
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlanar chiral palladacycle precatalysts for asymmetric synthesis†
Ross A.
Arthurs
,
David L.
Hughes
and
Christopher J.
Richards
 *
*
School of Chemistry, University of East Anglia, Norwich Research Park, Norwich, NR4 7TU, UK. E-mail: Chris.Richards@uea.ac.uk
First published on 2nd July 2020
Abstract
Chiral non-racemic palladacycles were employed as precatalysts for Pd(0) mediated asymmetric synthesis. Addition of HPAr2/base to a ferrocenyloxazoline planar chiral palladacycle resulted in ligand synthesis and palladium capture to give a bidentate Phosferrox/Pd(0) complex. A series of these complexes were generated in situ and applied successfully as catalysts for asymmetric allylic alkylation.
Introduction
Air-stable palladacycles containing a bidentate carbon-heteroatom ligated palladium(II) core have been employed extensively in catalysis.1 Following the discovery that high turn-over numbers may be achieved on application of the Herrmann complex to the Heck reaction,2 this and other palladacycles have also found application in a number of cross-coupling processes including the Suzuki–Miyaura3 and Buchwald–Hartwig amination reactions.4 In addition, related chiral non-racemic palladacycles have been applied successfully to the catalysis of asymmetric reactions.5 Examples include the allylic imidate rearrangement,6 hydrophosphination,7 imine arylation,8 the aza-Morita–Baylis–Hillman reaction9 and Michael type reactions.10 Palladium(II) mediated activation of the substrate and retention of the palladacycle C–Pd bond in the chiral catalyst is either demonstrated11 or invoked.7–10 A recent report on palladacycle catalysed asymmetric Suzuki cross-coupling was explained by the operation of a Pd(II)/Pd(IV) catalytic cycle.12 In contrast, the use of chiral non-racemic palladacycles as precatalysts for asymmetric reactions proceeding via a Pd(0)/Pd(II) catalytic cycle does not appear to have been demonstrated.13In this paper we describe the use of chiral non-racemic palladacycles as precatalysts for the formation of Pd(0)/chiral ligand coordinated complexes, followed by the application of these as catalysts in asymmetric synthesis. This first report on simultaneous bidentate ligand synthesis/palladium(0) capture provides a new and versatile approach to the generation of Pd/chiral ligand complexes, with potential applications across a range of palladium catalysed asymmetric transformations.14
Results and discussion
Palladacycles are known to undergo ligand phosphination on reaction with MPPh2 (M = Li, K), where initial phosphide coordination to palladium followed by reductive elimination results in C–P bond formation.15,16 In this way a number of N–P, S–P and P–P bidentate ligands have been synthesised from the corresponding S, N or P precursor palladacycles. Many of these product ligands are chiral, including planar chiral examples displaying stereospecific replacement of the C–Pd bond by a new C–P bond.15a,b Formation of these ligands also results in the generation of Pd(0), but as far as we are aware, isolation of a resulting ligated Pd(0) complex in this way has not been reported, let alone used subsequently in catalysis.To address this challenge we chose to start with ferrocenyloxazoline (S)-1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 as this may be transformed readily into planar chiral palladacycles (S,Sp)-2a/b (Scheme 1).18 Furthermore, the Phosferrox ligand (S,Sp)-319 is also derived from (S)-1, this bidentate ligand having been applied successfully in several palladium catalysed asymmetric reactions.20 It was anticipated that this would aid the identification and comparative application of a corresponding palladacycle derived Pd(0) complex of (S,Sp)-3.
17 as this may be transformed readily into planar chiral palladacycles (S,Sp)-2a/b (Scheme 1).18 Furthermore, the Phosferrox ligand (S,Sp)-319 is also derived from (S)-1, this bidentate ligand having been applied successfully in several palladium catalysed asymmetric reactions.20 It was anticipated that this would aid the identification and comparative application of a corresponding palladacycle derived Pd(0) complex of (S,Sp)-3.
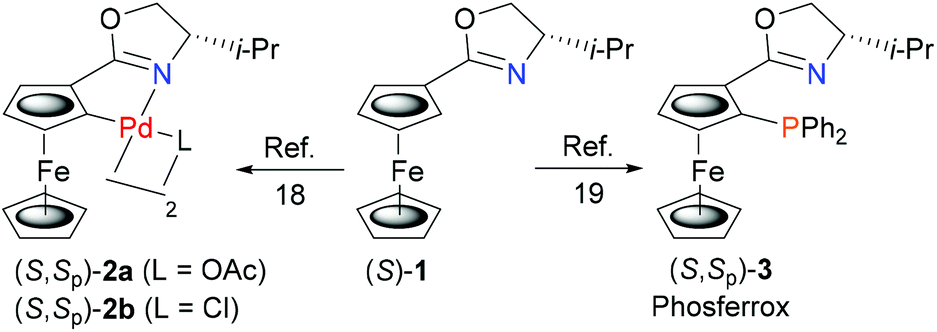 | ||
| Scheme 1 Palladacycles and a chiral bidentate ligand derived from (S)-1. | ||
We reasoned that identification of in situ formed P–N ligated Pd(0) would be aided by addition of a second ligand to give a readily characterised complex. As maleic anhydride (MA) is known to result in stable low valent complexes of palladium,21 we first used this electron deficient alkene with (S,Sp)-3 and Pd2(dba)3 for the synthesis of (S,Sp)-4 (Scheme 2). This air-stable Pd(0) complex was purified readily by column chromatography. Its 31P NMR spectrum22 contained characteristic major and minor signals at 14.39 and 15.81 ppm respectively due to the two possible (exo/endo) orientations of the cyclic unsaturated ligand.
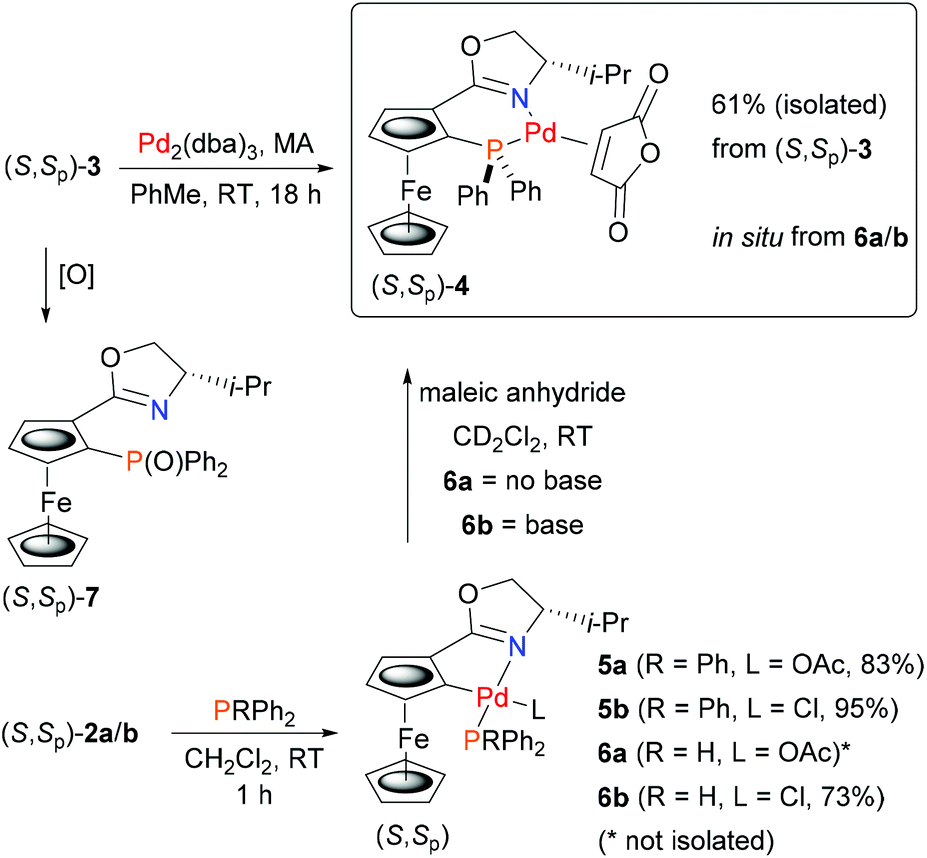 | ||
| Scheme 2 Synthesis of Pd(0) complex (S,Sp)-4 by ligand exchange and ligand synthesis/palladium capture. | ||
Phosphine complexes of palladacycles (S,Sp)-2a/b were first investigated using triphenylphosphine. On combination in CH2Cl2 at room temperature monomeric adducts 5a/b were formed cleanly (Scheme 2). The phosphine is assigned a trans to nitrogen orientation in the resulting complexes in accord with the geometry of related monomeric phosphine coordinated palladacycles.15b,23 The 31P NMR signals22 observed for 5a (36.5 ppm) and 5b (37.5 ppm), compared to PPh3 (−5.4 ppm), were used to aid the assignment of the corresponding adducts obtained from HPPh2 (−40.1 ppm). Addition of this secondary phosphine to chloride ligated 2b gave stable and isolable adduct 6b (8.4 ppm). In contrast, addition to acetate ligated 2a resulted in immediate darkening to brown/black of the initially orange solution, and after stirring for 1 hour, no phosphine adduct was obtained.
To investigate the possibility that this colour change was indicative of reductive elimination from an initially formed phosphine adduct, the experiment was repeated in CD2Cl2 and followed by 31P NMR spectroscopy. Following addition of HPPh2 to 2a at room temperature, a major signal observed in the 31P NMR spectrum obtained after ca. 10 minutes (7.5 ppm) is indicative of adduct 6a (Fig. 1a). Significantly, following the addition of MA, subsequent NMR analysis revealed the disappearance of this complex and the formation of (S,Sp)-4 (Fig. 1b).
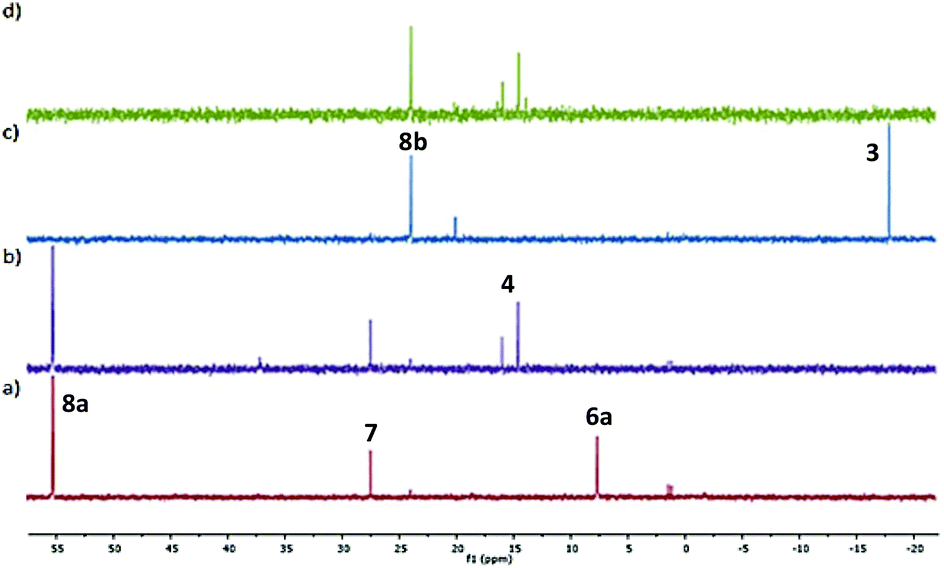 | ||
| Fig. 1 31P NMR spectra (CD2Cl2): (a) following the addition of HPPh2 (2 eq.) to 2a, (b) ca. 10 minutes after the addition of MA (2 eq.), (c) following the addition of NaCH(CO2Me)2 (10 eq.) to 6b, (d) ca. 1 h after the addition of MA (4 eq.). | ||
Two other major signals at 27.3 and 55.2 ppm were observed in the 31P NMR spectra obtained before and after the addition of MA. The first corresponds to (S,Sp)-7, a phosferrox derived phosphine oxide for which the identity was confirmed by independent synthesis. The second is assigned to a Pd(II)/PPh2 complex (vide infra). Formation of 4 was not observed when MA was added to palladacycle 2a before the addition of PHPh2 due to fast competitive Michael addition.24 The formation of 4 and 7, as a single diastereoisomer in both cases, confirms that replacement of palladium by phosphorus proceeds with no erosion of stereochemical integrity.25
In contrast to 6a, the corresponding experiment with chloride complex 6b (formed in situ from (S,Sp)-2b and HPPh2) required the addition of a base to promote reductive elimination and formation of the MA adduct 4. Nitrogen bases were employed initially, with triethylamine resulting in the highest conversion (14%) as determined by 31P NMR spectroscopy.26 With N,O-bis(trimethylsilyl)acetamide (BSA) the conversion was improved (23%), and the best result (29%) was obtained with sodium dimethyl malonate. Following addition of this base 31P NMR spectroscopy revealed two major signals at −18.1 and 23.8 ppm (Fig. 1c). The former corresponds to Phosferrox ligand 3, and on addition of MA this signal disappeared and was replaced predominantly by a corresponding amount of 4 (Fig. 1d). Following isolation of the other complex by column chromatography it was identified by X-ray crystallography as Pd(II)/PPh2 dimer 8b (Scheme 3 and Fig. 2).27 Addition of silver acetate to 8b gave 8a (31P NMR = −55.6 ppm), revealing that this corresponding acetate-bridged dimer is formed from the addition of HPPh2 to 2a (Fig. 1a and b). Addition of maleic anhydride to a solution of 8b in CD2Cl2 did not result in the formation of 4. Dimerisation to give 8a/b is an alternative and apparently irreversible pathway to reductive elimination and formation of ligand 3. It is of note that Pd(0) does not complex with 3 to a significant extent until maleic anhydride is added, an outcome consistent with initial and reversible formation of palladium nanoparticles.28 No oxidation to phosphine oxide (S,Sp)-7 was observed, in contrast to the appreciable formation of this compound from acetate-bridged palladacycle 2a, revealing a significant advantage of starting with chloride-bridged palladacycle 2b.29
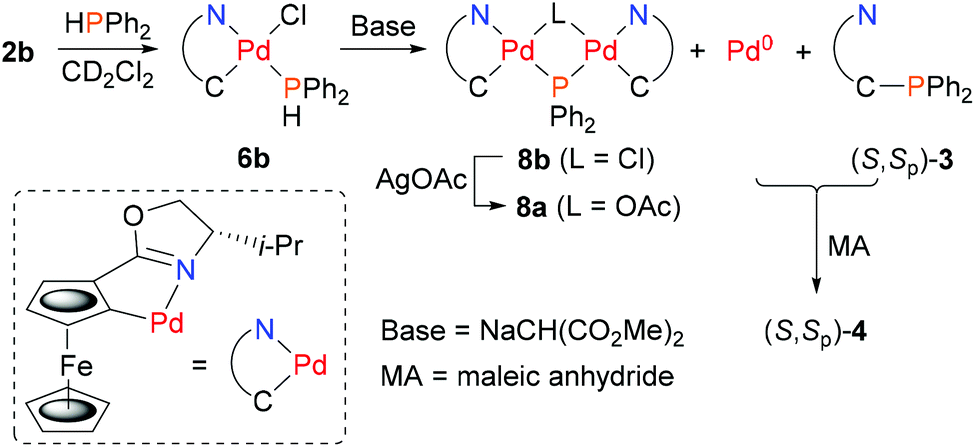 | ||
| Scheme 3 The generation of (S,Sp)-4. by ligand synthesis/palladium capture from palladacycle 2b. | ||
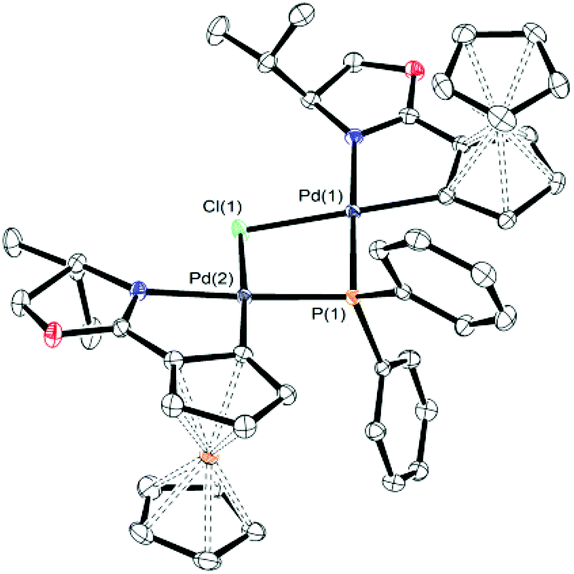 | ||
| Fig. 2 A representation of the X-ray structure of 8b (hydrogen atoms omitted for clarity). Principal bond lengths [Å] include: Pd(1)–Cl(1) = 2.4308(6), Pd(2)–Cl(1) = 2.4366(6), Pd(1)–P(1) = 2.2447(6), Pd(2)–P(1) = 2.2457(6). Principal bond angles [deg] include: Pd(1)–Cl(1)–Pd(2) = 86.641(18), Pd(1)–P(1)–Pd(2) = 96.09(2). Thermal ellipsoids are drawn at the 50% probability level. Flack parameter = −0.002(5). | ||
The successful formation of (S,Sp)-4 from palladacycle precursors prompted us to investigate the use of these as precatalysts in asymmetric synthesis. The allylic alkylation of trans-1,3-diphenylallyl acetate 9 was chosen as an exemplar reaction due to its experimental simplicity and the potential to employ the basic reaction conditions for in situ ligand/Pd(0) generation (Table 1). Furthermore, ligand (S,Sp)-3 is known to promote high enantioselectivity in this reaction, and in our hands repetition of conditions reported previously20a using BSA as base (3 eq.) gave (S)-10 in 95% ee (conditions A, Table 1, entry 1). Replacing [Pd(C3H5)Cl]2 with preformed Pd(0) complex (S,Sp)-4 resulted in some loss of enantioselectivity, and a very significant reduction in activity due to inhibition by maleic anhydride (entry 2). This precluded the use of this π-acidic ligand in any subsequent runs. Instead, a catalytic asymmetric allylic alkylation was attempted directly following the addition of HPPh2 to 2a, and after stirring for 10 minutes, to resulting in situ generated 6a was added sequentially rac-9, dimethyl malonate, BSA and KOAc (entry 3). Although slow, we noted with interest the formation of product 10, subsequently isolated as the S enantiomer in 85% ee. Use in the same way of chloride 2b resulted in a similar outcome (entry 4), and on repetition in the absence of KOAc the product ee increased to 94% (entry 5). No product was formed in the absence of added PHPh2 (entry 6). These initial results are consistent with the in situ formation of (S,Sp)-3 ligated Pd(0) as the sole catalytically active species, albeit in low concentration. Replacement of CH2Cl2 as solvent with THF or toluene resulted in no product formation.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Pd source (mol%) | Conditions | Time (h) | Con. (%) | ee (%) |
| a 1 mmol rac-9 (0.6 M) unless otherwise stated. b Lit. value 90% ee.20a c 0.33 mmol scale. d No KOAc. e Reaction essentially complete within 1 h (tlc). | |||||
| 1 | (S,Sp)-3 (2.5) + [Pd(C3H5)Cl]2 (1) | A | <1 | 100 | 95b |
| 2 | (S,Sp)-4 (1) | A | 96 | 6 | 86 |
| 3 | (S,Sp)-2a (1) + HPPh2 (2) | A | 96 | 51 | 85 |
| 4 | (S,Sp)-2b (1) + HPPh2 (2) | A | 96 | 77 | 85 |
| 5 | (S,Sp)-2b (1) + HPPh2 (2) |
Ac,d |
96 | 20 | 94 |
| 6 | (S,Sp)-2a (1) or (S,Sp)-2b (1) | A | 96 | 0 | na |
| 7 | (S,Sp)-3 (2.5) + [Pd(C3H5)Cl]2 (1) | B | 3.5 | 100 | 88 |
| 8 | (S,Sp)-2b (1) + HPPh2 (2) | B | 2.5 | 100 | 77 |
| 9 | (S,Sp)-2b (1) + HPPh2 (2) | C | 1 | 99 | 86 |
| 10 | 8b (1) | A | 24 | 0 | na |
| 11 | (S,Sp)-2b (1) + 11a (2) | C | 24 | 18 | 85 |
| 12 | (S,Sp)-2b (1) + 11b (2) | C | 24e | 100 | 84 |
| 13 | (S,Sp)-2b (1) + 11c (2) | C | 24e | 100 | 83 |

As sodium dimethyl malonate gave the highest and cleanest conversion to (S,Sp)-4, this was employed next as both a base and a nucleophile, and the process referenced initially by use of (S,Sp)-3/[Pd(C3H5)Cl]2 (3 fold excess, conditions B). The lower product ee, compared to that obtained from the use as base of BSA (95% – entry 1 vs. 88% – entry 7) is consistent with the superiority of the former conditions noted previously.30 Significantly, addition of a three-fold excess of sodium dimethyl malonate to rac-9 and 2 mol% of in situ generated 6b resulted in complete conversion within 2.5 h to allylic alkylation product (S)-10 formed in 77% ee (entry 8). Modifying these conditions further (conditions C) to the use sodium dimethyl malonate (10 mol%) as the base for in situ catalyst formation, and then the addition of BSA/dimethyl malonate (3 equivalents of each) further decreased the reaction time to 1 hour and increased the ee of (S)-10 to 86% (entry 9). The use of conditions C provided an in situ generated catalyst with comparable activity and selectivity to that obtained using a preformed ligand, and consistent with this outcome is the absence of any catalysis with Pd(II) complex 8b (entry 10).
Finally, this led us to explore the use of asymmetric allylic alkylation to test the viability of using electronically contrasting diarylphosphines in the ligand synthesis/Pd(0) capture protocol (Scheme 4). For this reaction previous work with related PHOX ligands containing PAr2 units with various para substituents revealed little difference in enantioselectivity compared to the parent PPh2 containing ligand,31 albeit with faster conversion with electron-withdrawing substituents.31a Accordingly, secondary phosphines 11a–c were added separately to palladacycle (S,Sp)-2b followed by sodium dimethyl malonate, and to the resulting solutions were added rac-9 (50 equivalents w.r.t. Pd) and dimethyl malonate/BSA (i.e. conditions C). Use of electron-deficient phosphine 11a resulted in a similar level of product enantioselectivity albeit in reduced yield (Table 1, entry 11). Further examination by 31P NMR spectroscopy (CD2Cl2) of the addition of 11a (−40.8 ppm) to 2b confirmed the formation of the phosphine coordinated adduct (10.9 ppm), but little Pd0 complex was observed following the addition of sodium dimethyl malonate and MA. In this instance catalyst formation appears to be limited by the inefficiency of reductive elimination.32 In contrast, the catalysts derived from phosphines 11b (entry 12) and 11c (entry 13) gave essentially complete conversion after 1 h and ee values very similar to that obtained from diphenylphosphine.
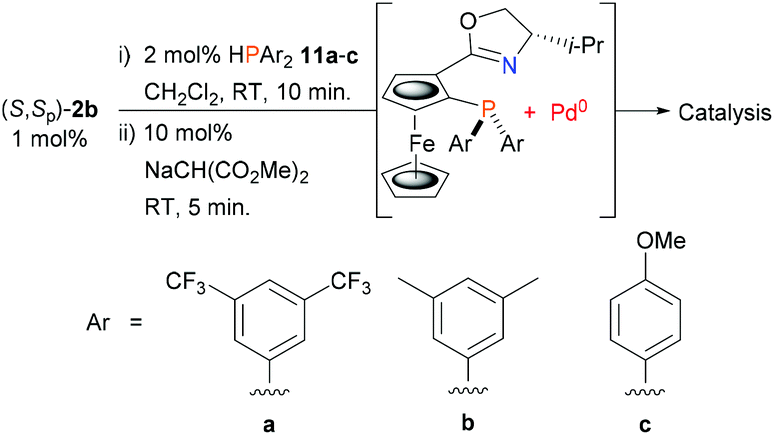 | ||
| Scheme 4 Phosphine variation for in situ ligand synthesis/Pd(0) capture from palladacycle (S,Sp)-b2. | ||
Conclusion
We have established that ferrocenyloxazoline-based planar chiral palladacycles 2a/b, on combination with diphenylphosphine (and for 2b a base), are viable precatalysts for in situ catalyst generation using a new ligand synthesis/palladium(0) capture approach. This was demonstrated by formation of a stable Phosferrox/maleic anhydride coordinated Pd(0) complex (S,Sp)-4a, and by application of Phosferrox/Pd(0) as a catalyst for asymmetric allylic alkylation in the absence of maleic anhydride. The other palladium(II) complex formed in this process is not a competitive catalyst or catalyst precursor. Product configuration and enantioselectivity using a palladacycle derived catalyst are similar to that obtained using a preformed Phosferrox ligand and a source of Pd(0). In addition to being a precursor to an active and selective catalyst generated in one step, palladacycle 2b may also be used for multiple catalyst generation with other diarylphosphines. It is anticipated that this approach will aid significantly the generation and identification of palladium based catalysts for application in other areas of asymmetric synthesis.Experimental
General information
Tetrahydrofuran was distilled over sodium and benzophenone ketyl and dichloromethane distilled over calcium hydride. All reactions were carried out under an inert atmosphere of either nitrogen or argon. Silica gel (60 Å pore size, 40–63 μm technical grade) was used for chromatography. All starting materials not commercially available are specifically referenced.Synthesis of (S,Sp)-4
(S,Sp)-319 (0.087 g, 0.18 mmol), tris(dibenzylideneacetone)-dipalladium(0) (0.076 g, 0.08 mmol) and maleic anhydride (0.024 g, 0.24 mmol) were added to a flame dried Schlenk tube and dissolved in toluene (15 mL). The resulting deep purple solution was stirred vigorously overnight resulting in the formation of a dark suspension. The reaction was filtered through Celite™ using toluene as the eluent and the solvent removed in vacuo. Purification by column chromatography (SiO2, 40% EtOAc/hexane) yielded an orange solid (0.07 g, 61%); mp decomposed ∼200 °C (under argon); HRMS (AS) [M + H]+ anal. calcd for C32H31FeNO4PPd 686.0389, found 686.0389; [α]20.5 °CD = −538 (c = 0.46, CHCl3); νmax (film)/cm−1 3056, 3016, 2960, 2923, 2873, 1790, 1723, 1626; 1H NMR (500 MHz, CDCl3) major isomer (ratio of isomers = 1.6:1) δ 7.80 (2H, ddd, J = 11.6, 7.8, 1.5 Hz), 7.56–7.46 (3H, m), 7.33–7.27 (3H, m), 7.08 (2H, ddd, J = 11.0, 6.6, 3.2 Hz), 5.04 (1H, brs), 4.70 (1H, t, J = 2.6 Hz), 4.41–4.38 (1H, m), 4.35 (1H, t, J = 3.7 Hz), 4.33–4.21 (3H, m), 4.07 (1H, dd, J = 10.5, 3.8 Hz), 3.96 (5H, s), 2.80 (1H, dtd, J = 13.8, 6.9, 3.3 Hz), 1.07 (3H, d, J = 7.1 Hz), 0.99 (3H, d, J = 6.9 Hz); minor isomer 7.76–7.68 (2H, m), 7.56–7.46 (3H, m), 7.33–7.27 (3H, m), 7.21–7.14 (2H, m), 5.04 (1H, brs), 4.72 (1H, t, J = 2.6 Hz), 4.50–4.46 (1H, m), 4.33–4.21 (3H, m), 3.86 (5H, s), 3.87–3.79 (2H, m), 2.67 (1H, dtd, J = 13.7, 6.9, 3.4), 1.04 (3H, d, J = 7.0 Hz), 0.95 (3H, d, J = 6.8 Hz); 13C NMR (125 MHz, CDCl3) major isomer δ 172.6 (d, J = 2.0 Hz), 171.5 (d, J = 5.2 Hz), 169.1, 135.5 (d, J = 36.7 Hz), 134.9 (d, J = 16.7 Hz), 134.7 (d, J = 40.1 Hz), 131.6 (d, J = 13.3 Hz), 131.2 (d, J = 2.0 Hz), 129.4 (d, J = 1.6 Hz), 128.6 (d, J = 10.8 Hz), 128.5 (d, J = 9.7 Hz), 75.5 (d, J = 2.6 Hz), 74.0 (d, J = 2.3 Hz), 73.5 (d, J = 46.2 Hz), 73.4 (d, J = 1.5 Hz), 73.1, 71.8 (d, J = 0.5 Hz), 71.4, 67.7, 47.4 (d, J = 31.7 Hz), 46.4 (d, J = 1.8 Hz), 29.2, 19.2, 14.7; minor isomer 172.9 (d, J = 1.5 Hz), 171.2 (d, J = 5.0 Hz), 169.1, 137.3 (d, J = 37.8 Hz), 135.1 (d, J = 16.6 Hz), 133.7 (d, J = 40.2 Hz), 131.4 (d, J = 13.1 Hz), 131.4 (d, J = 2.1 Hz), 129.1 (d, J = 1.5 Hz), 128.8 (d, J = 11.2 Hz), 128.5 (d, J = 9.7 Hz), 75.9 (d, J = 2.7 Hz), 75.1 (d, J = 1.8 Hz), 74.0 (d, J = 33.0 Hz), 74.0 (d, J = 2.1 Hz), 73.4 (d, J = 1.3 Hz), 73.1, 71.6, 67.7, 47.8 (d, J = 1.7 Hz), 46.7 (d, J = 32.7 Hz), 29.2, 19.2, 14.9; 31P NMR (202 MHz, CDCl3) major isomer δ 14.39; minor isomer 15.81.
General procedure for the synthesis of 5a, 5b and 6b
(S,Sp)-2a18a,b or (S,Sp)-2b18b (0.029 mmol) and either triphenylphosphine or diphenylphosphine (0.057 mmol) were added to a flame dried Schlenk tube and dissolved in dichloromethane (1 mL) and stirred at room temperature for 1 hour. The solvent was removed in vacuo yielding the product.
Synthesis of (S,Sp)-7
(S)-117 (0.059 g, 0.2 mmol) was added to a flame dried Schlenk tube under an atmosphere of argon and dissolved in dry THF (5 mL). The resulting orange solution was cooled to −78 °C and stirred for 5 min after which n-butyl lithium (2.5 M in hexanes) (0.11 mL, 0.28 mmol) was added slowly. After stirring for 2 hours the mixture was warmed to 0 °C and chlorodiphenylphosphine oxide (0.076 mL, 0.4 mmol) was added and the reaction was allowed to warm to room temperature. After an additional 15 min, the reaction was diluted with diethyl ether and then quenched with saturated sodium hydrogen carbonate solution. The organics were separated with H2O, washed with brine, dried over MgSO4 and the solvent removed in vacuo giving a crude product containing a 2:1 mixture of diastereoisomers. The diastereoisomers were separated by column chromatography (SiO2, 2% MeOH/Et2O) yielding the desired product as a yellow brown solid (0.0255 g, 26%): mp 197–198 °C; HRMS (ES) [M + H]+ anal. calcd for C28H29FeNO2P 498.1280, found 498.1263; [α]19.4 °CD = +114 (c = 0.2, CHCl3); νmax (film)/cm−1 3080, 3062, 2963, 2930, 2871, 1648, 1120; 1H NMR (500 MHz, CDCl3) δ 7.76–7.71 (2H, m), 7.69–7.64 (2H, m), 7.50–7.46 (1H, m), 7.45–7.36 (5H, m), 5.06 (1H, dt, J = 2.6, 1.4 Hz), 4.49 (5H, s), 4.45 (1H, dd, J = 4.4, 2.4 Hz), 4.22–4.18 (1H, m), 3.93 (1H, dd, J = 3.9, 2.5 Hz), 3.72 (1H, td, J = 9.3, 6.3 Hz), 3.13 (1H, t, J = 8.8 Hz), 1.53–1.47 (1H, m), 0.81 (3H, d, J = 6.8 Hz), 0.61 (3H, d, J = 6.8 Hz); 13C NMR (125 MHz, CDCl3) δ 164.6, 134.86 (d, J = 107 Hz), 134.8 (d, J = 110.4 Hz), 131.6 (d, J = 9.3 Hz), 131.4 (d, J = 9.7 Hz), 131.3 (d, J = 2.6 Hz), 131.0 (d, J = 2.9 Hz), 128.2 (d, J = 4.3 Hz), 128.1 (d, J = 4.8 Hz), 78.5 (d, J = 14.6 Hz), 75.7 (d, J = 109.9 Hz), 74.5 (d, J = 9.1 Hz), 73.5 (d, J = 8.2 Hz), 72.4, 71.5 (d, J = 10.9 Hz), 71.4, 70.1, 32.3, 19.2, 18.0; 31P NMR (202 MHz, CDCl3) δ 26.40.
Synthesis of (S,S,Sp,Sp)-8b
To a flame dried Schlenk tube (tube-1) was added sodium tert-butoxide solution (2 M in THF) (0.05 mL, 0.10 mmol) followed by a solution of dimethyl malonate (22.6 μl, 0.20 mmol) in THF (1 mL) at 0 °C. After stirring for 30 min the solvent was removed under high vacuum to give a beige residue. In a separate flame dried Schlenk tube (tube-2), (S,Sp)-6b (0.0125 g, 0.020 mmol) was dissolved in CH2Cl2 (1.7 mL) and then the contents of Schlenk tube (2) were transferred to Schlenk tube (1) and stirred at room temperature for 10 min. The solvent was then removed in vacuo and the residue purified by column chromatography (SiO2, 10% EtOAc/hexane) to give an orange solid (0.007 g, 70%): mp > 230 °C (under argon); HRMS (AS) [M − Cl]+ anal. calcd for C44H46Fe2N2O2PPd2 991.0103, found 991.0093; [α]20.1 °CD = −2000 (c = 0.23, CHCl3); νmax (film)/cm−1 3096, 2958, 2923, 2967, 1617, 1495; 1H NMR (500 MHz, CDCl3) δ 8.12–8.05 (4H, m), 7.38–7.34 (6H, m), 4.49–4.47 (4H, m), 4.36 (2H, d, J = 1.8 Hz), 4.08 (2H, td, J = 8.0, 4.6 Hz), 4.04 (10H, s), 4.02 (2H, t, J = 2.3 Hz), 3.22 (2H, d, J = 1.6 Hz), 2.51–2.43 (2H, m), 1.06 (6H, d, J = 6.8 Hz), 0.97 (6H, d, J = 7.0 Hz); 13C NMR (125 MHz, CDCl3) δ 177.9, 137.2 (d, J = 32.8 Hz), 135.4 (d, J = 12.7 Hz), 128.8, 127.8 (d, J = 10.5 Hz), 91.4 (d, J = 2.9 Hz), 75.1 (d, J = 8.6 Hz), 73.3 (d, J = 1.6 Hz), 72.2, 70.0, 69.0, 66.9, 64.5, 29.3, 19.6, 16.0; 31P NMR (202 MHz, CDCl3) δ 23.40.Synthesis of (S,S,Sp,Sp)-8a
Addition of excess AgOAc to an NMR tube containing (S,S,Sp,Sp)-8b showed the formation of acetate bridged palladacycle (S,S,Sp,Sp)-8a; νmax (film)/cm−1 3096, 2958, 2919, 2849, 1621, 1556, 1495; 1H NMR (500 MHz, CDCl3) δ 8.12–7.97 (4H, m), 7.33–7.27 (6H, m), 4.51–4.46 (4H, m), 4.34 (2H, brs), 4.09–4.03 (2H, m), 4.00 (10H, s), 3.97 (2H, brs), 3.00 (2H, brs), 2.70–2.60 (2H, m), 2.01 (3H, brs), 1.04 (6H, d, J = 6.8 Hz), 0.95 (6H, d, J = 7.1 Hz); 31P NMR (202 MHz, CDCl3) δ 55.63.Phosphine insertion studies
Using a glove box, 0.01 mmol of either (S,Sp)-2a or (S,Sp)-2b was dissolved in CD2Cl2 (0.7 mL) in an NMR tube fitted with a J. Young valve under an inert atmosphere. After addition of the reagents approximately 10 minutes elapsed before the first NMR spectrum was recorded.In situ catalyst preparation and asymmetric allylic alkylation (conditions C)
To a flame dried Schlenk tube (tube-1) was added sodium tert-butoxide solution (2 M in THF) (0.05 mL, 0.10 mmol) followed by a solution of dimethyl malonate (22.6 μl, 0.20 mmol) in THF (1 mL) at 0 °C. After stirring for 30 min the solvent was removed under high vacuum to give a beige residue. In a separate flame dried Schlenk tube (tube-2), (S,Sp)-2b (0.0088 g, 0.010 mmol) was dissolved in CH2Cl2 (1.7 mL) and phosphine (0.020 mmol) was added and stirred for 10 min. 1,3-Diphenylallyl acetate (0.252 g, 1.0 mmol) was dissolved in CH2Cl2 (1.7 mL) and then added to Schlenk tube (2) followed by dimethyl malonate (0.34 mL, 3.0 mmol). The contents of Schlenk tube (2) were transferred to Schlenk tube (1) and stirred for 5 min after which N,O-bis(trimethylsilyl)acetamide (0.74 mL, 3.0 mmol) was added and the reaction allowed to stir at room temperature. After completion or 24 hours (whichever came first), the reaction was quenched with saturated ammonium chloride, separated with diethyl ether, dried with magnesium sulphate and the solvent removed in vacuo. A 1H NMR was performed at this point to determine the conversion. Purification by column chromatography (SiO2, 5% EtOAc/hexane) gave the product (S)-10 as a colourless oil for which the 1H/13C NMR spectra matched previously reported data.33 The enantiomeric excess was determined by HPLC analysis using a CHIRALCEL OD-H column; eluent = 98:2 (hexane:IPA); flow rate = 0.5 mL min−1; concentration = 0.0015 g mL−1, injection volume = 5 μl, wavelength = 254 nm. (S)-Enantiomer RT = 20.5 min, (R)-enantiomer RT = 18.9 min.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The EPSRC (EP/N019393/1) (R. A. A.) is thanked for financial support. We also thank the EPSRC National Mass Spectrometry Centre (University of Wales, Swansea).Notes and references
- (a) J. Dupont, C. S. Consorti and J. Spencer, Chem. Rev., 2005, 105, 2527 CrossRef CAS PubMed; (b) Palladacycles: Synthesis Characterization and Applications, ed. J. Dupont and M. Pfeffer, Wiley-VCH, Weinheim, 2008 Search PubMed; (c) Palladacycles: Catalysis and Beyond, ed. A. R. Kapdi and D. Maiti, Elsevier, 2019 Search PubMed.
- W. A. Herrmann, C. Brossmer, C.-P. Reisinger, T. H. Riermeier, K. Ó¦fele and M. Beller, Chem. – Eur. J., 1997, 3, 1357 CrossRef CAS.
- F. Bellina, A. Carpita and R. Rossi, Synthesis, 2004, 2419 CAS.
- B. P. Fors and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 15914 CrossRef CAS PubMed.
- V. V. Dunina, O. N. Gorunova, P. A. Zykov and K. A. Kochetkov, Russ. Chem. Rev., 2011, 80, 51 CrossRef CAS.
- (a) H. Nomura and C. J. Richards, Chem. – Asian J., 2010, 5, 1726 CrossRef CAS PubMed; (b) J. S. Cannon and L. E. Overman, Acc. Chem. Res., 2016, 49, 2220 CrossRef CAS PubMed.
- R. J. Chew and P.-H. Leung, Chem. Rec., 2016, 16, 141 CrossRef CAS PubMed.
- C. Schrapel and R. Peters, Angew. Chem., Int. Ed., 2015, 54, 10289 CrossRef CAS PubMed.
- K. Hyodo, S. Nakamura and N. Shibata, Angew. Chem., Int. Ed., 2012, 51, 10337 CrossRef CAS PubMed.
- (a) M. Weber, S. Jautze, W. Frey and R. Peters, J. Am. Chem. Soc., 2010, 132, 12222 CrossRef CAS PubMed; (b) M. Weber and R. Peters, J. Org. Chem., 2012, 77, 10846 CrossRef CAS.
- (a) M. P. Watson, L. E. Overman and R. G. Bergman, J. Am. Chem. Soc., 2007, 129, 5031 CrossRef CAS PubMed; (b) J. S. Cannon, S. F. Kirsch, L. E. Overman and H. F. Sneddon, J. Am. Chem. Soc., 2010, 132, 15192 CrossRef CAS PubMed.
- O. N. Gorunova, Y. K. Grishin, M. M. Ilyin (Jr.), K. A. Kochetkov, A. V. Churakov, L. G. Kuz’mina and V. V. Dunina, Russ. Chem. Bull., Int. Ed., 2017, 66, 282 CrossRef CAS.
- A number of chiral non-racemic Pd(II)-based pincer complexes have also been applied to asymmetric catalysis, but again there appears to be no evidence that these proceed via a Pd(0)/Pd(II) catalytic cycle. For a recent review see: J.-K. Liu, J.-F. Gong and M.-P. Song, Org. Biomol. Chem., 2019, 17, 6069 RSC.
- (a) L. F. Tietze, H. Ila and H. P. Bell, Chem. Rev., 2004, 104, 3453 CrossRef CAS PubMed; (b) Q.-A. Chen, Z.-S. Ye, Y. Duan and Y.-G. Zhou, Chem. Soc. Rev., 2013, 42, 497 RSC.
- (a) V. I. Sokolov, L. L. Troitskaya and O. A. Reutov, J. Organomet. Chem., 1980, 202, C58 CrossRef CAS; (b) C. Bolm, K. Wenz and G. Raabe, J. Organomet. Chem., 2002, 662, 23 CrossRef CAS; (c) V. A. Stepanova, V. V. Dunina and I. P. Smoliakova, Organometallics, 2009, 28, 6546 CrossRef CAS; (d) V. A. Stepanova, V. V. Dunina and I. P. Smoliakova, J. Organomet. Chem., 2011, 696, 871 CrossRef CAS; (e) N. J. Korte, V. A. Stepanova and I. P. Smoliakova, J. Organomet. Chem., 2013, 745–746, 356 CrossRef CAS; (f) G. C. Dickmu, N. J. Korte and I. P. Smoliakova, J. Organomet. Chem., 2015, 797, 13 CrossRef CAS; (g) J. E. Kukowski, V. A. Stepanova and I. P. Smoliakova, J. Organomet. Chem., 2017, 830, 155 CrossRef CAS.
- In addition, C–P bonds have also been formed by palladium catalysed ortho-directed C–H activation via an intermediate palladacycle. See: L. Bonnafoux, R. Gramage-Doria, F. Colobert and F. R. Leroux, Chem. – Eur. J., 2011, 17, 11008 CrossRef CAS PubMed.
- (a) T. Sammakia, H. A. Latham and D. R. Schaad, J. Org. Chem., 1995, 60, 10 CrossRef CAS.
- (a) J.-B. Xia and S.-L. You, Organometallics, 2007, 26, 4869 CrossRef CAS; (b) R. A. Arthurs, D. L. Hughes and C. J. Richards, Organometallics, 2019, 38, 4271 CrossRef CAS.
- (a) C. J. Richards, T. Damalidis, D. E. Hibbs and M. B. Hursthouse, Synlett, 1995, 74 CrossRef CAS; (b) Y. Nishibayashi and S. Uemura, Synlett, 1995, 79 CrossRef CAS.
- (a) K. H. Ahn, C.-W. Cho, J. Park and S. Lee, Tetrahedron: Asymmetry, 1997, 8, 1179 CrossRef CAS; (b) A. J. Hennessy, Y. M. Malone and P. J. Guiry, Tetrahedron Lett., 2000, 41, 2261 CrossRef CAS; (c) M. Lautens, J.-L. Renaud and S. Hiebert, J. Am. Chem. Soc., 2000, 122, 1804 CrossRef CAS; (d) S.-L. You, X.-L. Hou, L.-X. Dai, Y.-H. Yu and W. Xia, J. Org. Chem., 2002, 67, 4684 CrossRef CAS; (e) C. Ma, Y. Huang and Y. Zhao, ACS Catal., 2016, 6, 6408 CrossRef CAS; (f) C. Zhu, D. Wang, Y. Zhao, W.-Y. Sun and Z. Shi, J. Am. Chem. Soc., 2017, 139, 16486 CrossRef CAS PubMed; (g) M. Okumura, A. S. Shved and D. Sarlah, J. Am. Chem. Soc., 2017, 139, 17787 CrossRef CAS PubMed.
- H. Minematsu, Y. Nonaka, S. Takahashi and N. Hagihara, J. Organomet. Chem., 1973, 59, 395 CrossRef CAS.
- 31P NMR spectral data from the use of CD2Cl2 as solvent excepting (S,Sp)-4, 8b, PPh3 and HPPh2 (CDCl3); (S,Sp)-5a/b and (S,Sp)-6b (MeCN-d3).
- D. S. Black, G. B. Deacon and G. L. Edwards, Aust. J. Chem., 1994, 47, 217 CrossRef CAS.
- J. A. Van Doorn and R. L. Wife, Phosphorus, Sulfur Silicon Relat. Elem., 1990, 47, 253 CrossRef CAS.
- The (S,Rp) epimer of (S,Sp)-7 is known (31P NMR = 36.10 ppm/CDCl3). R. A. Arthurs and C. J. Richards, Org. Lett., 2017, 19, 702 CrossRef CAS PubMed.
- Full details including 31P NMR spectra are available in the ESI.†.
- CCDC 1983850 contains the supplementary crystallographic data for this paper.†.
- A. J. Reay and I. J. S. Fairlamb, Chem. Commun., 2015, 51, 16289 RSC.
- Acetate-mediated phosphine oxidation with concomitant Pd(II) reduction is known, but in this instance Pd(0) formation is consistent with reductive elimination. See: C. Amatore, A. Jutand and M. A. M'Barki, Organometallics, 1992, 11, 3009 CrossRef CAS.
- B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS PubMed.
- (a) K. Tani, D. C. Behenna, R. M. McFadden and B. M. Stoltz, Org. Lett., 2007, 9, 2529 CrossRef CAS PubMed; (b) D. C. Behenna, J. T. Mohr, N. H. Sherden, S. C. Marinescu, A. M. Harned, K. Tani, M. Seto, S. Ma, Z. Novák, M. R. Krout, R. M. McFadden, J. L. Roizen, J. A. Enquist Jr., D. E. White, S. R. Levine, K. V. Petrova, A. Iwashita, S. C. Virgil and B. M. Stoltz, Chem. – Eur. J., 2011, 17, 14199 CrossRef CAS PubMed.
- J. F. Hartwig, Inorg. Chem., 2007, 46, 1936 CrossRef CAS.
- (a) Y. Tanaka, T. Mino, K. Akita, M. Sakamoto and T. Fujita, J. Org. Chem., 2004, 69, 6679 CrossRef CAS PubMed; (b) D. Liu, F. Xie and W. Zhang, J. Org. Chem., 2007, 72, 6992 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental detail, copies of the 1H, 13C and 31P NMR spectra, HPLC traces and details of the X-ray crystal structure determination. CCDC 1983850. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ob01331e |
| This journal is © The Royal Society of Chemistry 2020 |