
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Defluorination of 4-fluorothreonine by threonine deaminase†
Linrui
Wu
and
Hai
Deng
 *
*
Department of Chemistry, University of Aberdeen, Aberdeen AB24 3UE, UK. E-mail: h.deng@abdn.ac.uk
First published on 24th July 2020
Abstract
4-Fluorothreonine (4-FT) is the only naturally occurring fluorinated amino acid antibiotic. Although two conserved proteins in the 4-FT pathway have been found to be involved in self-detoxification mechanisms, the 4-FT-producing strains may also require an alternative pathway to degrade the intracellular 4-FT. In this study, we examined the possible degradation role of three enzymes involved in threonine metabolite pathways toward 4-FT as a possible degradation route to avoid in vivo 4-FT accumulation. Among these three enzymes, threonine deaminase was found to catalyse a defluorination reaction to generate 4-hydroxy-α-ketobutyrate, which is supposed to be further metabolised by an aldolase that likely is a unique occurrence in the 4-FT-producing strains. Our finding may constitute a 4-FT degradation pathway as a complementary resistance mechanism.
Introduction
For decades, bacteria have been known as the most proliferative producers of natural products (NPs), which account for over 40% of lead small molecules in drug discovery.1 In most cases, in bacteria, genes responsible for their synthesis are clustered together to form biosynthetic gene clusters (BGCs), enabling a more efficient expression regulation and functional assembly.2 To guard the host strain from the generated NPs, it is a common strategy for BGCs to incorporate a resistance gene, conferring a detoxification mechanism for self-protection.3 This knowledge in turn has facilitated the genome mining process for new NP discovery.4Despite the similarities of the steric properties between fluorine and hydrogen atoms, fluorinated drug molecules are often endowed with advantages compared to their non-fluorinated counterparts, such as a low susceptibility towards oxidative metabolism by cytochrome P450 monooxygenases and degrading hydrolytic effects.5 In contrast to the widely distributed synthetic fluorinated compounds, naturally occurring fluorometabolites are rare. However, their occurrence has attracted new research interest from the blooming fluorinated drug discovery industry using fluorine biocatalysts under mild conditions for stereo- and regio-selective fluorination.6 The most well-studied fluorometabolites, the toxin fluoroacetate (FAc) 1 and the antibiotic 4-fluorothreonine (4-FT) 2, were first identified in Streptomyces cattleya.7 The biosynthetic pathways of these two fluorometabolites have been well studied with the hallmark studies of the first naturally occurring fluorination enzyme, fluorinase, the first committed enzyme in fluorometabolism.8–11 Later on, genome mining revealed several other putative fluorometabolite-producing strains including Nocardia brasiliensis, Actinoplanes sp. N902-109, and the marine-derived Streptomyces xinghaiensis.12,13 In 2012, we identified the prolific fluorometabolite-producing strain Streptomyces sp. MA37 in Ghana, Africa that also produces a wide range of fluorinated NPs, including 5-fluoro-2,3,4-trihydroxypentanoic acid (FHPA) 3 (Fig. 1A).12,14 Unlike most of the microbial natural products, many of the genes responsible for bacterial fluorometabolites are spread in the genomes of the corresponding fluorometabolite-producing strains.15 Enzymes from primary metabolism, such as 5-methylthio-5-deoxy-ribose isomerase, may also act on the intermediates in the fluorometabolite pathway, due to the structural similarity among the fluorinated intermediates and intermediates from primary metabolism.10
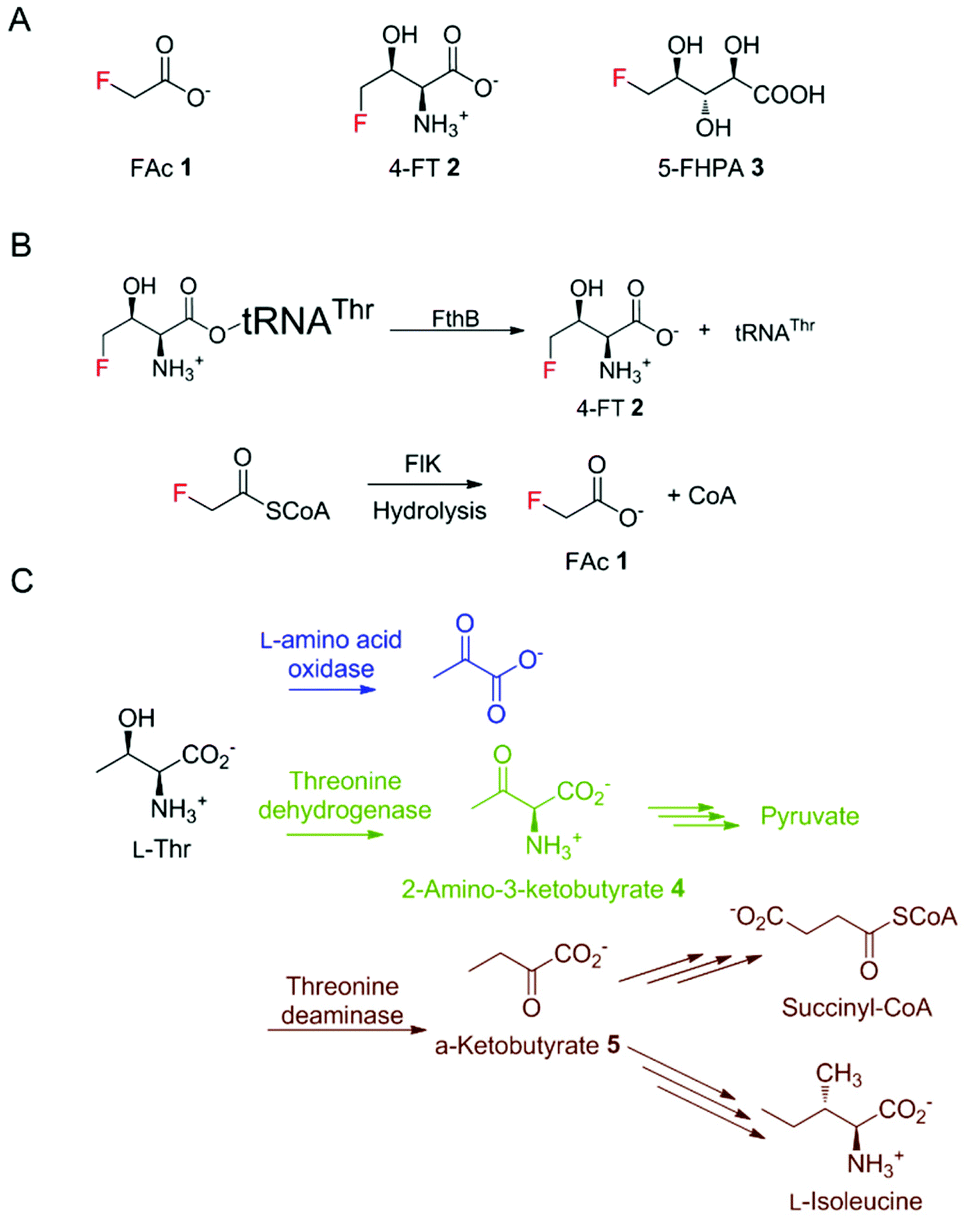 | ||
| Fig. 1 (A) The three fluorometabolites produced by MA37. (B) The detoxification of 4-FT and FAc in S. cattleya. (C) The enzymatic reactions on L-Thr in varisous organisms. | ||
Due to the similar steric properties between fluorine and hydrogen, both FAc 1 and 4-FT 2 can enter various primary metabolite pathways related to acetate and L-Thr, respectively. These misincorporations can be disruptive and toxic to the producing strains, which in turn have evolved to counteract with potential detoxification strategies.16 The resistance mechanism of host strains towards FAc 1 has been characterized. The toxicity of 1 arises from the transformation of 1, via fluoroacetyl-CoA, to the potent toxin (2R,3R)-fluorocitrate, which is a potent inhibitor of aconitase, a key enzyme of the citric acid cycle. In the case of 1 in the producing strains, a specialised fluoroacetyl-CoA thioesterase, FlK, can selectively hydrolyse the misacylated fluoroacetyl-CoA to prevent the activation of fluoroacetate (Fig. 1B).17–19 The detoxification mechanism of 4-FT 2 employed by S. cattleya has also been extensively elaborated by the study of FthB and FthC, the fluorothreonyl-tRNA deacylase and 4-FT transporter, respectively. The structural similarity between 4-FT 2 and L-Thr allows 2 to form a misacylated tRNA, 4-FT-tRNA, hence entering the ribosomal protein biosynthesis. FthB, the fluorothreonyl-tRNA deacylase, can hydrolyse the 4-FT misacylated tRNA and reduce the misincorporation of 4-FT (Fig. 1B).20 Interestingly, the genetic knock-out studies on both S. cattleya and S. MA37 showed that its inactivation does not result in the fluctuation of fluorometabolite biosynthesis or the strain's phenotype alteration.21 The inactivation of the 4-FT transporter FthC, which led to increased intracellular 4-FT 2 accumulation, also does not interfere with cell growth,20 leaving an unanswered question of the metabolic fate of these intracellular 4-FT 2.
In different organisms, there exist various enzymes in primary metabolism taking up L-Thr as a substrate for its transformation (Fig. 1C). Considering the structural similarity of 4-FT and L-Thr, these primary enzymes might also be used for the degradation of intracellular 4-FT 2, counteracting its poisonous effect towards the host strain.
Here we report the biochemical defluorination reaction of 4-FT 2, catalysed by threonine deaminase (TDA), which may serve an alternative pathway of 4-FT 2 to prevent its intracellular accumulation. The products, resulting from this defluorination reaction, were determined to be inorganic fluoride and 4-hydroxy-α-ketobutyrate through 19F-NMR analysis, chemical derivatization and high-resolution mass spectroscopic analysis, and the latter product was proposed to be further degraded into pyruvate by the promiscuous enzyme 2-keto-4-hydroxyglutarate aldolase through comparative genomic analysis.
Results and discussion
Three enzymes that can metabolise L-Thr, threonine dehydrogenase (TDH), L-amino acid oxidase (LAAO) and threonine deaminase (TDA), were chosen to be tested on 4-FT 2. LAAO (Sigma-Aldrich product no. A9253), mostly found in snake venom, catalyses the deamination of L-amino acids to generate the corresponding α-keto acid, ammonia and hydrogen peroxide22. LAAO has relatively broad substrate tolerance towards different amino acids and was found to transform L-Thr to pyruvate (Fig. 1C), albeit with less efficiency compared to amino acids with hydrophobic side chains.23 In our previous studies, we successfully overexpressed the 4-fluorothreonine transaldolase FTaseMA from Streptomyces sp. MA37 and characterized its function in the last step of the transaldol reaction of 4-FT biosynthesis. It utilizes L-Thr and fluoroacetaldehyde (FAd) as substrates and pyridoxal phosphate (PLP) as a cofactor to generate 4-FT.24 To test whether LAAO acts on 4-FT 2, we performed a coupling reaction. An assay of LAAO with FTaseMA using L-Thr and fluoroacetaldehyde (FAd) to generate 4-FT 2in situ was carried out (Fig. S1†). The reaction was monitored by 19F-NMR analysis. However, there was no apparent new organofluorine signal appearing in the 19F-NMR spectrum while 4-FT 2 from the FTaseMA biochemical reaction accumulated, suggesting that the fluorine atom at the C4 position of 4-FT 2 strongly inhibits the reactivity of LAAO.Next, we overexpressed the genes, those that encode TDH and TDA from E. coli, in E. coli BL21(DE3) and purified them to near homogeneity (Fig. S2A and S3A†). TDH is responsible for catalysing the oxidation of L-Thr to L-2-amino-3-oxobutanoate 4 in amino acid metabolism25 while TDA catalyses the initial step from L-Thr to α-ketobutyrate 5 (Fig. 1C). TDA is a crucial enzyme leading to the branched pathway of amino acid degradation and L-isoleucine biosynthesis26 (Fig. 1C). The catalytic activities of the recombinant proteins have been confirmed (Fig. S2B and S3B†). To examine the catalytic capacity of TDH towards 4-FT 2, an assay of TDH coupled with FTaseMA was performed overnight, and the reaction mixture was analysed by 19F-NMR. No new organofluorine signals were observed, demonstrating that 4-FT is not the substrate of TDH (Fig. S4†). We then carried out the coupling assay of TDA with FTaseMA and monitored the biotransformation by 19F-NMR. Interestingly, the level of 4-FT 2 in this coupling reaction was reduced compared to the control reaction in which TDA was inactivated (Fig. S5†). However, no new organofluorine signals were observed in the 19F-NMR spectrum, indicating that no organofluorines were produced. Instead, we found a fluorine signal at −121.35 ppm, corresponding to the fluoride ion. To further demonstrate the defluorination reaction by TDA, we developed a two-step biotransformation in a time-course fashion. 4-FT 2 was first synthesised by FTaseMA with PLP and L-Thr, followed by removing FTaseMA by heat denaturing. After the depletion of FTaseMA, TDA was supplemented to the system. As shown in Fig. 2A, 4-FT 2 was constantly consumed as observed throughout the time scale, and the decrease of 4-FT 2 was only accompanied by the emergence of the inorganic fluoride ion in the 19F-NMR spectrum (Fig. 2B).
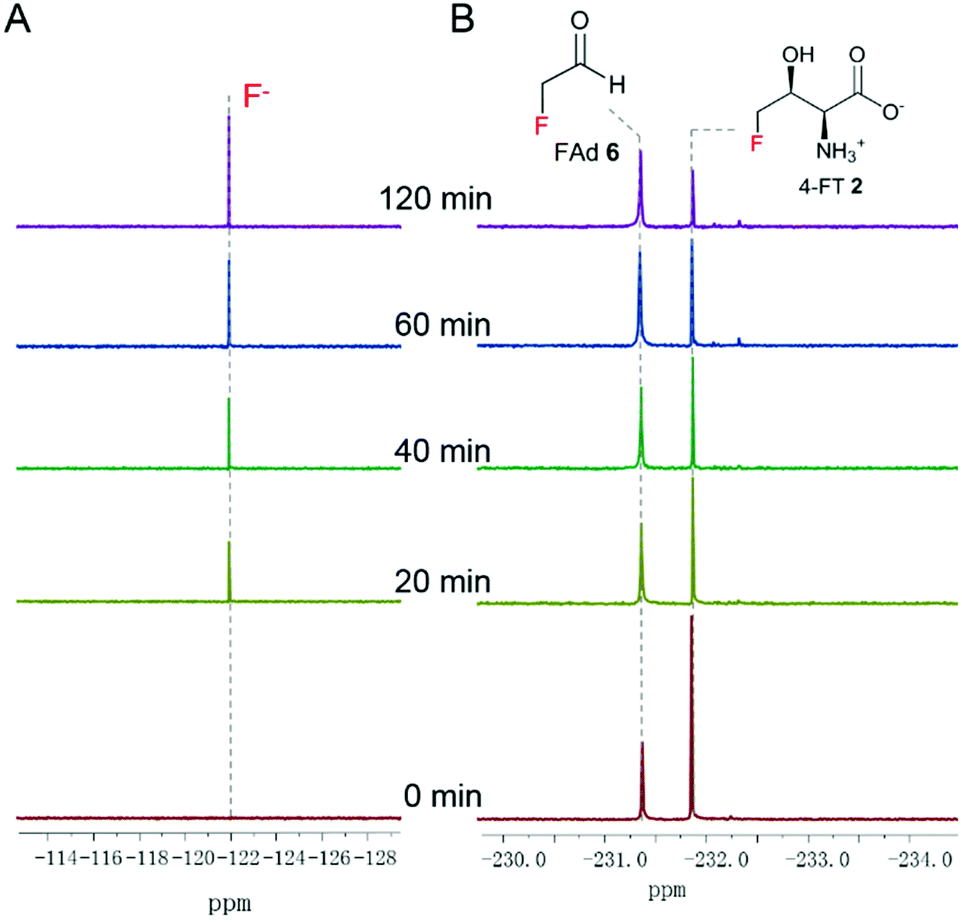 | ||
| Fig. 2 The time course of the coupling reaction of FTaseMA and TDA, showing the generation of the fluoride ion (A) and the consumption of 4-FT 2 (B). FAd 1 and 4-FT 2 were the substrate and the product left in the FTaseMA biotransformation reaction, respectively. | ||
To examine what products were generated during the defluorination catalysed by TDA, we applied the derivatization analytical method that we used in our previous study.24 2,4-Dinitrophenylhydrazine (DNPH) was used as a derivatization agent for monitoring aldehyde and ketone species in the reaction mixture. The reaction mixture was derivatized by DNPH, followed by HPLC analysis of keto-DNPH derivatives.27 The derivatized mixtures were also subjected to LC-high-resolution MS (HR-MS) analysis. In the coupling assay (Fig. 3A), three expected DNPH-derived compounds containing keto functional groups, such as α-ketobutyrate 5 (Fig. S6A†), fluoroacetaldehyde (FAd) 7 (Fig. S6B†), and acetaldehyde (Ad) 7 (Fig. S6C†), were observed. FAd 6 and Ad 7 are the unreacted components from the FTaseMA biotransformation system, while α-ketobutyrate 5 is the product of TDA which acts on the residue of L-Thr from the FTaseMA biotransformation system. Interestingly, we also observed the presence of a new peak at a retention time (RT) of 9.8 min. HR-MS analysis indicated that an ion with an m/z value of 299.0629 (calcd m/z = 299.0622; calcd MF = C10H11O7N4+, Δ = 0.675 ppm) corresponds to this DNPH-derived compound 8. In the control experiments where either a combination of both denatured FTaseMA and TDA (Fig. 3B and C) or a combination of active FTaseMA and denatured TDA was used in the presence of substrates and co-factors, this new peak was absent, demonstrating that 9 is generated by the coupling reaction in which both FTaseMA and TDA were active. As a result, it is likely that TDA catalyses a defluorination reaction on 4-FT 2 to yield the inorganic fluoride ion and 2-keto-4-hydroxybutyric acid 8.
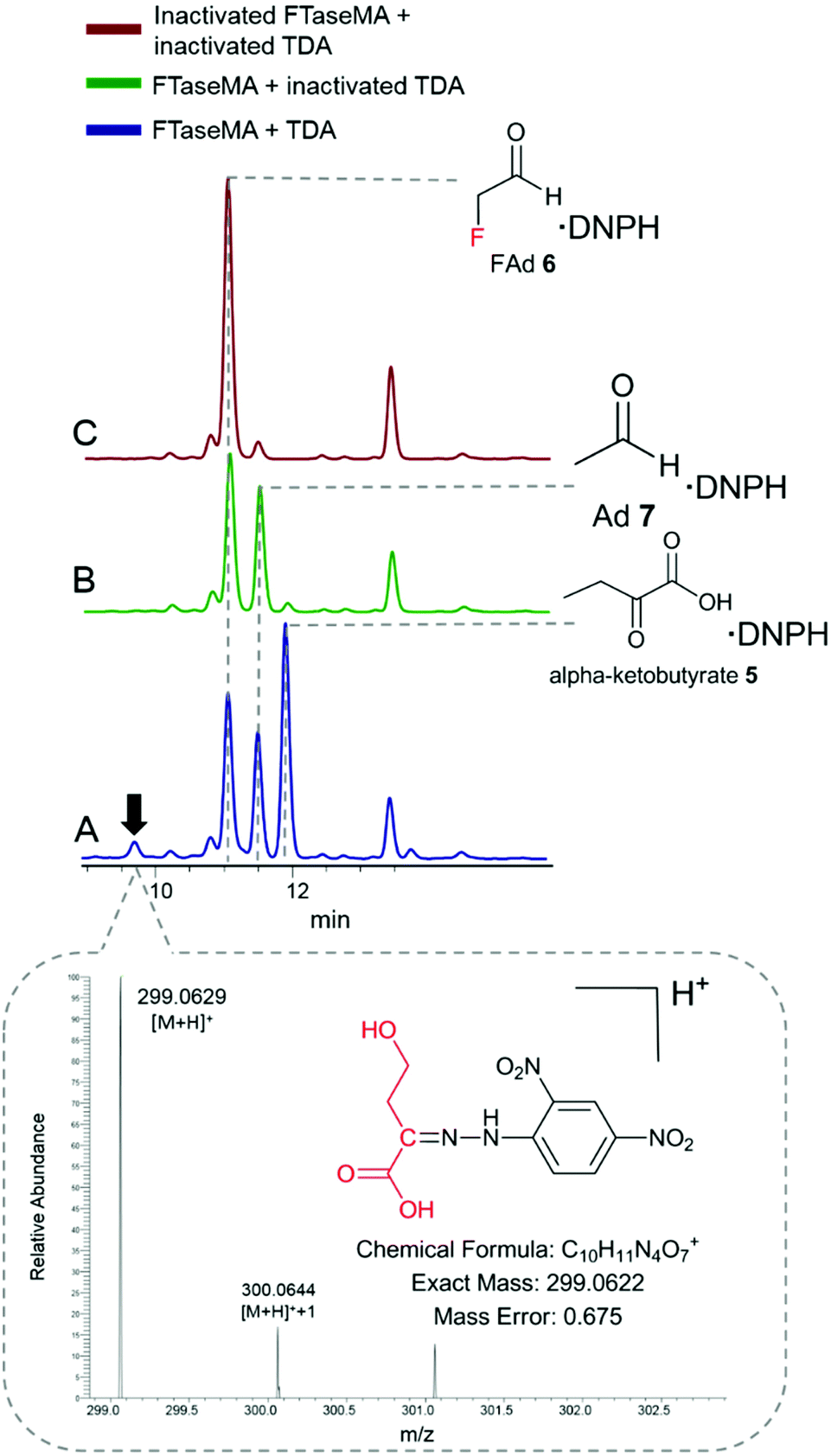 | ||
| Fig. 3 HPLC analysis of the reaction mixture after DNPH derivatization, showing the production of a new compound (A) that corresponds to 2-keto-4-hydroxybutyric acid 8, using the inactivated TDA (B), and the inactivated FTaseMA/TDA (C) as the controls. The identity of DNPH-8 was confirmed by HRMS analysis (the blowup). | ||
Taken together, a catalytic mechanism of the defluorination on 4-FT, catalysed by TDA based on the current knowledge of PLP-dependent biochemistry, is shown in Fig. 4. Both L-Thr and 4-FT 2 undergo the same pathway of β-dehydration mediated by TDA to generate the key intermediate 9. In the case of L-Thr, hydrolysis of 9 would yield the imine molecule 10 which is further hydrolysed into α-keto-butyrate 5 (Fig. 4, blue curly arrow). In the case of 4-FT 2, it is likely that the hydrolysis, however, would result in using the fluoride ion as the leaving group to produce 2-iminobut-3-enoic acid 11. Subsequent Michael addition by water results in 2-keto-4-hydroxybutyric acid 8.
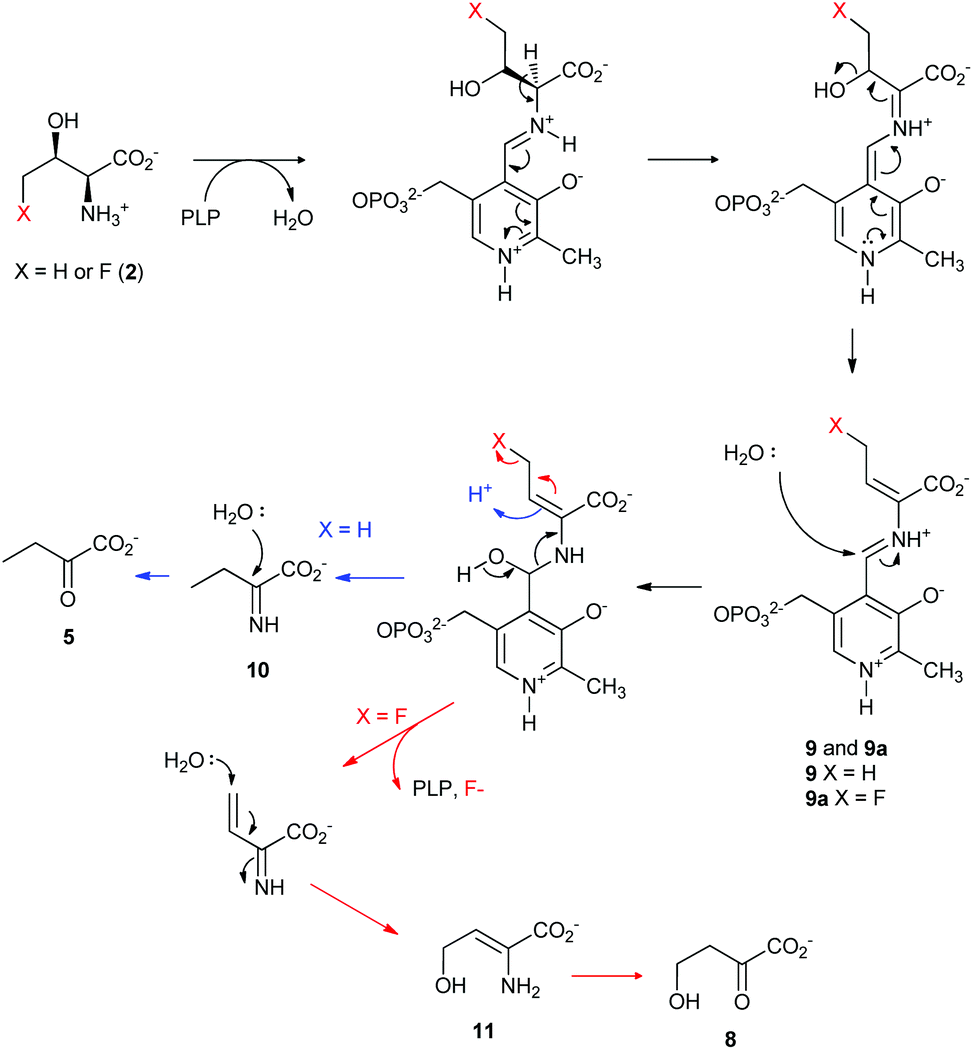 | ||
| Fig. 4 The proposed defluorination mechanism of 4-FT, catalysed by TDA based on the current knowledge of PLP-dependent biochemistry. The release from PLP can branch in two ways depending on the substitution. | ||
The observed defluorination catalysed by TDA is considerably interesting as it unusually cleaves the C–F bond, the strongest covalent bond in organic chemistry. Very few enzymes discovered thus far have such capacity, one of which is fluoroacetate dehalogenases from Burkholderia sp. and Delftia acidovorans.28,29 These enzymes recruit Asp in their active sites to displace the fluoride ion in an SN2 manner to generate an enzyme-glycolate ester intermediate, followed by subsequent hydrolysis of this ester by water to yield glycolate and recover the enzymes to their original states.30
The identified defluorination reaction of 4-FT 2 suggested that the host strain may recruit TDA from the primary metabolism for the intracellular 4-FT degeneration, preventing a toxic level of 4-FT 2 accumulation in vivo. The examination of the metabolic fate of 2-keto-4-hydroxybutyric acid 8 revealed that, although not present as a common primary metabolite, it can be accepted by the promiscuous 2-keto-4-hydroxyglutarate aldolase to be converted into formaldehyde and pyruvate as presented by previous studies.31 A copy of gene-encoded 2-keto-4-hydroxyglutarate aldolase was found in the genome of S. cattleya (accession no. AEW93759). BLAST search using its sequence as a query against the annotated MA37 draft genome in the RAST server revealed the presence of one gene encoding 2-keto-4-hydroxyglutarate aldolase in MA37. Interestingly, 2-keto-4-hydroxyglutarate aldolase is rarely found in Streptomyces including Nocardia brasiliensis and Actinoplanes sp. N902-109 that contain fluorinases.12,32 Only twelve homologous genes were found among thousands of Streptomyces genomes in the NCBI database. It is worth noting that Nocardia brasiliensis cannot produce any fluorometabolites under the laboratory conditions,12,32 and Actinoplanes sp N902-109 is not available in the public domain so that whether this strain has the capacity to produce fluorometabolites has not been assessed.12Streptomyces xinghaiensis, on the other hand, is the fluorometabolite-producing strain that originated from marine environments which only produces FAc 1. Bioinformatics analysis of the fluorometabolite biosynthetic gene cluster in S. xinghaiensis suggested that the putative FTase gene is truncated, meaning that the strain has no capacity for producing 4-FT 2.16 Coincidently, S. xinghaiensis lacks the 2-keto-4-hydroxyglutarate aldolase gene in its genome as well. Taken together, TDA and 2-keto-4-hydroxyglutarate aldolase may constitute a 4-FT degradation pathway in the producing strains to recycle formaldehyde and pyruvate in primary metabolism. It may serve as a complementary mechanism in addition to FthB to maintain a minimal level of intracellular 4-FT 2 for a normal state of ribosomal protein biosynthesis. The potential 4-FT degradation pathway of 4-FT catalysed by putative TDA and 2-keto-4-hydroxyglutarate aldolase from the 2 producing strains will be the focus of our future studies.
Conclusions
In this study, we tested several L-Thr metabolism related enzymes and conducted biochemical assays to determine their activity against 4-FT 2. Incubation of 4-FT 2 generated by FTaseMA with the recombinant TDA leads to the identification of an interesting 4-FT defluorination mechanism. Bioinformatics analysis found a copy of the 2-keto-4-hydroxyglutarate aldolase gene in both genomes of S. cattleya and S. MA37, indicating that there exists a new 4-FT degradation pathway for its detoxification and recycling. Combined with the fluorothreonyl-tRNA deacylase, they may serve as the 4-FT resistance mechanism employed by the fluorometabolite-producing strains to avoid homeostasis disruption. This discovery would also facilitate our understanding of the biochemistry of fluorometabolites.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was funded by an IBioIC PhD studentship (LW) and the Royal Society-NSFC Newton Mobility Grant Award (IEC\NSFC\170617 to HD).Notes and references
- G. M. Cragg and D. J. Newman, Biochim. Biophys. Acta, 2013, 1830, 3670–3695 CrossRef CAS PubMed.
- H.-W. Nützmann, C. Scazzocchio and A. Osbourn, Annu. Rev. Genet., 2018, 52, 159–183 CrossRef PubMed.
- Y. Yan, N. Liu and Y. Tang, Nat. Prod. Rep., 2020 Search PubMed.
- Y. Yan, Q. Liu, X. Zang, S. Yuan, U. Bat-Erdene, C. Nguyen, J. Gan, J. Zhou, S. E. Jacobsen and Y. Tang, Nature, 2018, 559, 415–418 CrossRef CAS PubMed.
- B. K. Park, N. R. Kitteringham and P. M. O'Neill, Annu. Rev. Pharmacol. Toxicol., 2001, 41, 443–470 CrossRef CAS PubMed.
- L. Wu, F. Maglangit and H. Deng, Curr. Opin. Chem. Biol., 2020, 55, 119–126 CrossRef CAS PubMed.
- M. Sanada, T. Miyano, S. Iwadare, J. M. Williamson, B. H. Arison, J. L. Smith, A. W. Douglas, J. M. Liesch and E. Inamine, J. Antibiot. Res., 1986, 39, 259–265 CrossRef CAS PubMed.
- D. O'Hagan, C. Schaffrath, S. L. Cobb, J. T. Hamilton and C. D. Murphy, Nature, 2002, 416, 279–279 CrossRef PubMed.
- C. Dong, F. Huang, H. Deng, C. Schaffrath, J. B. Spencer, D. O'hagan and J. H. Naismith, Nature, 2004, 427, 561–565 CrossRef CAS PubMed.
- H. Deng, S. M. Cross, R. P. McGlinchey, J. T. Hamilton and D. O'Hagan, Chem. Biol., 2008, 15, 1268–1276 CrossRef CAS PubMed.
- M. Onega, R. P. McGlinchey, H. Deng, J. T. Hamilton and D. O'Hagan, Bioorg. Chem., 2007, 35, 375–385 CrossRef CAS PubMed.
- H. Deng, L. Ma, N. Bandaranayaka, Z. Qin, G. Mann, K. Kyeremeh, Y. Yu, T. Shepherd, J. H. Naismith and D. O'Hagan, ChemBioChem, 2014, 15, 364–368 CrossRef CAS PubMed.
- S. Huang, L. Ma, M. H. Tong, Y. Yu, D. O'Hagan and H. Deng, Org. Biomol. Chem., 2014, 12, 4828–4831 RSC.
- L. Ma, A. Bartholome, M. H. Tong, Z. Qin, Y. Yu, T. Shepherd, K. Kyeremeh, H. Deng and D. O'Hagan, Chem. Sci., 2015, 6, 1414–1419 RSC.
- F. Huang, S. F. Haydock, D. Spiteller, T. Mironenko, T.-L. Li, D. O'Hagan, P. F. Leadlay and J. B. Spencer, Chem. Biol., 2006, 13, 475–484 CrossRef CAS PubMed.
- D. O'Hagan and H. Deng, Chem. Rev., 2015, 115, 634–649 CrossRef PubMed.
- A. M. Weeks, N. S. Keddie, R. D. Wadoux, D. O'Hagan and M. C. Chang, Biochemistry, 2014, 53, 2053–2063 CrossRef CAS PubMed.
- M. V. Dias, F. Huang, D. Y. Chirgadze, M. Tosin, D. Spiteller, E. F. Dry, P. F. Leadlay, J. B. Spencer and T. L. Blundell, J. Biol. Chem., 2010, 285, 22495–22504 CrossRef CAS PubMed.
- A. M. Weeks, N. Wang, J. G. Pelton and M. C. Chang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E2193–E2201 CrossRef CAS PubMed.
- J. L. McMurry and M. C. Chang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 11920–11925 CrossRef CAS PubMed.
- L. Wu, M. H. Tong, K. Kyeremeh and H. Deng, Biomolecules, 2020, 10, 1023 CrossRef PubMed.
- K. K. Tan, B. H. Bay and P. Gopalakrishnakone, Toxicon, 2018, 144, 7–13 CrossRef CAS PubMed.
- D. H. Souza, L. M. Eugenio, J. E. Fletcher, M.-S. Jiang, R. C. Garratt, G. Oliva and H. S. Selistre-de-Araujo, Arch. Biochem. Biophys., 1999, 368, 285–290 CrossRef CAS PubMed.
- L. Wu, M. H. Tong, A. Raab, Q. Fang, S. Wang, K. Kyeremeh, Y. Yu and H. Deng, Appl. Microbiol. Biotechnol., 2020, 1–12 Search PubMed.
- K. Ishikawa, N. Higashi, T. Nakamura, T. Matsuura and A. Nakagawa, J. Mol. Biol., 2007, 366, 857–867 CrossRef CAS PubMed.
- L. Chen, Z. Chen, P. Zheng, J. Sun and A.-P. Zeng, Appl. Microbiol. Biotechnol., 2013, 97, 2939–2949 CrossRef CAS PubMed.
- X. Guan, E. Rubin and H. Anni, Alcohol.: Clin. Exp. Res., 2012, 36, 398–405 CrossRef CAS PubMed.
- C. Donnelly and C. D. Murphy, Biotechnol. Lett., 2009, 31, 245 CrossRef CAS PubMed.
- T. Kurihara, T. Yamauchi, S. Ichiyama, H. Takahata and N. Esaki, J. Mol. Catal. B: Enzym., 2003, 23, 347–355 CrossRef CAS.
- J. Jin and U. Hanefeld, Chem. Commun., 2011, 47, 2502–2510 RSC.
- C. Wang, J. Ren, L. Zhou, Z. Li, L. Chen and A.-P. Zeng, ACS Synth. Biol., 2019, 8, 2483–2493 CrossRef CAS PubMed.
- Y. Wang, Z. Deng and X. Qu, F1000Research, 2014, 3 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob01358g |
| This journal is © The Royal Society of Chemistry 2020 |