
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of the reducing-end anomeric configuration of the Man9 epitope on DC-SIGN recognition†
Noelia
de la Cruz
a,
Javier
Ramos-Soriano
*a,
José J.
Reina
a,
José L.
de Paz
a,
Michel
Thépaut
b,
Franck
Fieschi
b,
Ana
Sousa-Herves
a and
Javier
Rojo
 *a
*a
aGlycosystems Laboratory, Instituto de Investigaciones Químicas (IIQ), CSIC – Universidad de Sevilla, Av. Américo Vespucio 49, Seville 41092, Spain. E-mail: javiramossoriano@gmail.com; javier.rojo@iiq.csic.es
bUniv. Grenoble Alpes, CNRS, CEA, Institut de Biologie Structurale, 38000 Grenoble, France
First published on 27th July 2020
Abstract
High-mannose (Man9GlcNAc2) is the main carbohydrate unit present in viral envelope glycoproteins such as gp120 of HIV and the GP1 of Ebola virus. This oligosaccharide comprises the Man9 epitope conjugated to two terminal N-acetylglucosamines by otherwise rarely-encountered β-mannose glycosidic bond. Formation of this challenging linkage is the bottleneck of the few synthetic approaches described to prepare high mannose. Herein, we report the synthesis of the Man9 epitope with both alpha and beta configurations at the reducing end, and subsequent evaluation of the impact of this configuration on binding to natural receptor of high-mannose, DC-SIGN. Using fluorescence polarization assays, we demonstrate that both anomers bind to DC-SIGN with comparable affinity. These relevant results therefore indicate that the more synthetically-accesible Man9 alpha epitope may be deployed as ligand for DC-SIGN in both in vitro and in vivo biological assays.
Introduction
Carbohydrates are ubiquitous in nature and participate in many biological events relevant to health and disease, such as cell growth and differentiation, fertilization, inflammation, tumour progression and metastasis, viral infection, and many others.1 The function of carbohydrates in these processes results from interactions with specific receptors, mainly proteins known as lectins.2 Carbohydrate–protein interactions are characterised by a high selectivity and a low affinity, the latter of which is compensated by multivalent interactions resulting from the presentation of multiple copies of the carbohydrate epitopes and receptors in nature.3 Understanding carbohydrate-mediated processes therefore requires the accessibility of multivalent carbohydrate platforms capable of intervening with these processes.4–6High-mannose (Man9GlcNAc2) is a complex relevant N-glycan present in several viral envelope glycoproteins (Fig. 1). This sugar plays a crucial role during the attachment of pathogens to cells in the first stages of the infection process through the interaction with the DC-SIGN (Dendritic Cell-Specific ICAM-3 Grabbing Non-integrin) receptor, found in lipid patches at the dendritic cell (DC) surface.7 In particular, gp120 of HIV-1 has an average of 24 N-linked glycans clustered on the protein surface, of which 53–76% are high-mannose derivatives.8 This cluster presentation is fundamental to the efficient interaction with DC-SIGN.
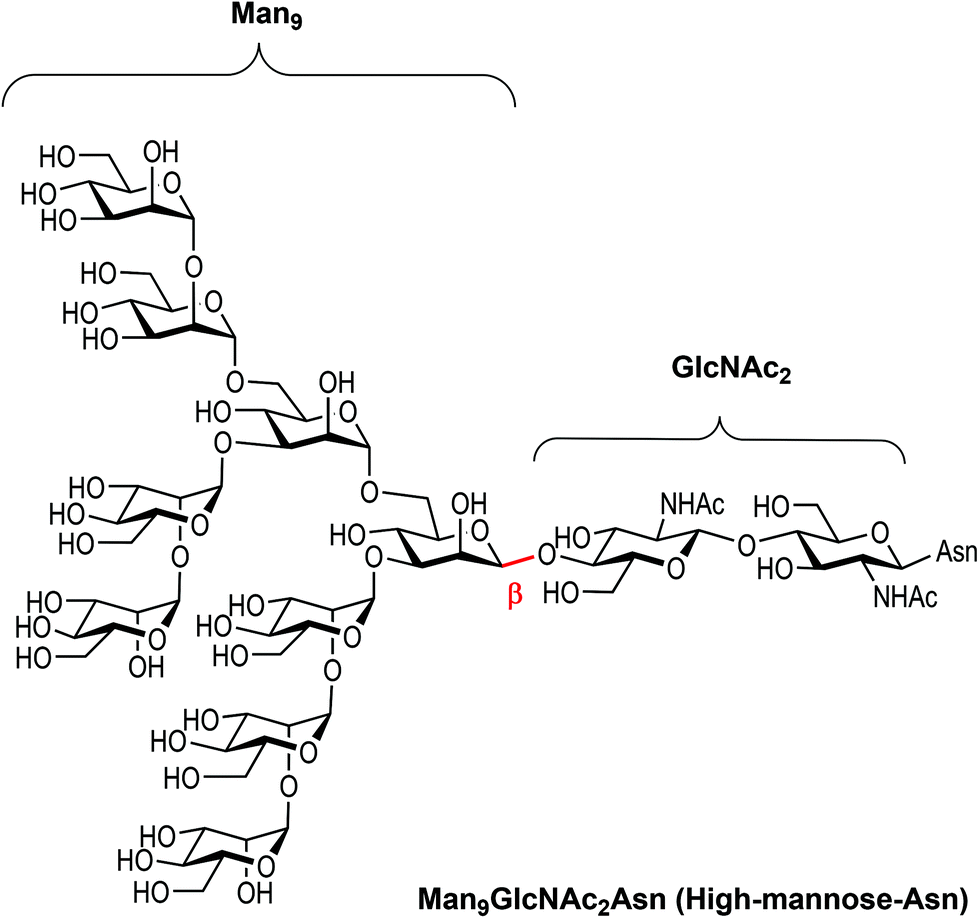 | ||
| Fig. 1 Chemical structure of high-mannose and its constitutive parts, the beta Man9 and the GlcNAc2 epitopes. | ||
Towards better understanding the recognition process between high-mannose and DC-SIGN, several multivalent systems have been envisaged. However, the structural complexity of the constituent sugars and the difficulty of obtaining sufficient quantities of pure material from natural sources renders the preparation of multivalent systems carrying the natural beta epitope rather challenging. In fact, very few examples are reported in the literature describing the synthesis of Man9 and high-mannose multivalent systems.9–11 In this context, different approaches have been developed for the preparation of simpler compounds that mimic the multivalent presentation of the sugar found in nature. Small fragments of the Man9 epitope have been synthesised and evaluated in different glycomimetic systems in pursuit of sugars simpler than Man9 but with reasonable binding affinities for DC-SIGN.12–15 Recently, we have developed a straightforward synthetic strategy for the preparation of the Man9 epitope with the natural beta configuration at the reducing end.16 Although the synthesis that we have described is competitive with those published previously by other laboratories, the preparation of the β-mannose unit remains the limiting step. The synthesis of the appropriately-functionalised beta-mannose monosaccharide building block, required to perform the necessary glycosylation steps towards the preparation of the nonasaccharide, demands eleven synthetic steps with a global yield of 43% from S-tolyl-α-D-mannopyranoside.16 As a possible solution, we considered whether the more synthetically-accessible α-mannose analogue would provide a similar affinity to DC-SIGN than the beta anomer, therefore circumventing the requirement for the complicated β-mannose building block, in the preparation of the corresponding carbohydrate multivalent systems.
To evaluate our proposed strategy, we have synthesised the alpha and beta anomers of the Man9 epitope and the corresponding trivalent glycocluster to examine the differences in binding affinities to the DC-SIGN receptor using a fluorescence polarization assay.
Results and discussion
Preparation of Man9 epitopes and glycoclusters
The synthesis of beta Man9 using a convergent straightforward synthetic strategy was previously described by our laboratory.16 In this work, we used the same strategy to prepare the corresponding alpha anomer.The di-, penta-, and trisaccharides building blocks were common intermediates for both anomers, (Fig. 2) being the mannose of the reducing end with the alpha configuration in the anomeric position prepared as described in Scheme 1.
 | ||
| Fig. 2 Retrosynthetic scheme for the preparation of αMan9 and βMan9. | ||
 | ||
| Scheme 1 Consecutive synthesis of mannose derivative 6. | ||
The synthesis of the appropriately-protected reducing end mannose was achieved as depicted in Scheme 1, starting from the previously described mannosyl derivative 5.17 Mannose 5 was orthogonally protected using a consecutive approach, performing purification only at the final step. Positions 4 and 6 were protected via the formation of the benzylidene acetal by subjecting 5 to benzaldehyde dimethyl acetal and camphorsulfonic acid (CSA). Subsequently, position 3 was protected as the p-methoxybenzy ether, via the initial formation of a tin acetal between position 2 and 3, followed by reaction with p-methoxybenzyl chloride (PMBCl) and tetrabutyl ammonium iodine (TBAI). The free OH in position 2 was then protected by benzoylation with benzoyl anhydride in the presence of triethyl amine and a catalytic amount of dimethylamino pyridine (DMAP). Finally, the position 3 was selectively deprotected with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) to yield the target mannose derivative 6 in 26% overall yield from mannose 5 following final chromatographic purification.
With the monosaccharide 6 in hand, glycosylation with the previously synthesised trisaccharide 7 and pentasaccharide 8,16 enabled the formation of the protected Man9 nonasaccharide 11 (Scheme 2).
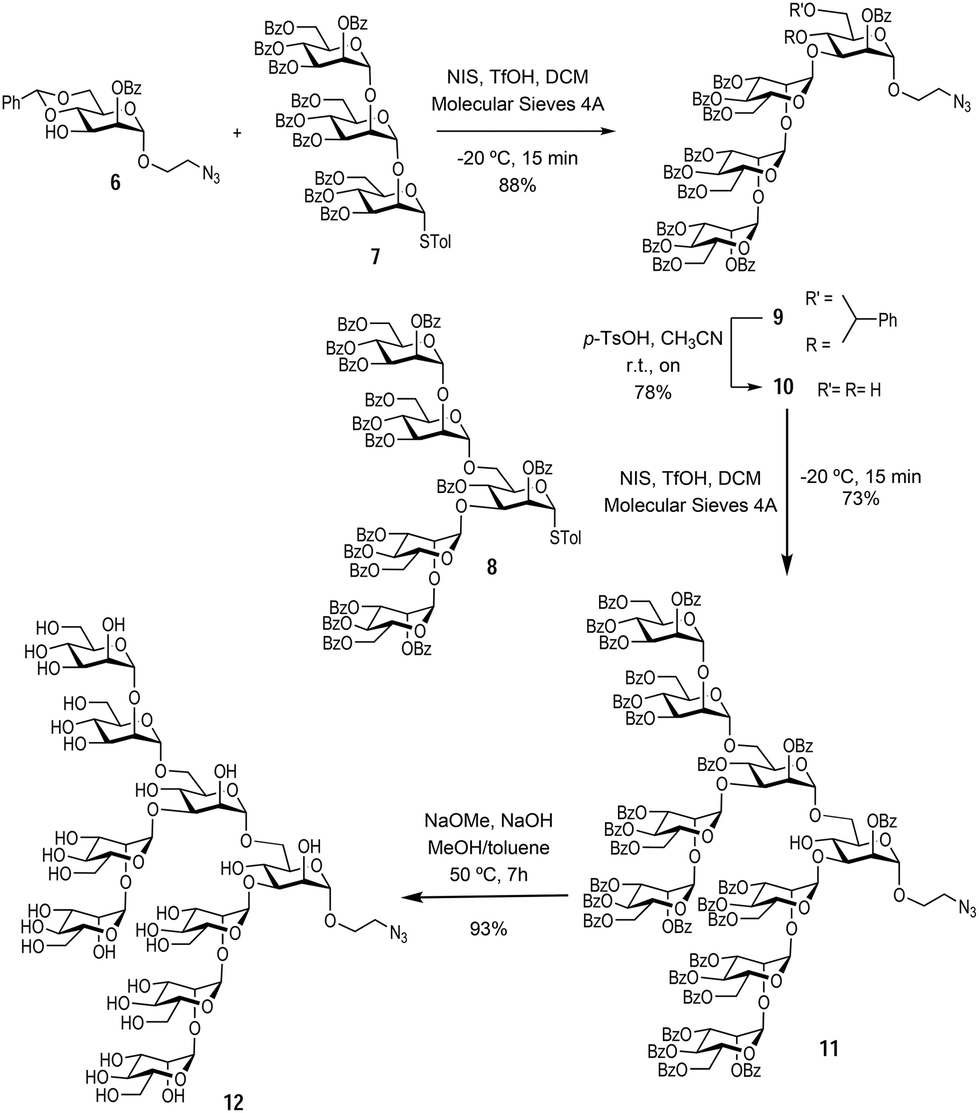 | ||
| Scheme 2 Synthesis of the αMan912. | ||
The glycosylation between trisaccharide 7 and mannose derivative 6 was performed using N-iodo succinimide and trifluoromethanesulfonic acid as the glycosylation promotor to obtain tetrasaccharide 9 in 88% yield. Next, the benzylidene acetal was removed in acidic media using p-TsOH to afford the tetrasaccharide 10 (bearing unprotected hydroxyl groups at positions 4 and 6 of the terminal mannose at the reducing end) in 78% yield. The final glycosylation between tetrasaccharide 10 and pentasaccharide 8 took place only at the position 6 hydroxyl group of the acceptor, since this is more reactive than the sterically hindered hydroxyl group at position 4. This glycosylation was carried out using the same conditions as above to yield the protected nonasaccharide 11 in 73% yield. The nonasaccharide 11 was characterised by 1H and 13C NMR and ESI-MS. Finally, global deprotection step with NaOMe and 2 M NaOH in MeOH and toluene cleaved all O-benzoyl groups affording the αMan912 in excellent yield (Scheme 2). In this way, the synthesis of the alpha anomer of Man9 was achieved in an expedient manner through a convergent strategy. Both Man9 epitopes (alpha and beta) were prepared with a short spacer at the anomeric position functionalised with a terminal azido group. This group permits conjugation to multivalent scaffolds using the Cu(I) azide–alkyne cycloaddition (CuAAC) reaction.20,21
To examine the effect that the multivalent presentation has on the binding DC-SIGN to both anomers, we prepared the trivalent glycoclusters with the alpha and beta Man9 epitopes (Scheme 3).
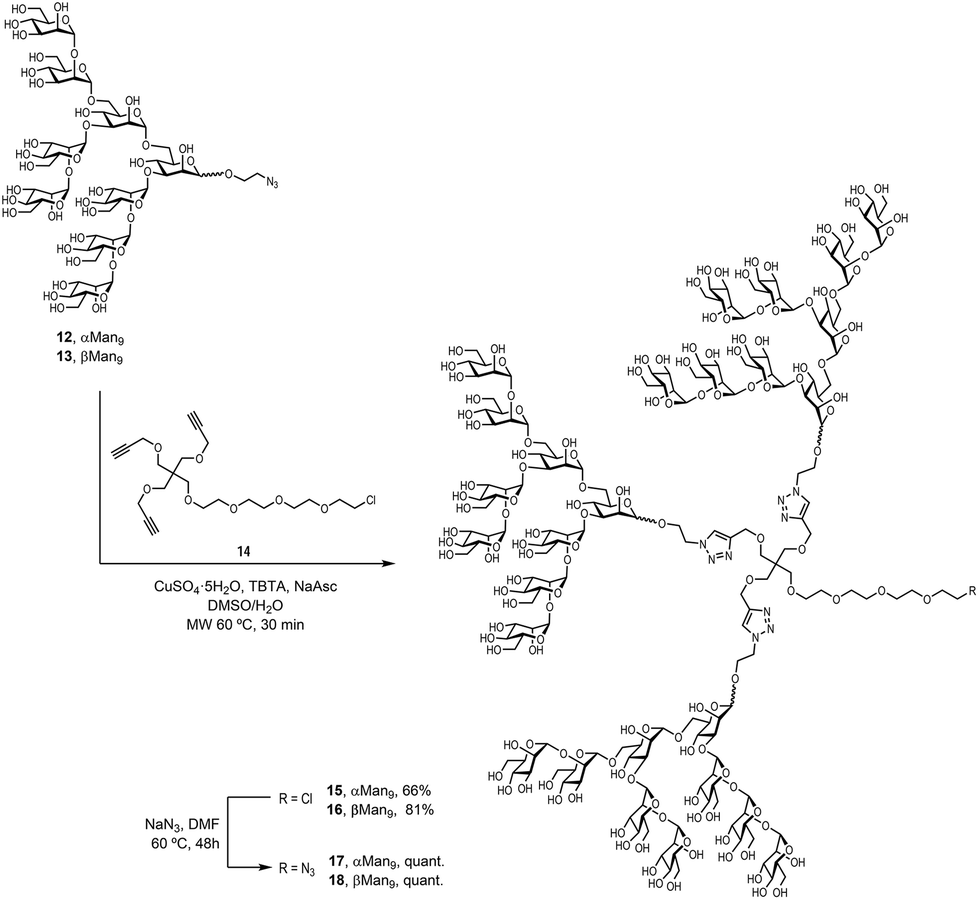 | ||
| Scheme 3 Synthesis of the trivalent glycoclusters 17 and 18. | ||
For this purpose, we employed a trialkynylated pentaerythritol scaffold, frequently used for the preparation of carbohydrate multivalent systems by our group. This scaffold 14 was prepared in two steps from pentaerythritol as previously reported.18 The coupling of αMan9 and βMan9 ligands was then carried out via a click chemistry19 CuAAC reaction promoted by CuSO4, sodium ascorbate and tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) under mild conditions to obtain the corresponding trivalent glycoclusters. These multivalent systems were purified by treatment with Quadrasil MP resin to remove the copper catalyst and by G50 Sephadex chromatography to afford the glycoclusters 15 and 16 in good yields (Scheme 3). The final step was the substitution of the chlorine atom for an azido group at the end of the linker at the focal position of the scaffold. This reaction was performed using an excess of NaN3 in DMF at 70 °C for two days furnish azido-functionalised glycoclusters 17 and 18 in excellent yields.
Finally, it was necessary to introduce a chromophore for the fluorescence polarization assays. An alkynyl derivative of fluorescein, the commercially available FAM-alkyne 6-isomer 19, was therefore conjugated to the αMan912, the βMan913, and their corresponding trivalent glycoclusters 17 and 18, respectively, by a CuAAC click reaction promoted by CuBr and TBTA in DMSO at room temperature (Scheme 4).
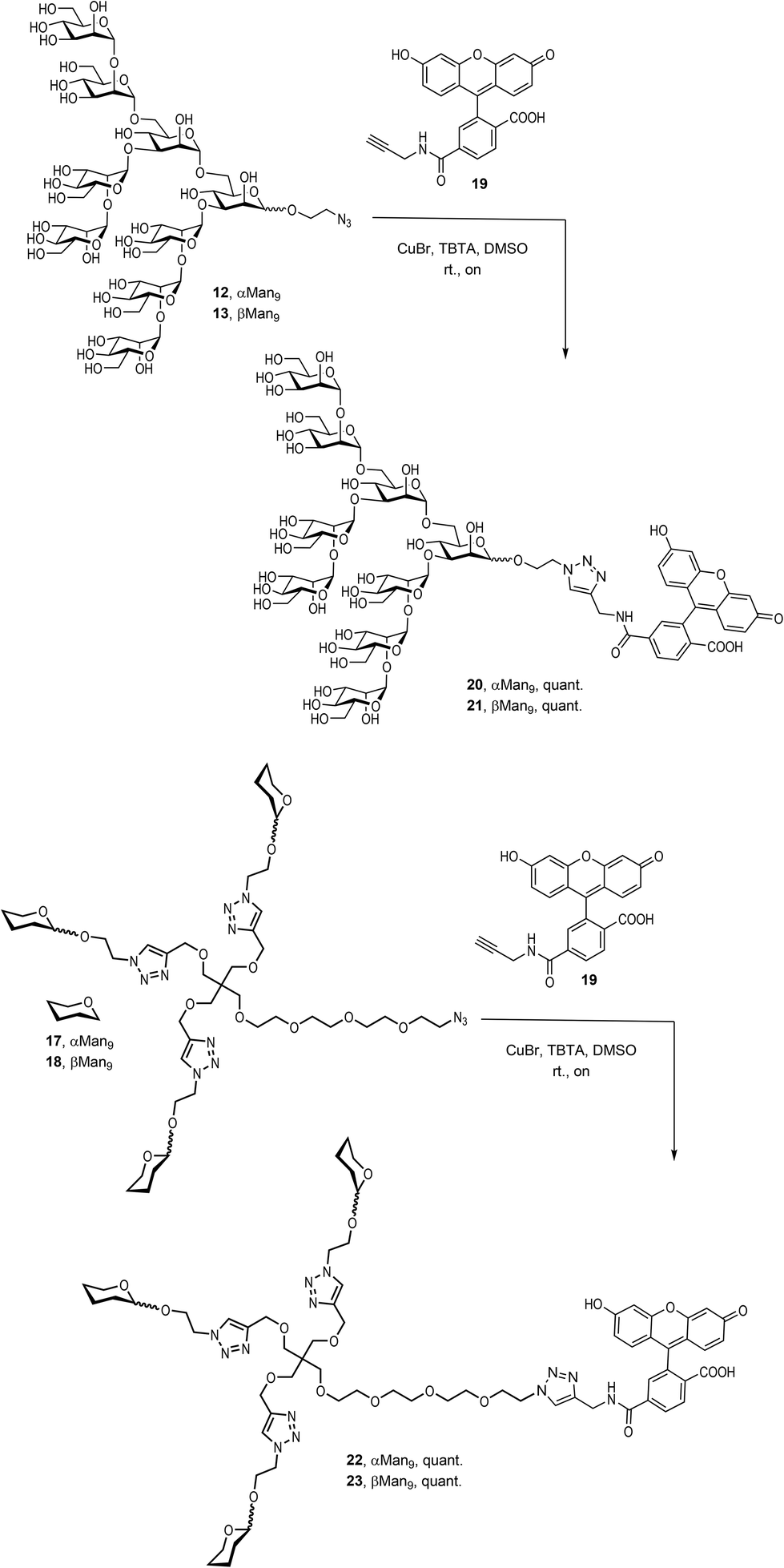 | ||
| Scheme 4 Synthesis of fluorescence labelled compounds 20, 21, 22 and 23. | ||
The compounds were treated with Quadrasil MP resin and then, submitted to a LH20 Sephadex chromatography to deliver the fluorescently-labelled compounds 20, 21, 22 and 23 in excellent yields.
With the fluorescent tool compounds prepared, we evaluated their capacity to interact with DC-SIGN, the natural receptor of this Man9 ligand.
Fluorescence polarization assays
Fluorescence polarization is widely used to study binding events owing to several advantages of this technique.22,23 The assay requires only small quantities of ligands and receptors, is well suited for high-throughput screening and does not require the attachment of any of the partners to a surface, allowing ligands and receptors diffuse freely in solution. In particular, fluorescence polarization experiments have been successfully employed in the analysis of carbohydrate–protein interactions.24–27 Thus, we selected this assay to evaluate the binding affinity of our alpha and beta epimers of Man9 (20 and 21) and their corresponding multivalent systems (22 and 23) for DC-SIGN using the tetravalent Extracellular Domain (ECD) of the lectin. For this, we have used a microtiter plate where different concentrations of the protein (DC-SIGN ECD), ranging from 75 nM to 28 μM in Tris buffer, were added to the wells containing a fixed final 10 nM concentration of appropriate fluorescent ligands (20–23). We observed an increase in the fluorescence polarization value with increasing protein concentrations, demonstrating that the fluorescence ligands bound to DC-SIGN ECD. The resulting Langmuir isotherm curves are represented in Fig. 3. From these curves, the KD of the binding process for each ligand was calculated. For the α and β anomers of Man9, similar values of KD, (5.2 ± 1.2 and 4.6 ± 1.3 μM, respectively) were found demonstrating that the configuration of the anomer in the mannosyl unit at the reducing end of the oligosaccharide does not play a critical role in the binding process. | ||
| Fig. 3 Langmuir isothermal curves of binding between DC-SIGN ECD and fluorescence compounds: (a) αMan9 (20); (b) βMan9 (21); (c) trivalent glycocluster 22; and (d) trivalent glycocluster 23. | ||
In order to evaluate the influence of the anomer configuration when the Man9 ligand is presented in a multivalent scaffold, fluorophore labelled glycoclusters 22 and 23 were tested in the fluorescence polarization assays (Fig. 3). In this case, the KD were one order of magnitude lower than the data found for the monovalent systems, indicating a clear multivalent effect (0.53 ± 0.09 and 0.37 ± 0.04 μM for the trivalent αMan9 and βMan9, respectively). Again, no significant differences between the dissociation constants of the two anomers were found as in the case of the monovalent ligands.
Conclusions
The stereochemical configuration of sugars is fundamental to their selective recognition by lectins. Indeed, just a simple difference in the configuration of one position can drive the recognition by one specific lectin, e.g. glucose (with all OH groups in equatorial disposition) versus galactose (which bears an axial OH at C4). Furthermore, the configuration of the glycosidic bonds between the monosaccharidic units that form an oligosaccharide are relevant for their recognition and function. In the case of high mannose, all the mannoses of the Man9 epitope display the alpha configuration of the glycosidic bonds, as usual for this type of sugar. However, the linkage between the Man9 epitope and the GlcNAc disaccharide exhibits a beta configuration. This glycosidic bond configuration is rare for mannoses and presents a significant hurdle towards addressing this bond configuration via chemical synthesis. All N-glycans have this moiety as connection with the corresponding Asn in the N-glycan proteins. This specific arrangement is critical for the proper orientation of the mannosyl cluster, providing a rigid structure on the protein surface to optimize the binding to DC-SIGN. However, when new glycomultivalent systems are designed, the glycan epitope is normally conjugated to the multivalent scaffold with a flexible linker and therefore the specific orientation provided by the beta-mannose linkage is likely not necessary. To demonstrate this hypothesis, we have synthesised the Man9 epitope of high mannose with alpha and beta configuration in the terminal mannose at the reducing end (20 and 21) and the corresponding trivalent glycoclusters (22 and 23). These ligands were labelled with a fluorescein derivative and their binding to DC-SIGN evaluated by fluorescence polarization assays. Our findings have shown that the configuration, alpha or beta, of the anomeric position of the terminal mannose does not influence in the binding affinity for the natural receptor DC-SIGN and therefore they can be used interchangeably, meaning the more synthetically-accessible alpha-linked epimers can be deployed instead of the more challenging natural beta analogues. We anticipate this knowledge is of key relevance in the design of new glycosystems of high mannose, since notably reduces the complexity, the time and the cost of the glycan synthesis by circumventing the synthetic bottleneck of the beta mannose preparation.Experimental
Materials and methods
Solvents were HPLC grade and used as received unless otherwise stated. Size exclusion chromatography was performed with Sephadex LH20, G-25 and G-50 (GE Healthcare). Thin-layer chromatography (TLC) was performed on silica plates (Merck). Reagents and QuadraSil® MP were purchased from Sigma Aldrich. 2-Azidoethyl α-D-mannopyranoside17 and trialkynylated scaffold 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 were synthesised as previously described. NMR experiments were performed using a Bruker Advance DRX 400 instrument. NMR chemical shifts are reported in ppm (δ units) downfield from the CDCl3 signal or the HOD peak (D2O). 2D experiments (COSY and HSQC) were performed when necessary. NMR spectra were analysed with MestreNova software.
18 were synthesised as previously described. NMR experiments were performed using a Bruker Advance DRX 400 instrument. NMR chemical shifts are reported in ppm (δ units) downfield from the CDCl3 signal or the HOD peak (D2O). 2D experiments (COSY and HSQC) were performed when necessary. NMR spectra were analysed with MestreNova software.
Synthesis of ligands
:3) to give the compound 6 (320 mg, 0.72 mmol, 26%) as a colourless oil. [α]D = −37 (c 1.00, CHCl3). 1H-NMR (400 MHz, CDCl3) δ: 8.11 (m, 2H, H–Ar), 7.60 (m, 1H, H–Ar), 7.56–7.45 (m, 4H, H–Ar), 7.43–7.35 (m, 3H, H–Ar), 5.67 (s, 1H, Hacetal), 5.52 (dd, J2,3 = 3.6, J2,1 = 1.6 Hz, 1H, H-2), 5.01 (d, J1,2 = 1.6 Hz, 1H, H-1), 4.41 (dt, J3,4 = 9.6, J3,2 = 3.9 Hz, 1H, H-3), 4.33 (dd, J6a,6b = 9.9, J6a,5 = 4.4 Hz, 1H, H-6a), 4.06 (t, J4,3 = J4,5 = 9.5 Hz, 1H, H-4), 4.03–3.84 (m, 3H, H-5, H-6b, H-1′a), 3.73–3.63 (m, 1H, H-1′b), 3.55–3.40 (m, 2H, H-2′), 2.32 (d, JOH,3 = 4.2 Hz, 1H, OH). 13C-NMR (100 MHz, CDCl3) δ: 166.0, 137.1, 133.5, 129.9, 129.5, 129.3, 128.5, 128.3, 126.3, 102.2, 98.7, 79.2, 72.5, 68.7, 67.1, 66.9, 63.8, 50.4. ESI-MS m/z calcd for C22H23N3O7: 441.2; found: 464.2 [M + Na]+.
:2 to 1:1.25) to give the tetrasaccharide 9 (193 mg, 0.10 mmol, 88%) as a white solid. [α]D = −22 (c 1.00, CHCl3). 1H-NMR (400 MHz, CDCl3) δ: 8.22 (m, 1H, H–Ar), 8.19–8.12 (m, 2H, H–Ar), 8.09–7.92 (m, 7H, H–Ar), 7.92–7.81 (m, 7H, H–Ar), 7.76–7.68 (m, 2H, H–Ar), 7.64–7.19 under CDCl3 (m, 36H, H–Ar), 7.14–6.95 (m, 4H), 6.78 (m, 1H), 6.05–5.81 (m, 5H), 5.79–5.69 (m, 2H), 5.64 (dd, J = 9.2, J = 3.1 Hz, 1H), 5.57 (s, 1H, H-1), 5.49 (br s, 1H, Hacetal), 5.35 (s, 1H, H-1), 5.05 (d, J = 1.5 Hz, 1H, H-1), 4.91 (s, 1H, H-1), 4.72–4.50 (m, 4H), 4.47 (t, J = 2.8 Hz, 1H), 4.38 (m, 1H), 4.35–4.13 (m, 4H), 4.07–3.94 (m, 3H), 3.94–3.79 (m, 3H), 3.65 (m, 1H), 3.35 (m, 2H). 13C-NMR (100 MHz, CDCl3) δ: 166.4, 166.0, 165.9, 165.8, 165.5, 165.3, 165.3, 165.2, 165.2, 165.0, 164.8, 137.0, 133.7, 133.5, 133.3, 133.2, 133.1, 133.1, 133.1, 133.0, 132.8, 130.1–129.6, 129.3, 129.2, 129.2, 129.1, 128.9–128.3 101.9, 99.8, 90.1, 79.1, 71.9, 71.4, 70.4, 70.1, 69.8, 69.7, 69.5, 68.8, 67.6, 67.2, 67.0, 66.3, 64.1, 63.6, 63.5, 62.9, 62.5, 50.4. ESI-MS m/z calcd for C110H93N3O32: 1967.6; found: 1990.4 [M + Na]+. ESI-HRMS m/z calcd for C110H93N3O32Na [M + Na]+: 1990.5634; found: 1990.5609
:5) to give the tetrasaccharide 10 (130 mg, 0.07 mmol, 78%) as a white amorphous solid. [α]D = −15 (c 1.00, CHCl3). 1H-NMR (400 MHz, CDCl3) δ: 8.14 (m, 2H, H–Bz), 8.07–7.93 (m, 13H, H–Bz), 7.90–7.82 (m, 4H, H–Bz), 7.70 (m, 2H, H–Bz), 7.61–7.18 under CDCl3 (m, 34H, H–Bz), 6.13 (t, J = 10.2 Hz, 1H), 6.03–5.90 (m, 3H), 5.84–5.72 (m, 2H), 5.67–5.57 (m, 3H, 1H-1), 5.52 (d, J = 1.6 Hz, 1H, H-1), 5.09 (br s, 1H, H-1), 5.00 (d, J = 1.7 Hz, 1H, H-1), 4.73–4.31 (m, 12H), 4.31–4.15 (m, 2H), 4.01 (m, 2H), 3.95–3.78 (m, 3H), 3.59 (dt, J = 10.4, 4.8 Hz, 1H), 2.34 (m, 2H). 13C-NMR (100 MHz, CDCl3) δ: 166.4, 166.3, 166.0, 165.9, 165.7, 165.5, 165.2, 164.9, 133.6, 133.5, 133.4, 133.3, 133.2, 133.0, 130.2–129.8, 129.6, 129.5, 129.4, 129.3, 129.1, 129.0, 128.9, 128.6–128.4, 100.8, 99.4, 98.1, 76.4, 73.3, 72.4, 70.5, 70.3, 69.9, 69.7–69.6, 69.4, 68.2, 67.8, 67.0, 66.6, 63.9–63.8, 62.6, 62.3, 50.4 (C-2′). ESI-MS m/z calcd for C103H89N3O32: 1879.5; found: 962.5 [M + Na]+. ESI-HRMS m/z calcd for C103H89N3O32Na [M + Na]+: 1902.5321; found: 1902.5307.
:5 → 1:1) to give the nonasaccharide 11 (185 mg, 0.04 mmol, 73%) as a white solid. [α]D = −19 (c 1.00, CHCl3). 1H-NMR (400 MHz, CDCl3) δ: 8.30–7.67 (m, 52H, HBz), 7.56–7.18 (m, 80H, HBz), 6.99 (m, 2H, HBz), 6.81 (t, J = 7.5, 1H, HBz), 6.22–5.80 (m, 16H), 5.55 (s, 1H, H-1), 5.46 (m, 2H, 2H-1), 5.32 (s, 1H, H-1), 5.18 (s, 1H, H-1), 5.05 (m, 2H, 2H-1), 4.74–4.02 (m, 36H, H-1, H-1), 3.84–3.72 (m, 2H), 3.49–3.32 (m, 3H). 13C-NMR (100 MHz, CDCl3) δ: 166.5, 166.4, 166.4, 166.3, 166.1, 166.0, 165.8, 165.6, 165.6, 165.4, 165.4, 165.3, 165.0, 165.0, 164.8, 164.8, 164.6, 133.5, 133.4, 133.4, 133.2, 133.2, 133.1, 130.2–129.5, 129.5, 129.5, 129.3, 129.2, 129.2, 129.0, 129.0, 128.9, 128.9, 128.7–128.3, 101.1, 100.9, 100.3, 99.9, 99.5, 98.5, 97.7, 97.5, 78.2, 77.9, 75.6, 72.4, 72.2, 71.9, 71.1, 70.8, 70.5, 70.1–69.4, 68.9, 68.1, 68.0, 67.7, 67.3–67.0, 66.6, 66.4, 66.1, 63.7, 63.5, 63.1, 62.6, 62.4, 50.4. ESI-MS m/z calcd for C245H203N3O73: 4354.2; found: 2200.1 [M + 2Na]2+ and 1473.5 [M + 3Na]3+. ESI-HRMS m/z calcd for C245H203N3O73Na [M + 2Na]2+: 2189.1115; found: 2189.1088.
:1, 1.7 mL), NaOMe (11 mg, 0.20 mmol) and a NaOH 2 M solution (0.7 mL) were added and the reaction was stirred at 50 °C for 5 h. After neutralization with Amberlite IR-120H+, the solution was filtered and concentrated. The crude was purified by size-exclusion chromatography (Sephadex G-25, H2O/MeOH 9/1), giving Man9 (12) (55 mg, 0.04 mmol, 93%) as a white amorphous solid. 1H-NMR (400 MHz, D2O) δ: 5.40 (s, 1H, H-1), 5.33 (s, 1H, H-1), 5.30 (s, 1H, H-1), 5.15 (s, 1H, H-1), 5.08–5.01 (m, 3H, 3 × H-1), 4.88 (s, 1H, H-1), under D2O (1H, H-1), 4.19–3.60 (m, 58H). 13C-NMR (100 MHz, D2O) δ: 102.2, 100.8, 100.6, 100.0, 99.5, 98.0, 78.8–78.5, 73.2, 73.2, 72.7, 71.1, 71.1, 70.3–70.0, 69.6, 66.9, 66.8, 66.6, 65.7, 65.5, 65.2, 61.2–61.0, 50.2. ESI-MS m/z calcd for C56H95N3O46: 1545.5; found: 1568.2 [M + Na]+, 795.5 [M + 2Na]2+.
:1, 0.5 mL). Fresh solutions of CuSO4·5H2O (1.80 μmol), tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) (3.59 μmol) and sodium ascorbate (5.39 μmol) were added to a sealed microwave vial. The solution was heated at 60 °C in a microwave oven for 30 min. A metal scavenger resin, QuadrasilMP, was added to the reaction solution and stirred for 20 min at rt. After that, the mixture was filtered and the resulting solution was purified by size-exclusion chromatography (Sephadex G-50, H2O/MeOH 9:1) yielding the glycocluster 15 (12 mg, 2.37 μmol, 66%) as a white amorphous solid. 1H-NMR (400 MHz, D2O) δ: 8.06 (s, 3H, Htriazole), 5.41 (br s, 3H, 3H-1), 5.37–5.27 (m, 6H, 6H-1), 5.15 (br s, 3H, 3H-1), 5.09–5.02 (m, 9H, 9H-1), under D2O (6H, 6H-1), 4.67 (m, 3H, O CH2CHHN), 4.57 (br s, 3H, OCHHCtriazole), 4.14–3.62 (m, 198H). 13C-NMR (100 MHz, D2O) δ: 144.2, 125.4, 102.2, 100.7, 99.8, 99.5, 98.0, 78.8–78.4, 73.3–73.2, 72.7, 72.0, 71.1–71.0, 70.8, 70.3, 70.0, 70.0, 69.7, 69.6, 69.5, 66.9, 66.8, 65.9, 65.8, 65.6, 65.4, 65.2, 64.8, 63.4, 62.5, 61.0, 50.1, 43.2. ESI-MS m/z calcd for C191H320N9O145Cl: 5080.7; found: 2562.9 [M + 2Na]2+, 1715.5 [M + 3Na]3+, and 1295.7 [M + 4Na]4+.
:1, 0.5 mL). Fresh solutions of CuSO4·5H2O (1.80 μmol), TBTA (3.59 μmol) and sodium ascorbate (5.39 μmol) were added to a sealed microwave vial. The solution was heated at 60 °C in a microwave oven for 30 min. A metal scavenger resin, QuadrasilMP, was added to the reaction solution and stirred for 20 min at rt. After that, the mixture was filtered and the resulting solution was purified by size-exclusion chromatography (Sephadex G-50, H2O/MeOH 9:1) yielding the glycocluster 16 (14.8 mg, 2.91 μmol, 81%) as a white amorphous solid. 1H-NMR (400 MHz, D2O) δ: 8.05 (s, 3H, 3Htriazole), 5.42 (br s, 1H, 3H-1), 5.34–5.28 (m, 6H, 6H-1), 5.15 (br s, 3H, 3H-1), 5.09–5.03 (m, 9H, 9H-1), 4.87 (br s, 3H, 3H-1), 4.67 (m, 3H, OCH2CHHN), 4.61–4.56 (br s, 6H, H-1, OCHHCtriazole), 4.18–3.56 (m, 198H). 13C-NMR (100 MHz, D2O) δ: 144.1, 125.5, 102.3, 102.2, 100.8, 100.7, 100.6, 99.9, 99.5, 98.0, 80.9, 78.9–78.5, 74.0, 73.3–73.2, 72.7, 71.1, 70.8, 70.3–69.9, 69.7–69.5, 68.3–67.7, 66.9–66.8, 65.6–65.2, 63.5, 62.5, 61.1–61.0, 50.3, 44.7, 43.2. ESI-MS m/z calcd for C191H320N9O145Cl: 5080.7; found: 2561.1 [M + 2Na]2+, 1715.2 [M + 3Na]3+, and 1293.5 [M + 4Na]4+.
:1), yielding the fluorescent probe 20 (6.3 mg, quant.) as an orange amorphous solid. 1H-NMR (400 MHz, D2O) δ: 8.22–7.87 (m, 3H), 7.68 (br s, 1H), 7.30–7.07 (m, 2H), 6.87–6.54 (m, 4H), 5.40–5.24 (m, 3H, 3H-1), 5.11 (br s, 1H, 1H-1), 5.04 (br s, 3H, 3H-1), under D2O (2H, 2H-1), 4.64 (m, 2H), 4.12–3.57 (m, 58H). 13C-NMR (100 MHz, selected data obtained from HSQC, D2O) δ: 131.2, 128.2, 128.2, 127.9, 121.7, 103.0, 102.2, 100.8, 100.6, 100.0, 99.5, 98.0, 78.8–78.5, 73.2, 73.2, 72.7, 71.1, 71.1, 70.3–70.0, 69.6, 66.9, 66.8, 66.6, 65.7, 65.5, 65.2, 61.2–61.0, 50.2. ESI-MS: m/z calcd for C80H110N4O52: 1958.6, found: 978.1 [M − 2H]2−.
:1), yielding the fluorescent probe 21 (10.1 mg, quant.) as an orange amorphous solid. 1H-NMR (400 MHz, D2O) δ: 8.05–7.98 (m, 2H), 7.94 (m, 1H), 7.57 (br s, 1H), 7.15–7.07 (m, 2H), 6.69–6.63 (m, 4H), 5.37 (br s, 1H, H-1), 5.31–5.26 (m, 2H, 2H-1), 5.10 (br s, 1H, H-1), 5.06–5.00 (m, 4H, 4H-1), 4.65 (m, 2H, CH2Ctriazole), 4.59 (m, 2H, CH2CH2N), 4.49 (br s, 1H, H-1), 4.21 (m, 1H), 4.12–4.05 (m, 6H), 4.03–3.92 (m, 6H), 3.90–3.60 (m, 42H), 3.41 (m, 1H). 13C-NMR (100 MHz, selected data obtained from HSQC, D2O) δ: 131.0, 128.2, 128.2, 127.9, 121.7, 103.0, 102.3, 102.1, 100.7, 100.5, 99.9, 99.5, 97.9, 81.0, 78.9, 78.6, 78.5, 78.4, 74.0, 73.2–73.1, 72.6, 71.1, 70.3–69.9, 69.4, 68.4, 66.9–66.8, 65.6–65.4, 61.1–60.9, 50.3. ESI-MS: m/z calcd for C80H110N4O52: 1958.6, found: 1982.3 [M − H]−, and 1002.1 [M − 2H]2−.
:1), yielding the fluorescent probe 22 (8.7 mg, quant.) as an orange amorphous solid. 1H-NMR (400 MHz, D2O) δ: 8.06–7.78 (m, 6H), 7.57 (br s, 1H), 7.07–6.88 (m, 2H), 6.62–6.42 (m, 4H), 5.35–5.16 (m, 9H, 9H-1), 5.06 (br s, 3H, 3H-1), 4.96 (br s, 9H, 9H-1), under D2O (6H, 6H-1), 4.55–4.29 (m, 6H), 4.07–3.11 (m, 213H). 13C-NMR (100 MHz, selected data obtained from HSQC, D2O) δ: 126.8, 122.9, 102.9, 102.3, 100.7, 99.8, 99.5, 98.0, 78.8–78.4, 73.3–73.2, 72.0, 71.1–71.0, 70.3–69.9, 69.7–69.5, 66.9–66.8, 65.9, 65.6, 65.4, 65.2, 63.4, 62.5, 61.1–61.0, 50.1. ESI-MS: m/z calcd for C215H335N13O151: 5514.9, found: 2749.4 [M − 2H]2−, and 1833.1 [M − 3H]3−.
:1), yielding the fluorescent probe 23 (15.0 mg, quant.) as an orange amorphous solid. 1H-NMR (400 MHz, D2O) δ: 8.02–7.80 (m, 6H), 7.54 (br s, 1H), 7.01–6.87 (m, 2H), 6.58–6.46 (m, 4H), 5.40–5.30 (m, 6H, 6H-1), 5.21 (m, 6H, 6H-1), 5.06 (br s, 3H, 3H-1), 4.96 (m, 9H, 9H-1), 4.53–4.41 (m, 12H, 3H-1), 4.29 (br s, 6H), 4.07–3.05 (m, 204H). 13C-NMR (100 MHz, selected data obtained from HSQC, D2O) δ: 131.0, 129.6, 128.5, 124.0, 122.4, 103.8, 102.3, 102.2, 100.8, 100.7, 100.6, 99.9, 99.5, 98.0, 80.9, 78.9–78.4, 74.0, 73.3–73.2, 72.7, 72.0, 71.1, 70.3–69.9, 69.7–69.5, 69.2, 68.3, 67.7, 66.9–66.8, 65.6–65.2, 63.5, 62.4, 61.1–61.0, 50.1 (OCH2CH2N). ESI-MS: m/z calcd for C215H335N13O151: 5514.9, found: 2748.5 [M − 2H]2−, and 1830.8 [M − 3H]3−.
DC-SIGN ECD expression
DC-SIGN ECD expression and production was performed as previously described. Briefly, the DC-SIGN ECD protein is expressed as inclusion bodies in E. coli, refolded and purified using a mannose affinity chromatography step followed by a size exclusion.28,29 Both steps ensure respectively the selection of correctly folded and oligomerized protein.DC-SIGN binding assays by fluorescence polarization
The fluorescence polarization measurements were performed in 384-well microplates (black polystyrene, non-treated, Corning), using a TRIAD multimode microplate reader (from Dynex) with excitation and emission wavelengths of 485 and 535 nm, respectively. Fluorescent compounds 20–23 and DC-SIGN-ECD were dissolved in Tris buffer (25 mM, pH 8, 150 mM NaCl, 4 mM CaCl2). 15 μL of a 20 nM fluorescent ligand solution were transferred to each well. Then, 15 μL of protein solutions, with concentrations ranging from 75 nM to 28 μM, were added to the microplate wells. Therefore, the total sample volume in each well was 30 μL. The microplate was shaken in the dark for 5 min, before reading. Blank wells contained 15 μL of the DC-SIGN-ECD solution and 15 μL of Tris buffer, and their measurements were subtracted from all values. All experiments on samples were performed in replicates of three.Wells containing 15 μL of the 20 nM fluorescence compound solution and 15 μL of Tris buffer afforded the background polarization of the fluorescent molecule, in the absence of protein. This value was subtracted from the polarization values of all the samples, giving the increment in the fluorescence polarization (ΔP). The average ΔP values of three replicate wells were plotted against the concentration of DC-SIGN-ECD, and the resulting curve was fitted to the equation for a one-site binding model: y = ΔPmaxx/[KD + x] where ΔPmax is the maximal value of ΔP and KD is the dissociation constant of the interaction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by MINECO (CTQ2017-86265-P, PGC2018-099497-B-100 and IJCI-2015-23272), and ISCIII RETICS ARADyAL (RD16/0006/0011). Grants were co-funded by the European Regional Development Fund (ERDF). A. S.-H. and N. d. l. C thank MINECO for their Juan de la Cierva-Incorporacion and FPI contracts, respectively. This work used the Multistep Protein Purification Platform for human CLRs production of the Grenoble Instruct-ERIC Center (ISBG; UMS 3518 CNRS-CEA-UGA-EMBL) within the Grenoble Partnership for Structural Biology (PSB), supported by FRISBI (ANR-10-INBS-05-02) and GRAL, financed within the University Grenoble Alpes graduate school (Ecoles Universitaires de Recherche) CBH-EUR-GS (ANR-17-EURE-0003). F. F. also acknowledge the French Agence Nationale de la Recherche (ANR) PIA for Glyco@Alps (ANR-15-IDEX-02). We acknowledge Dr Michael P. O'Hagan for the revision of the English version of the manuscript.References
- A. Varki, Glycobiology, 2017, 27, 3–49 CrossRef CAS PubMed.
- H. Kaltner, J. Abad-Rodríguez, A. P. Corfield, J. Kopitz and H.-J. Gabius, Biochem. J., 2019, 476, 2623–2655 CrossRef CAS PubMed.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS PubMed.
- N. Jayaraman, Chem. Soc. Rev., 2009, 38, 3463–3483 RSC.
- V. Wittmann, Curr. Opin. Chem. Biol., 2013, 7, 982–989 CrossRef PubMed.
- B. Lepenies, J. Lee and S. Sonkaria, Adv. Drug Delivery Rev., 2013, 65, 1271–1281 CrossRef CAS PubMed.
- Y. van Kooyk and T. B. H. Geijtenbeek, Nat. Rev. Immunol., 2003, 3, 697–709 CrossRef CAS PubMed.
- L. Mathys and J. Balzarini, PLoS One, 2015, 10, e0130621, DOI:10.1371/journal.pone.013062.
- V. Y. Dudkin, M. Orlova, X. Geng, M. Mandal, W. C. Olson and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 9560–9562 CrossRef CAS PubMed.
- L. X. Wang, J. Ni, S. Singh and H. Li, Chem. Biol., 2004, 11, 127–134 CAS.
- S.-K. Wang, P.-H. Liang, R. D. Astronomo, T.-L. Hsu, S.-L. Hsieh, D. R. Burton and C.-H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3690–3695 CrossRef CAS PubMed.
- F. Chiodo, P. M. Enríquez-Navas, J. Angulo, M. Marradi and S. Penadés, Carbohydr. Res., 2015, 405, 102–109 CrossRef CAS PubMed.
- H.-C. Wen, C.-H. Lin, J.-S. Huang, C.-L. Tsai, T.-F. Chen and S.-K. Wang, Chem. Commun., 2019, 55, 9124–9127 RSC.
- H. Feinberg, R. Castelli, K. Drickamer, P. H. Seeberger and W. I. Weis, J. Biol. Chem., 2007, 282, 4202–4209 CrossRef CAS PubMed.
- J. Ramos-Soriano, J. J. Reina, B. M. Illescas, N. de la Cruz, L. Rodríguez-Pérez, F. Lasala, J. Rojo, R. Delgado and N. Martín, J. Am. Chem. Soc., 2019, 141, 15403–15412 CrossRef CAS PubMed.
- J. Ramos-Soriano, M. C. de la Fuente, N. de la Cruz, R. C. Figueiredo, J. Rojo and J. J. Reina, Org. Biomol. Chem., 2017, 15, 8877–8882 RSC.
- J. J. Reina and J. Rojo, Tetrahedron Lett., 2006, 47, 2475–2578 CrossRef CAS.
- R. Ribeiro-Viana, M. Sánchez-Navarro, J. Luczkowiak, J. R. Koeppe, R. Delgado, J. Rojo and B. G. Davis, Nat. Commun., 2012, 3, 1303 CrossRef PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3062 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- W. A. Lea and A. Simeonov, Expert Opin. Drug Discovery, 2011, 6, 17–32 CrossRef CAS PubMed.
- K. Kakehi, Y. Oda and M. Kinoshita, Anal. Biochem., 2001, 297, 111–116 CrossRef CAS PubMed.
- P. Sörme, B. Kahl-Knutsson, M. Huflejt, U. J. Nilsson and H. Leffler, Anal. Biochem., 2004, 334, 36–47 CrossRef PubMed.
- S. Maza, M. Mar Kayser, G. Macchione, J. Lopez-Prados, J. Angulo, J. L. de Paz and P. M. Nieto, Org. Biomol. Chem., 2013, 11, 3510–3525 RSC.
- C. Solera, G. Macchione, S. Maza, M. M. Kayser, F. Corzana, J. L. de Paz and P. M. Nieto, Chem. – Eur. J., 2016, 22, 2356–2369 CrossRef CAS PubMed.
- I. Joachim, S. Rikker, D. Hauck, D. Ponader, S. Boden, R. Sommer, L. Hartmann and A. Titz, Org. Biomol. Chem., 2016, 14, 7933–7948 RSC.
- J. J. Reina, I. Díaz, P. M. Nieto, N. E. Campillo, J. A. Páez, G. Tabarani, F. Fieschi and J. Rojo, Org. Biomol. Chem., 2008, 6, 2743–2754 RSC.
- G. Tabarani, M. Thépaut, D. Stroebel, C. Ebel, C. Vivès, P. Vachette, D. Durand and F. Fieschi, J. Biol. Chem., 2009, 284, 21229–21240 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra and selected HSQC and MS spectra. See DOI: 10.1039/D0OB01380C |
| This journal is © The Royal Society of Chemistry 2020 |