = 2.850(3) Å, as well as two intermolecular interactions, I(1)⋯O(2A) = 2.805(3) Å and I(1A)⋯O(3A) = 2.859(3) Å. The intramolecular and intermolecular interactions in 6a are slightly weaker with the triflate anion, I(1)⋯O(3) = 2.930(2) Å and I(1)⋯O(4) = 3.094(3) Å respectively. An intramolecular interaction (3.023(3) Å) between F(3) of the tetrafluoroborate counteranion and I(1) is also present in 7a.
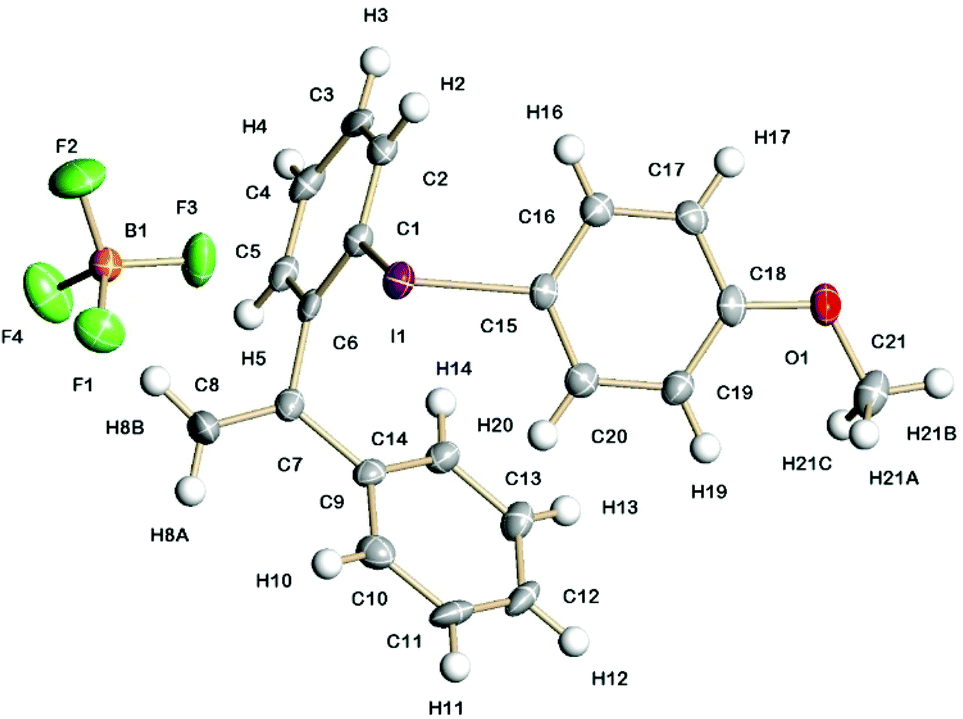 | ||
| Fig. 3 Molecular structure of 7a containing an o-styrene sidearm and showing 50% displacement ellipsoids. Selected bond lengths and angles: I(1)–C(1) 2.118(4) Å; I(1)–C(15) 2.099(4) Å; I(1)⋯F(3) 3.023(3) Å; C(15)–I(1)–C(1) 95.26(14)°; F(3)⋯I(1)–C(15) 171.38(13)°. | ||
Fluorinations
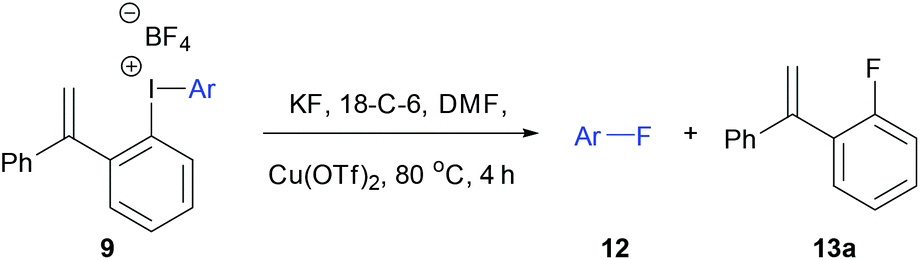
Initially, the unsymmetrical diaryliodonium salts 7a, 9a and 10a were reacted with potassium fluoride (1.1 equivalent) and 18-crown-6 (0.4 equivalent) in DMF at 60 °C for 18 hours both with and without [Cu(MeCN)4]BF4 (0.2 equivalent), as reported by Sanford in the fluorination of aryl(mesityl)iodonium salts (Table 2).10a Without the copper catalyst the ortho-effect dominated in the fluorination of 7a (entry 1) and the aryl group containing the ortho-styrene sidearm was fluorinated producing 1-fluoro-2-(1-phenylvinyl)benzene 13a in 53% yield with byproduct 4-iodoanisole formed in 72% yield. However, the ortho-effect was reversed in the copper-catalysed fluorination and the desired product, 4-fluoroanisole 12a, was delivered in a moderate 39% yield (entry 2). Surprisingly, when the unsymmetrical salts, 9a and 10a, were fluorinated in the absence of copper, there was very little fluorination but there was still a strong ortho-effect with 4-iodoanisole being obtained in 79% and 85% yield respectively (entries 3 and 5). Under the copper-catalysed conditions (entries 4 and 6) fluorination was observed, but the ortho-effect was not reversed and the aryl groups containing the ortho-sidearms were fluorinated in 74 and 65% yields respectively. Presumably, the presence of the oxygen atoms in the sidearms of 9a and 10a, which had strong interactions with the iodine atom in the solid-state structures, were affecting the selectivity of these fluorinations.|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Ar group | [Cu] (equiv.) | 12a (%) | 13 (%) |
| a Determined by GC and 19F NMR spectroscopy with internal standards. | |||||
| 1 | 7a |

|
0 | 0 | 53 |
| 2 | 7a | 0.2 | 39 | 7 | |
| 3 | 9a |
|
0 | 0 | 8 |
| 4 | 9a | 0.2 | 0 | 74 | |
| 5 | 10a |

|
0 | 0 | 6 |
| 6 | 10a | 0.2 | 0 | 65 | |
The fluorination of 7a was optimised by screening the reaction conditions, different solvents, copper catalysts and fluorinating reagents (see ESI† for full details). Increasing the reaction temperature to 80 °C in entry 1 (Table 3) improved both the yield of 4-fluoroanisole 12a to 45% and the selectivity of the reaction to 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10. Since the use of TEMPO has proved beneficial for the fluorinations of some diaryliodonium salts,7 1 equivalent of TEMPO was used in entry 2 but it had no effect on the reaction. Both [Cu(MeCN)4]OTf and Cu(OTf)2 were good catalysts with Cu(OTf)2 providing 4-fluoroanisole in 60% yield. The yield improved to 65% and the selectivity increased to 92:8 by reducing the reaction time to 4 hours in entry 5. The uncatalysed reaction in entry 6 proceeded in an excellent 80% yield but with the opposite selectivity (1:99). Finally, the effects of changing the counteranion from tetrafluoroborate 7a to triflate 6a (entry 7) and tosylate 11a (entry 8) were investigated under the optimised reaction conditions but only low yields of 4-fluoroanisole 12a were obtained.
:10. Since the use of TEMPO has proved beneficial for the fluorinations of some diaryliodonium salts,7 1 equivalent of TEMPO was used in entry 2 but it had no effect on the reaction. Both [Cu(MeCN)4]OTf and Cu(OTf)2 were good catalysts with Cu(OTf)2 providing 4-fluoroanisole in 60% yield. The yield improved to 65% and the selectivity increased to 92:8 by reducing the reaction time to 4 hours in entry 5. The uncatalysed reaction in entry 6 proceeded in an excellent 80% yield but with the opposite selectivity (1:99). Finally, the effects of changing the counteranion from tetrafluoroborate 7a to triflate 6a (entry 7) and tosylate 11a (entry 8) were investigated under the optimised reaction conditions but only low yields of 4-fluoroanisole 12a were obtained.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Cu catalyst | Time (h) | 12a (%) | 13a (%) |
12a:13a |
| a Determined by GC with an internal standard. b 1 Equivalent of TEMPO in the dark. | ||||||
| 1 | 7a | [Cu(MeCN)4]BF4 | 18 | 45 | 5 | 90:10 |
| 2b | 7a | [Cu(MeCN)4]BF4 | 18 | 43 | 5 | 90:10 |
| 3 | 7a | [Cu(MeCN)4]OTf | 18 | 50 | 7 | 88:12 |
| 4 | 7a | Cu(OTf)2 | 18 | 60 | 9 | 87:13 |
| 5 | 7a | Cu(OTf)2 | 4 | 65 | 6 | 92:8 |
| 6 | 7a | No catalyst | 4 | 1 | 80 | 1:99 |
| 7 | 6a | Cu(OTf)2 | 4 | 34 | 1 | 97:3 |
| 8 | 11a | Cu(OTf)2 | 4 | 13 | 1 | 93:7 |
Having established optimum conditions, each of the diaryliodonium tetrafluoroborates, 7a to 7h, were fluorinated using potassium fluoride and 20 mol% Cu(OTf)2 in DMF at 80 °C for 4 h (Table 4). The salts, 7a and 7b, produced the desired p-fluorinated aromatics, 12a and 12b, in good yields (61–65%) with excellent selectivities (91:9). However, p-fluorophenol 12c was only obtained in a low 20% yield in the fluorination of 7c but this was probably due to the free hydroxy group in the salt.20 In the copper-catalysed fluorinations of 7d to 7f, the fluorinated aromatics 12d to 12f were produced in good (66%) to excellent (82%–87%) yields and with high selectivities (93:7). As expected, the selectivity dropped to 75:25 in the fluorination of 7g because of the presence of the ortho-methyl group in the activated aromatic, but 2,4-dimethylfluorobenzene 12g was still formed in a good 63% yield. The selectivity was reduced further to 47:53 in the fluorination of 7h due to the bigger ethyl substituent in the ortho-position and the desired fluorinated aromatic 12h was only obtained in a low 23% yield.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Product | 12 (%) | 13a (%) |
12:13a |
| a Determined by 19F NMR spectroscopy with an internal standard. | |||||
| 1 | 7a |
|
65 | 6 | 92:8 |
| 2 | 7b |
|
61 | 6 | 91:9 |
| 3 | 7c |
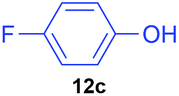
|
20 | 0 | 100:0 |
| 4 | 7d |

|
87 | 6 | 94:6 |
| 5 | 7e |
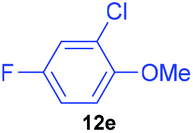
|
66 | 5 | 93:7 |
| 6 | 7f |
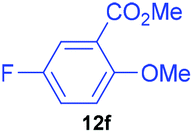
|
82 | 7 | 92:8 |
| 7 | 7g |
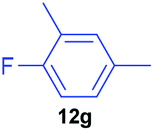
|
63 | 21 | 75:25 |
| 8 | 7h |
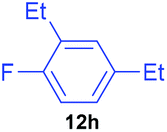
|
23 | 26 | 47:53 |
Conclusions
In summary, this paper describes a convenient synthesis of para-fluorinated aromatics from activated aromatics and fluoroiodane reagent 2. Initially, two new classes of unsymmetrical diaryliodonium salts containing ortho-sidearms were prepared by a site selective SEAr reaction between activated aromatics and two different fluoroiodane reagents 1 and 2 with triflic acid. With fluoroiodane 1 the unsymmetrical diaryliodonium salts contained an ortho-propan-2-ol sidearm, whereas the alcohol sidearm was eliminated to form an ortho-styrene sidearm in the reaction with fluoroiodane 2. After counteranion exchange, only the diaryliodonium tetrafluoroborates containing a styrene sidearm 7 reacted selectively with potassium fluoride under copper-catalysed conditions to form para-fluorinated aromatics 12 in good yields (61–87%).Conflicts of interest
There are no conflicts to declare.Acknowledgements
AMHA would like to thank The Higher Committee for Education Development in Iraq (HCED) and the University of Al-Qadisiyah for the PhD scholarship.Notes and references
- (a) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Acena, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef CAS PubMed; (b) J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (c) P. Jeschke, ChemBioChem, 2004, 5, 570 CrossRef CAS PubMed; (d) P. Kirsch, J. Fluorine Chem., 2015, 177, 29 CrossRef CAS; (e) M. Bremer, P. Kirsch, M. Klasen-Memmer and K. Tarumi, Angew. Chem., Int. Ed., 2013, 52, 8880 CrossRef CAS PubMed; (f) S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719 CrossRef CAS PubMed; (g) X. Deng, J. Rong, L. Wang, N. Vasdev, L. Zhung, L. Josephson and S. H. Liang, Angew. Chem., Int. Ed., 2019, 58, 2580 CrossRef CAS PubMed.
- M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612 CrossRef CAS PubMed.
- (a) Y. Ye, S. D. Schimler, P. S. Hanley and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 16292 CrossRef CAS PubMed; (b) M. Tredwell, S. M. Preshlock, N. J. Taylor, S. Gruber, M. Huiban, J. Passchier, J. Mercier, C. Génicot and V. Gouverneur, Angew. Chem., Int. Ed., 2014, 53, 7751 CrossRef CAS PubMed; (c) A. V. Mossine, A. F. Brooks, K. J. Makaravage, J. M. Miller, N. Ichiisi, M. S. Sanford and P. J. H. Scott, Org. Lett., 2015, 17, 5780 CrossRef CAS PubMed.
- K. J. Makaravage, A. F. Brooks, A. V. Mossine, M. S. Sanford and P. J. H. Scott, Org. Lett., 2016, 18, 5440 CrossRef CAS PubMed.
- (a) M. K. Narayanam, G. Ma, P. A. Champagne, K. N. Houk and J. M. Murphy, Angew. Chem., Int. Ed., 2017, 56, 13006 CrossRef CAS PubMed; (b) M. K. Narayanam, G. Ma, P. A. Champagne, K. N. Houk and J. M. Murphy, Synlett, 2018, 29, 1131 CrossRef CAS.
- (a) V. W. Pike and F. I. Aigbirhio, J. Chem. Soc., Chem. Commun., 1995, 2215 RSC; (b) V. W. Pike and F. I. Aigbirhio, J. Labelled Compd. Radiopharm., 1995, 37, 120 Search PubMed.
- (a) V. W. Pike, J. Labelled Compd. Radiopharm., 2018, 61, 196 CrossRef CAS PubMed; (b) M. S. Yusubov, D. Y. Svitich, M. S. Larkina and V. V. Zhdankin, ARKIVOC, 2013, 7(i), 364 Search PubMed.
- (a) T. L. Ross, J. Ermert and H. H. Coenen, J. Labelled Compd. Radiopharm., 2005, 48(Suppl. 1), S153 Search PubMed; (b) T. L. Ross, J. Ermert, C. Hocke and H. H. Coenen, J. Am. Chem. Soc., 2007, 129, 8018 CrossRef CAS PubMed; (c) M. A. Carroll, C. Jones and S.-L. Tang, J. Labelled Compd. Radiopharm., 2007, 50, 450 CrossRef CAS.
- V. W. Pike and J.-H. Chun, Org. Biomol. Chem., 2013, 11, 6300 RSC.
- (a) N. Ichiishi, A. J. Canty, B. F. Yates and M. S. Sanford, Org. Lett., 2013, 15, 5134 CrossRef CAS PubMed; (b) N. Ichiishi, A. F. Brooks, J. J. Topczewski, M. E. Rodnick, M. S. Sanford and P. J. H. Scott, Org. Lett., 2014, 16, 3224 CrossRef CAS PubMed; (c) M. S. McCammant, S. Thompson, A. F. Brooks, S. W. Krska, P. J. H. Scott and M. S. Sanford, Org. Lett., 2017, 19, 3939 CrossRef CAS PubMed.
- (a) A. Yamaguchi, H. Hanaoka, T. Higuchi and Y. Tsushima, J. Nucl. Med., 2018, 59, 815 CrossRef CAS; (b) A. Yamaguchi, H. Hanaoka, T. Higuchi and Y. Tsushima, J. Labelled Compd. Radiopharm., 2020, 63, 368 CrossRef CAS.
- Y.-D. Kwon, J. Son and J.-H. Chun, J. Org. Chem., 2019, 84, 3678 CrossRef CAS.
- (a) B. H. Rotstein, N. A. Stephenson, N. Vasdev and S. H. Liang, Nat. Commun., 2014, 5, 4365 CrossRef CAS; (b) J. Cardinale, J. Ermert, S. Humpert and H. H. Coenen, RSC Adv., 2014, 4, 17293 RSC; (c) M. B. Haskali, S. Telu, Y.-S. Lee, C. L. Morse, S. Lu and V. W. Pike, J. Org. Chem., 2016, 81, 297 CrossRef CAS PubMed; (d) Y.-D. Kwon, J. Son and J.-H. Chun, Org. Lett., 2018, 20, 7902 CrossRef CAS PubMed.
- (a) G. C. Geary, E. G. Hope, K. Singh and A. M. Stuart, Chem. Commun., 2013, 49, 9263 RSC; (b) G. C. Geary, E. G. Hope, K. Singh and A. M. Stuart, RSC Adv., 2015, 5, 16501 RSC; (c) G. C. Geary, E. G. Hope and A. M. Stuart, Angew. Chem., Int. Ed., 2015, 54, 14911 CrossRef CAS PubMed; (d) H. Minhas, W. Riley, A. M. Stuart and M. Urbonaite, Org. Biomol. Chem., 2018, 16, 7170 RSC.
- (a) N. O. Ilchenko, B. O. A. Tasch and K. J. Szabó, Angew. Chem., Int. Ed., 2014, 53, 12897 CrossRef CAS PubMed; (b) W. Yuan and K. J. Szabó, Angew. Chem., Int. Ed., 2015, 54, 8533 CrossRef CAS PubMed; (c) W. Yuan, L. Eriksson and K. J. Szabó, Angew. Chem., Int. Ed., 2016, 55, 8410 CrossRef CAS PubMed; (d) N. O. Ilchenko, M. Heberg and K. J. Szabó, Chem. Sci., 2017, 8, 1056 RSC; (e) A. Ulmer, C. Brunner, A. M. Arnold, A. Poething and T. Gulder, Chem. – Eur. J., 2016, 22, 3660 CrossRef CAS PubMed; (f) C. Brunner, A. Andries-Ulmer, G. M. Kiefl and T. Gulder, Eur. J. Org. Chem., 2018, 2615 CrossRef CAS.
- (a) B. Yang, K. Chansaenpak, H. Wu, L. Zhu, M. Wang, Z. Li and H. Lu, Chem. Commun., 2017, 53, 3497 RSC; (b) M. A. Cortés González, P. Nordeman, A. Bermejo Gómez, D. N. Meyer, G. Antoni, M. Schou and K. J. Szabó, Chem. Commun., 2018, 54, 4286 RSC.
- (a) T. Yan, B. Zhou, X.-S. Xue and J.-P. Cheng, J. Org. Chem., 2016, 81, 9006 CrossRef CAS PubMed; (b) B. Zhou, T. Yan, X.-S. Xue and J.-P. Cheng, Org. Lett., 2016, 18, 6128 CrossRef CAS PubMed; (c) J. Zhang, K. J. Szabó and F. Himo, ACS Catal., 2017, 7, 1093 CrossRef CAS.
- A. Yoshimura, M. T. Shea, O. Guselnikova, P. S. Postnikov, G. T. Rohde, A. Saito, M. S. Yusubov, V. N. Nemykin and V. V. Zhdankin, Chem. Commun., 2018, 54, 10363 RSC.
- L. Wang, Z. Jian, C. G. Daniliuc, G. Kehr and G. Erker, Dalton Trans., 2018, 47, 10853 RSC.
- When the fluorination of 7c was attempted under the same reaction conditions, but without the copper catalyst, there was no evidence of any fluorination at all.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra. CCDC 2013356–2013360. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ob01401j |
| This journal is © The Royal Society of Chemistry 2020 |