
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccess to a stabilized i-motif DNA structure through four successive ligation reactions on a cyclopeptide scaffold†
Alexandre
Devaux
,
Laureen
Bonnat
,
Thomas
Lavergne
 * and
Eric
Defrancq
*
* and
Eric
Defrancq
*
Univ. Grenoble Alpes, Département de Chimie Moléculaire, CNRS, 570 rue de la chimie, CS 40700, Grenoble 38000, France. E-mail: thomas.lavergne@univ-grenoble-alpes.fr; eric.defrancq@univ-grenoble-alpes.fr
First published on 31st July 2020
Abstract
i-Motifs are largely underexplored tetraplex nucleic acid structures which have been suggested to perform essential biological functions and might constitute future therapeutic targets. i-Motifs generally require acidic conditions to fold in vitro, a particularity which significantly complicates the use of native i-motif forming sequences for interaction studies with potential ligands and biological components (e.g. proteins). In this context, we report herein on the assembly of a peptide–DNA conjugate capable of folding at room temperature into a stable i-motif structure at neutral pH. To achieve the controlled assembly of the i-motif forming conjugate, we developed a new synthetic pathway of four successive orthogonal ligation reactions between bifunctional C-rich DNA strands and a tetrafunctional cyclopeptide scaffold.
Introduction
Over half a century ago, the seminal contributions of Watson, Crick, Wilkins and Franklin established the double-helical structure of DNA in which two antiparallel strands are held together through canonical A/T and G/C base pairing. However, the past decades have brought accumulating evidence of the existence of a wide variety of alternative nucleic acid topologies including hairpin, triplex, cruciform and tetraplex structures. In particular, G- and C-rich sequences have been shown to form tetraplex architectures, respectively, G-quadruplex (G4) and i-motif structures.1,2 Unlike duplex DNA, G4 and i-motif formation does not involve canonical Watson–Crick base pairing: indeed a G4 DNA is formed from the stacking of Hoogsteen hydrogen bonded guanine tetrads whereas for i-motif formation two anti-parallel duplexes are held together by hemi-protonated C–C+ base pairs intercalated with each other (Fig. 1).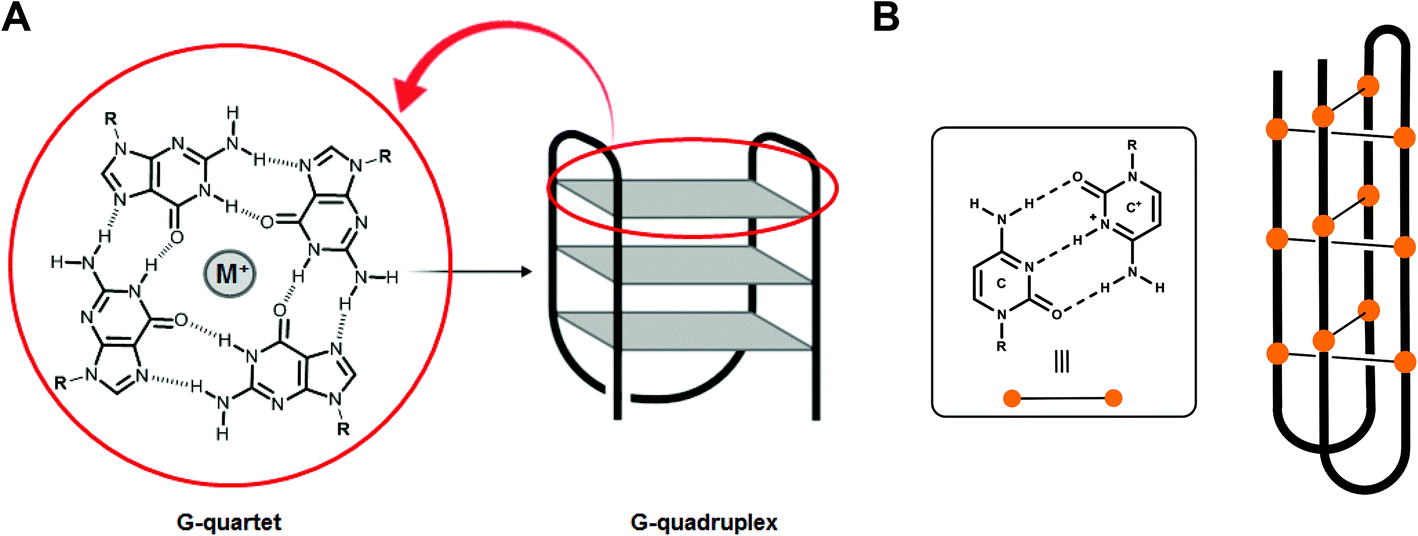 | ||
| Fig. 1 (A) G-quartet with Hoogsteen hydrogen bonded guanine and schematic drawing of a G4 structure and (B) hemi-protonated C–C+ base pairs and schematic representation of an i-motif structure. | ||
G4 DNA structures have been the subject of intensive investigations. Sequencing and bioinformatics analyses of the human genome indicate that it contains as many as 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 sequences having the potential to form stable G-quadruplex structures3 and a large number of studies have reported on their central biological functions within physiological and pathological processes.4 Although i-motif DNA structures have attracted less attention, several recent studies suggest that they may also perform essential biological functions. By using the Quadparser algorithm, more than 5000 putative i-motif forming sequences have been found in the human genome. Interestingly, more than 600 were found to be located in promotor sequences of genes such as c-myc, bcl-2 or c-kit.5 Recent studies have proposed that i-motifs are involved in the regulation of transcription6 and the integrity of telomeric DNA.7 Proteins such as hnRNP LL have been identified to interact with i-motif structures acting as an activating transcription factor.8 More recently, two independent studies have demonstrated the presence of i-motif structures in human cells through in cellulo NMR spectroscopy9 and immunofluorescence by using a specific antibody against the i-motif.10 In addition to the regulation of cellular processes in human cells, an i-motif forming sequence has been identified in the long terminal repeat promoter of the HIV-1 proviral genome where it has been shown to modulate viral transcription.11
000 sequences having the potential to form stable G-quadruplex structures3 and a large number of studies have reported on their central biological functions within physiological and pathological processes.4 Although i-motif DNA structures have attracted less attention, several recent studies suggest that they may also perform essential biological functions. By using the Quadparser algorithm, more than 5000 putative i-motif forming sequences have been found in the human genome. Interestingly, more than 600 were found to be located in promotor sequences of genes such as c-myc, bcl-2 or c-kit.5 Recent studies have proposed that i-motifs are involved in the regulation of transcription6 and the integrity of telomeric DNA.7 Proteins such as hnRNP LL have been identified to interact with i-motif structures acting as an activating transcription factor.8 More recently, two independent studies have demonstrated the presence of i-motif structures in human cells through in cellulo NMR spectroscopy9 and immunofluorescence by using a specific antibody against the i-motif.10 In addition to the regulation of cellular processes in human cells, an i-motif forming sequence has been identified in the long terminal repeat promoter of the HIV-1 proviral genome where it has been shown to modulate viral transcription.11
Encouraged by these inspiring reports, several groups have engaged in successful efforts to identify small molecule ligands that interact with C-rich sequences, stabilizing or destabilizing the corresponding i-motif structures.12 Some of these ligands have been shown to modulate gene regulation and expression and they now constitute promising probes and drug candidates targeting i-motif sequences within cancer-related genes or viral genomes.6
Beyond their biological occurrences, i-motifs have also attracted huge interest in the field of functional nanostructures. i-Motif structures which can fold and unfold reversibly depending on their environments have been manipulated by chemists to create nanotools such as in cell pH sensors13 or pH responsive shape-memory hydrogels.14 Indeed, the formation and the stability of i-motif DNA structures are affected in vitro by a number of parameters such as the length and the nature of the sequence, the solute concentration and the pH. In particular, i-motifs are generally formed in vitro at acidic pH, a particularity that complicates the use of native i-motif sequences to study the interactions of potential ligands.
As a consequence, numerous strategies have been explored to stabilize the i-motif structure and to promote its formation at physiological pH. Extending the number of C–C+ base pairs and adjusting the sequence of the loop region can help to stabilize the folded structure.15 For a given sequence of interest, modifications of the sugar,16 base17 and phosphate moieties18 can promote the formation of the i-motif. However, all of these modifications are prone to affect the recognition of the native structure of interest by putative targeting ligands.
In this context, the design of biomolecular conjugates capable of folding into well-defined and stable native i-motif structures would be of interest as they would find applications in the development of structure-specific i-motif binding ligands as well as in the identification of their structure-specific binding partners (e.g., proteins) via pull-down strategies.
In the past, our group, along with others, have proposed the use of peptide scaffolds to assemble stable mimics of tetraplex DNA structures (as in Fig. 2).19 Our strategy is based on the use of regioselectively addressable cyclopeptide platforms for directing the intramolecular assembly of the attached oligonucleotides into the desired tetraplex topology. By site-specifically fixing DNA and/or RNA G4-forming sequences onto cyclopeptide scaffolds, we have been able to trigger the formation of topologically controlled and thermally stabilized G4 architectures such as parallel tetramolecular DNA and RNA G4, antiparallel dimeric DNA G4 and DNA:RNA hybrid G4 structures.19a,20,21 For the preparation of the parallel DNA (or RNA) tetrameric systems (Fig. 2A), four oxime ligation reactions were simultaneously performed between an aminooxy-containing cyclopeptide platform and DNA (or RNA) strands that bear aldehyde complementary functions, whereas the controlled anchoring of nucleic acid strands to form antiparallel structures such as the i-motif structure (Fig. 2B) required two sequential ligations and for that purpose oxime ligation (OL) and copper-catalyzed azide–alkyne cycloaddition (CuAAC) reactions were performed between an aminooxy- and azide-containing cyclopeptide and DNA strands that bore either aldehyde or alkyne functions.22 More recently, a more sophisticated system consisting of a biologically relevant viral G4 (Fig. 2C) has been elaborated with the site-specific attachment of two distinct oligonucleotides in three successive ligations involving the OL, the CuAAC and a third compatible ligation, namely the thiol-iodoacetamide (TC) SN2 reaction. The latter ligation was found to fulfil the criteria of chemical orthogonality associated with the synthesis of such a bioconjugate and the resulting thioether linkage had no perturbative effects on the folding of the G4 structure.23 All the resulting tetraplex mimics obtained so far displayed increased stabilities and reduced structural polymorphism. Owing to these properties, the different G4 motifs have found applications in the selection and characterization of structure-specific ligands.24
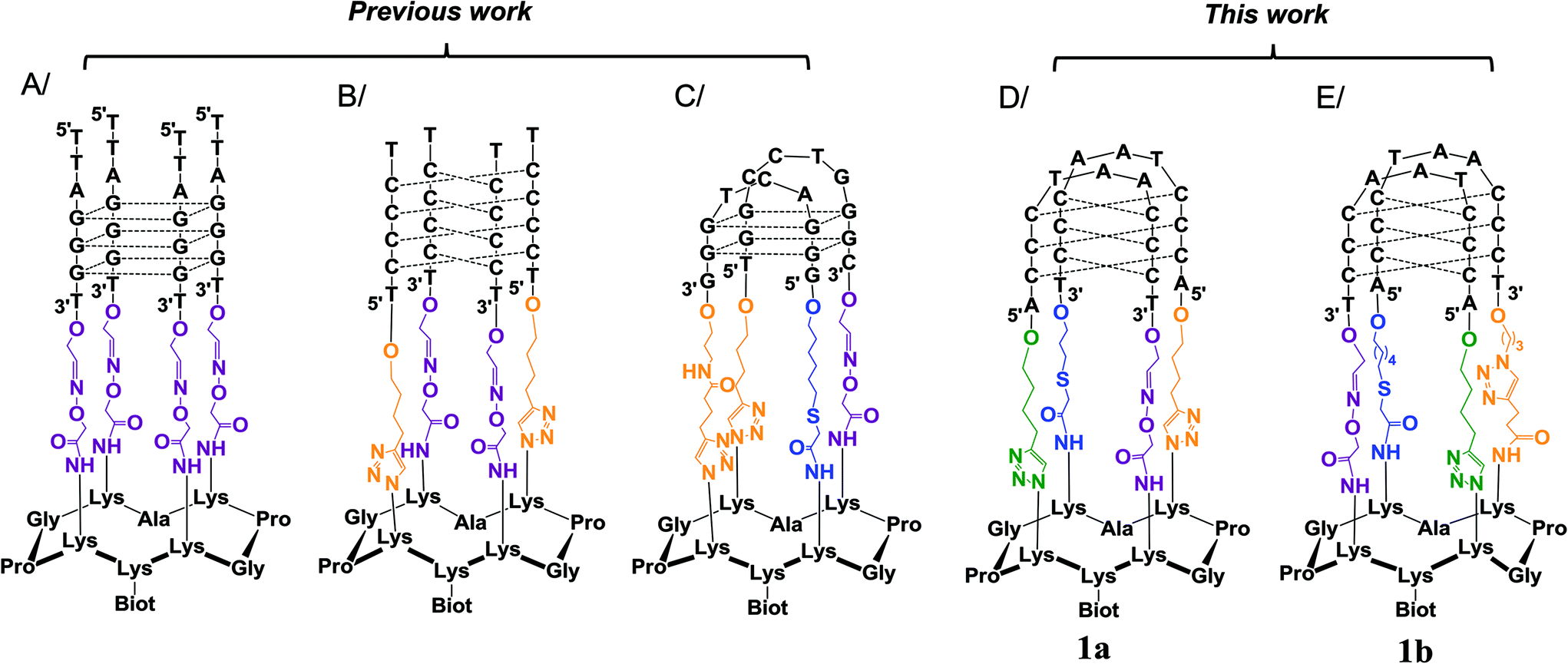 | ||
| Fig. 2 Use of the cyclopeptide scaffold for the design of stabilized tetraplex DNA structures: (A) parallel G4-DNA,19a (B) i-motif DNA structure without loops,22 (C) antiparallel G4 structure from the HIV virus sequence,23 (D) telomeric i-motif mimic 1a and (E) telomeric i-motif mimic 1b. For the sake of clarity, the positive charges shared by the two cytosines involved in the C–C base pairs of structures B, D and E are not displayed. Dotted lines represent the base pairing. | ||
To further expand this approach and to access more sophisticated biomolecular systems mimicking nucleic acid structures of high biological interest, we explored strategies to fully control the assembly of DNA–peptide conjugates via four successive ligations steps. As a proof of concept, we report herein on our unsuccessful (1a, Fig. 2D) and successful (1b, Fig. 2E) optimization efforts to assemble and stabilize a biologically relevant i-motif structure derived from the human telomeric sequence (CCCTAA)4. Unlike the i-motif structure in Fig. 2B, which consisted of four tracts of four cytosines forming a tetramolecular i-motif structure without biological relevance, the i-motif structure within 1b was designed to form via the association of four tracts of three cytosines forming a bimolecular i-motif structure displaying two 5′TAA3′ loops. This new conjugate therefore precisely mimics the biologically relevant structure of the human telomeric i-motif and in particular the loop organization, a feature that is crucial for the subsequent selection and characterization of structure-specific ligands.
To assemble the i-motif conjugates 1a and 1bvia four successive orthogonal reactions, we have chosen to make use of the three previously exploited ligations (vide supra), i.e. the oxime ligation (OL), the copper-catalyzed azide–alkyne cycloaddition (CuAAC) and the thiol-iodoacetamide SN2 reaction (TC), with the objective of using one of them twice. Oxime ligation has already been used for the successive incorporation of reporters for the preparation of ODN bis-conjugates.25 However both partners for the OL (i.e. aldehyde and aminooxy functions) are not very stable and thus, if one would want to perform two OL stepwise for the synthesis of sophisticated systems such as 1a/b, non-trivial protection strategies should be implemented (i.e. use of different protecting groups for the aldehyde or aminooxy groups). In our hand, TC reactions have been found to be more sluggish than OL and CuAAC during the formation of DNA–peptide conjugates and therefore repeating twice this reaction in the course of the assembly of these conjugates may prove problematic. On the other hand, CuAAC reactions have been repeatedly used for the successive incorporation of reporters at internal positions or at 3′,5′ extremities of nucleic acids.26 Furthermore, protected alkynes can be used along with a free alkyne to allow for sequential CuAAC reactions after mild deprotections.26a CuAAC can also be performed in the presence of free amines which can later be converted into azides via a mild diazotransfer reaction for subsequent CuAAC reactions.27 Finally, both the alkyne and the azide are stable under a wide range of non-reductive experimental conditions. Based on the robustness and flexibility of the CuAAC reaction, we decided to explore the formation of the i-motif structural mimic via the stepwise assembly of peptide–DNA conjugates through four successive ligations with one OL, one TC and two CuAAC reactions.
Two strategies were explored and involved the use of two diversely functionalized cyclopeptides and three distinct bifunctional C-rich oligonucleotides. The first strategy (Route A, Scheme 1) consists of starting with the oxime ligation followed by a first CuAAC reaction to attach bifunctional oligonucleotide 2. Subsequent thiol-iodoacetamide SN2 (TC) and CuAAC reactions are performed to incorporate the second bifunctional oligonucleotide 3 toward the final conjugate (OL → CuAAC → TC → CuAAC). Unfortunately, Route A was found to be inefficient at affording the expected target 1a (Fig. 2D). A second route (Route B, Scheme 2) was thus explored consisting of starting the assembly with the least yield-efficient TC chemistry using oligonucleotide 10 and achieving the assembly by the most efficient OL reaction. The following synthetic pathway was therefore used: TC → CuAAC → CuAAC → OL. This route allowed us to access conjugate 1b (Fig. 2E). The resulting structural mimic was found to fold into a stable i-motif structure at physiological pH and room temperature paving the way to functional applications.
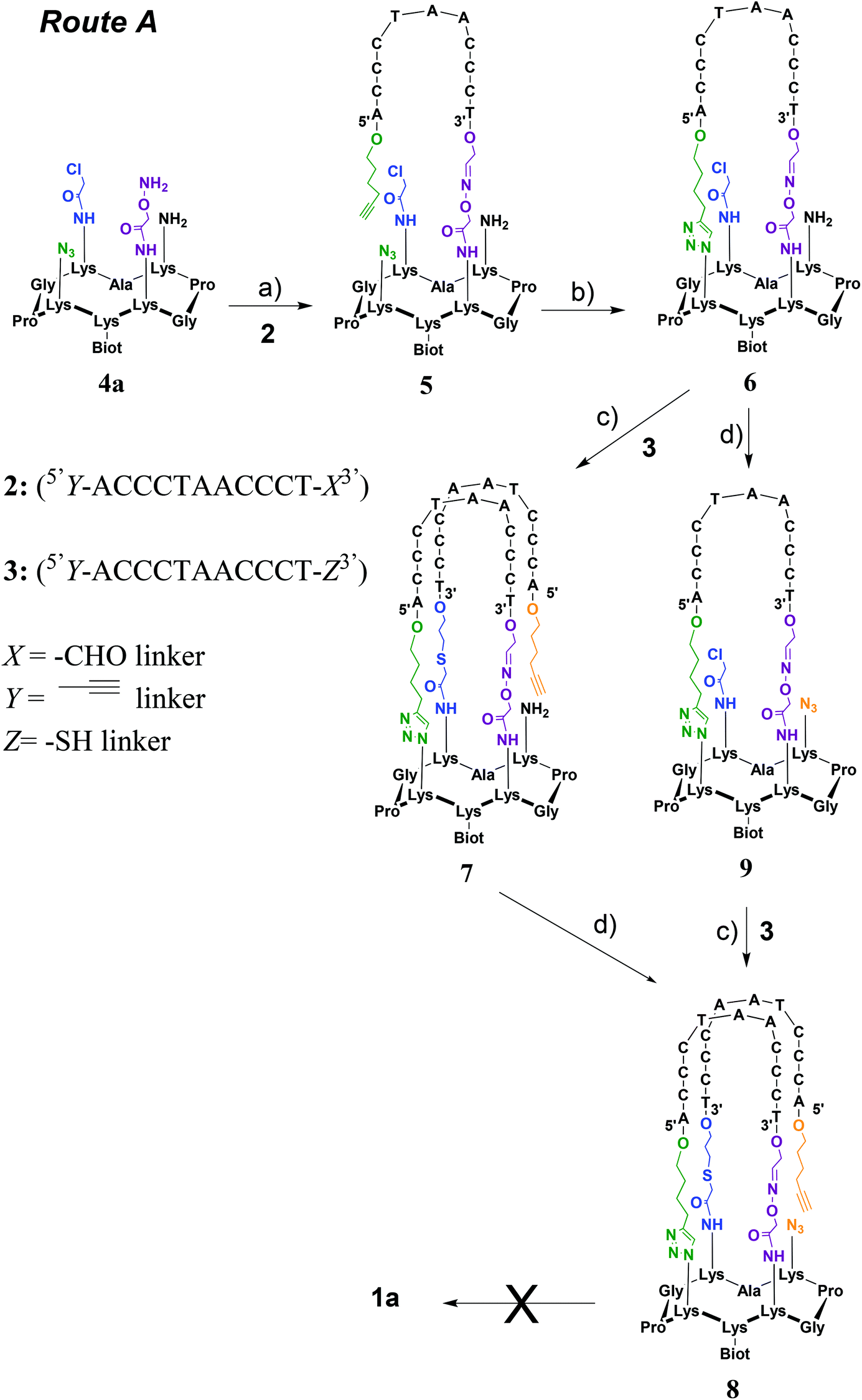 | ||
| Scheme 1 Synthesis of i-motif 1a through Route A. (a) Oligonucleotide 2 in 0.4 M ammonium acetate buffer (pH 4.5) at 55 °C for 45 min; (b) 100 mM HEPES buffer (pH 7.4), CuSO4 (6 equiv.), THPTA (30 equiv.), sodium ascorbate (30 equiv.) at 37 °C for 2 h; (c) oligonucleotide 3 in H2O/CH3CN (9:1, v/v), TCEP (2 equiv.), 500 mM KCl, DIEA (45 equiv.), KI (60 equiv.) at RT for 3 h; (d) 50 mM NaHCO3 buffer/MeOH, CuSO4 (2.8 equiv.), imidazole-1-sulfonyl azide hydrochloride (ISAHC, 20 equiv.), 30 min at 60 °C. | ||
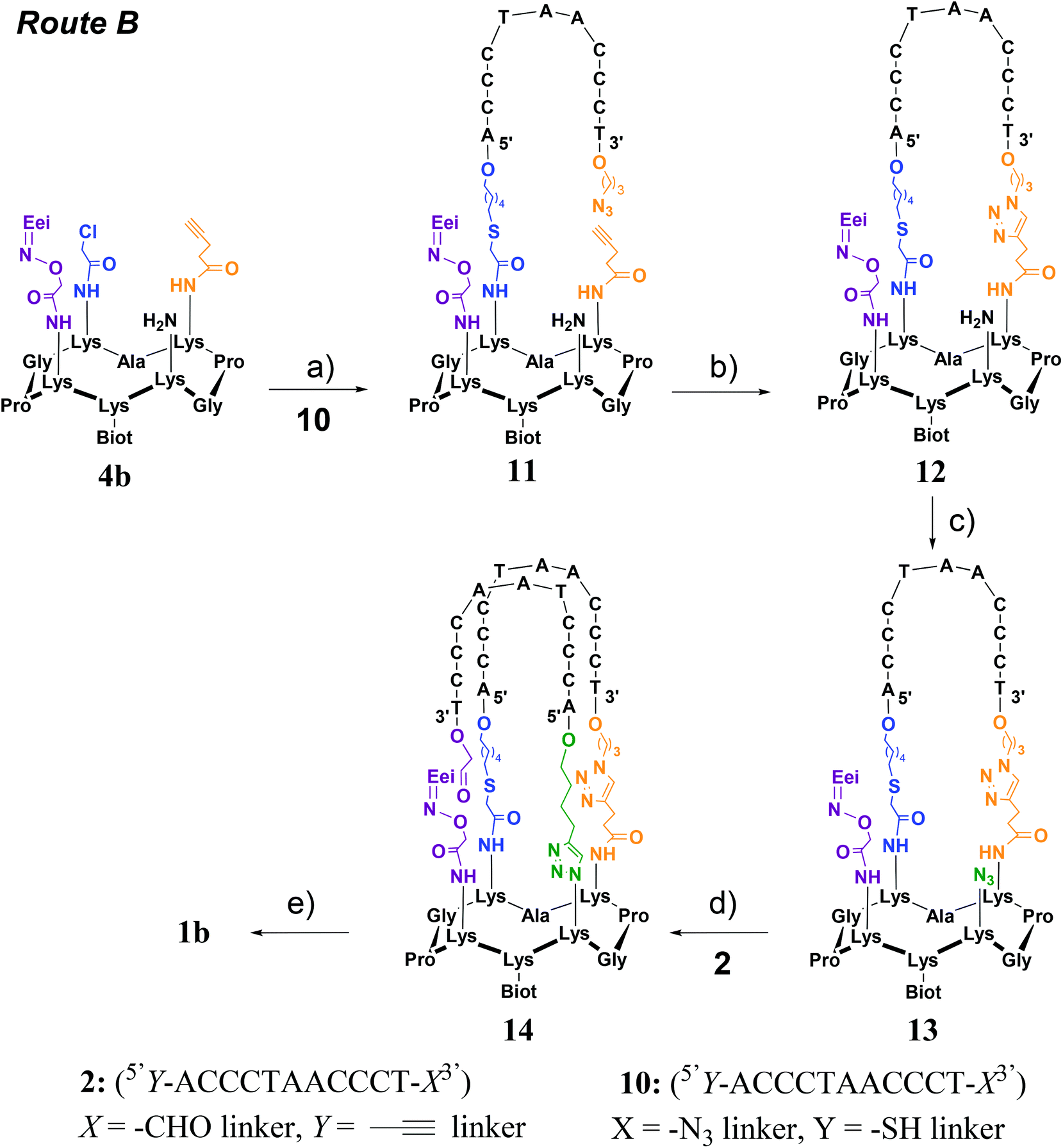 | ||
| Scheme 2 Synthesis of i-motif 1b through Route B. (a) Oligonucleotide 10 in water, TCEP (1 equiv.), DIEA (60 equiv.), KI (60 equiv.), pH 8.5, 90 min at 37 °C; (b) 50 mM PBS buffer, 10% DMF, CuSO4 (3 equiv.), THPTA (30 equiv.), sodium ascorbate (30 equiv.), 500 mM KCl pH 7.4, 2 h at 37 °C; (c) 50 mM NaHCO3 buffer/MeOH, CuSO4 (3 equiv.), imidazole-1-sulfonyl azide hydrochloride (ISAHC, 30 equiv.), 1 h at 60 °C; (d) oligonucleotide 2 in 50 mM PBS buffer, 10% DMF, CuSO4 (3 equiv.), THPTA (30 equiv.), sodium ascorbate (30 equiv.), 500 mM KCl pH 7.4, 1 h at 60 °C; (e) 1% TFA in water, 1 h at room temperature. | ||
Results and discussion
Route A (OL → CuAAC → TC → CuAAC)
This strategy requires the attachment of oligonucleotides 2 (5′Y-ACCCTAACCCT-X3′) and 3 (5′Y-ACCCTAACCCT-Z3′) in which X represents a 3′-aldehyde linker, Y a 5′-alkynyl linker and Z a 3′-thiol linker through OL, CuAAC and TC reactions onto the corresponding peptide scaffold 4a bearing the complementary functional groups (Scheme 1).Cyclopeptide scaffold 4a was prepared using standard fluorenylmethoxycarbonyl (Fmoc)/tBu solid-phase peptide synthesis (SPPS) on a 2-chlorotrityl resin followed by a cyclization reaction in solution according to the reported procedures (Scheme S1†).28 Briefly, the biotin, aminooxy and azido moieties were incorporated during SPPS by using an Nε-biotin-functionalized lysine building block, an Nε-ethoxyethylidene (Eei) protected aminooxy functionalized lysine building block and the L-azidonorleucine building block, respectively. To equip the cyclopeptide with the chloroacetamide function and the second azido group, two other orthogonally protected lysines (i.e. Nε-Boc and Nε-Dde protected lysine) were introduced during SPPS. After the cleavage from the resin under slightly acidic conditions, to prevent the cleavage of the ethoxyethylidene (Eei) protecting group, and cyclization of the linear peptide under dilute conditions, the chloroacetamide group was anchored on the lysine side chain using chloroacetic anhydride after selective removal of the bis-N-[1-(4,4-dimethyl-2,6-dioxocyclohexylidene)ethyl] (Dde) lysine protecting group with hydrazine. Final treatment under acidic conditions allowed the removal of tert-butoxycarbonyl (Boc) as well as the ethoxyethylidene protections and afforded the desired cyclopeptide scaffold 4a. To synthesize oligonucleotides 2 and 3, solid-phase oligonucleotide syntheses (SPOS), using phosphoramidite chemistry, were performed on a commercially available glyceryl CPG solid support and a 3′-thiol modifier C3 S–S CPG solid support, respectively. The alkynyl function was introduced at the 5′-terminus of oligonucleotides 2 and 3 by using commercially available 5′-hexynyl (β-cyanoethyl) phosphoramidite. Following the cleavage from the solid support and deprotection steps, oligonucleotide 2 was obtained after oxidative cleavage of the 3′-diol group. To obtain oligonucleotide 3, a 100 mM DTT treatment was performed to reduce the disulfide linkage generated upon the cleavage from the solid support.
Toward the incorporation of the first oligonucleotide, 2 was grafted on cyclopeptide scaffold 4a through oxime bond formation to generate intermediate 5. The coupling reaction was carried out in ammonium acetate buffer (pH 4.5) at 55 °C using a slight excess of aminooxy-containing peptide 4a. The reaction was monitored by RP-HPLC and proceeded to completion within 45 min. Purification through RP-HPLC followed by desalting using size-exclusion chromatography (SEC) afforded pure conjugate 5 with an isolated yield of 65%. A first intramolecular CuAAC reaction was then performed in HEPES buffer (pH 7.4) in the presence of CuSO4, tris(benzyltriazolylmethyl)amine (THPTA) ligand and sodium ascorbate at 37 °C for 2 h to afford, following desalting using size-exclusion chromatography (SEC) and RP-HPLC purification, conjugate 6 in 72% yield. Conjugates 5 and 6 were characterized by ESI MS analysis, which showed an excellent agreement between the experimentally determined molecular weight and the calculated value (Fig. S15 and S17†).
From intermediate 6, we envisioned to perform first the TC reaction and to complete the assembly with a CuAAC reaction. This decision was mainly guided by the instability of the thiol function during the CuAAC reaction. Toward 1a from 6, two alternative pathways have been investigated that consist of performing the diazo transfer reaction either after (a) or before (b) the TC reaction (Scheme 1). Previous studies have shown a poor reactivity for the TC reaction when the first oligonucleotide was already anchored on the cyclopeptide scaffold and consequently an optimization of the TC reaction had to be performed.23 The assembly of conjugate 6 with thiol containing oligonucleotide 3 was thus carried out in water at room temperature in the presence of tris(2-carboxyethyl)phosphine (TCEP) to prevent the formation of disulfide species, potassium iodide (KI) for in situ activation of the reaction through the transient formation of a reactive iodoacetamide group and DIEA to obtain a pH value around 8.5. Furthermore, under these optimized conditions, the addition of KCl is absolutely required for cation shielding to neutralize unfavorable charge repulsions between conjugate 6 and non-complementary oligonucleotide 3. Intermediate 7 was obtained in 74% yield after RP-HPLC purification and then subjected to a diazo transfer reaction with the ISAHC (imidazole-1-sulfonylazide hydrochloride) reagent by using previously reported conditions27 to afford conjugate 8 in 80% yield. The other alternative (way b) has also been explored by performing first the diazo transfer reaction on conjugate 6 to give intermediate 9 and the subsequent TC reaction with oligonucleotide 3 under the same optimized conditions as above to afford conjugate 8 in 30% overall yield. Conjugate 8 obtained by both pathways was characterized by ESI-MS analysis which showed a good agreement between the experimentally determined molecular weight and the calculated value (Fig. S21†). It is noteworthy that the diazo transfer reaction was found to be efficient and compatible with the sophisticated conjugates 6 and 7.
Toward 1a, a second CuAAC reaction had to be achieved through the reaction of the alkyne moiety on the 5′-extremity of the grafted oligonucleotide and the azide function carried by the cyclopeptide. By using the standard and above-mentioned CuAAC conditions as well as in the presence of 500 mM KCl with the aim to neutralize unfavorable charge repulsions between the oligonucleotide strands, no transformation was observed as evidenced by RP-HPLC monitoring (data not shown). We only observed the recovery of the starting material 8. Variation of reactant equivalents and temperatures did not prove to be beneficial for promoting the formation of 1a. As i-motifs are known to form at acidic pH, we thus envisioned to perform the CuAAC reaction under slightly acidic conditions compatible with CuAAC. At slightly acidic pH, the DNA strands of conjugate 8 were expected to adopt an antiparallel topology (pre-organization to an i-motif structure) and thus to hold the azide and alkyne groups close to one another thereby lowering the entropic cost of the CuAAC reaction. CD analysis of the starting conjugate 8 indeed revealed the formation of the antiparallel topology at pH 4, 5 and 6 whereas at pH 7 no structuring was noticed (Fig. S39†). CuAAC reactions were thus carried out at pH 4, 5, 6 and 7 under the above conditions (with and without KCl) and again no conversion toward 1a could be observed by RP-HPLC monitoring regardless of the conditions: only the starting material 8 was observed (data not shown). The absence of conversion could be explained by the ineffectiveness of the CuAAC reaction under acidic conditions.
Although Route A was found to be unsuitable to afford the desired constrained i-motif 1a it provided precious information to design the second strategy (Route B). Indeed, a major drawback with Route A is that during the last synthetic step, i.e. CuAAC reaction, the molecular weights of the starting material 8 and final product 1a are the same thus precluding the use of mass spectrometry to monitor the transformation and identify the final conjugate. Also, the TC reaction was again shown to be the least efficient and hard to carry out when an oligonucleotide is already grafted on the molecular scaffold. We thus decided for Route B to use the least efficient TC reaction at an early stage of the synthetic process while the most efficient OL reaction would be used in the last step. Furthermore, the intramolecular OL reaction results in the formation of a water by-product leading to a final product whose molecular weight is distinct from that of its precursor, thus allowing the use of MS for the monitoring of the reaction and identification of the final product 1b. Moreover, the OL reaction is known to be more efficient under acidic conditions and the formation of the i-motif is also known to be favored at acidic pH. We thus anticipated a pre-organization of the i-motif leading to a spatial proximity of the two reactive functions (i.e. aldehyde and aminooxy moieties) to promote the last intramolecular reaction.
Route B (TC → CuAAC → CuAAC → OL)
This strategy requires the successive attachment of oligonucleotide 10 (5′Y-ACCCTAACCCT-X3′) in which X represents a 3′-azide linker and Y a 5′-thiol linker and the above-described oligonucleotide 2 through TC, CuAAC and OL reactions onto the corresponding peptide scaffold 4b bearing the complementary functional groups (Scheme 2). Cyclopeptide 4b thus contains a protected aminooxy group for the OL reaction, an alkyne function for the first CuAAC reaction, a chloroacetamide moiety for the TC reaction and an amino function to be transformed into an azide for the second CuAAC reaction. It was prepared in the same manner as peptide 4a using standard fluorenylmethoxycarbonyl (Fmoc)/tBu solid-phase peptide synthesis (SPPS) on a 2-chlorotrityl resin and the cyclization reaction in solution (see Scheme S2†). Again the biotin and aminooxy moieties were incorporated during SPPS by using an Nε-biotin-functionalized lysine building block and an Nε-ethoxyethylidene (Eei) protected aminooxy functionalized lysine building block and two other orthogonally protected lysines (i.e. Nε-Alloc and Nε-Dde protected lysine) were introduced during SPPS for the introduction of chloroacetamide and azide moieties as above. The alkyne group was introduced as a building block during SPPS by using an Nε-pentyne functionalized lysine building block. After the cleavage from the resin under slightly acidic conditions (i.e. to prevent the removal of the Eei aminooxy protecting group) and cyclization of the linear peptide, the chloroacetamide group was anchored on the lysine side chain using chloroacetic anhydride after selective removal of the Dde protecting group with hydrazine. It should be noted that allylic alcohol should be used during the Dde cleavage to prevent the reduction of Alloc protection. The Alloc protecting group was next removed through treatment with Pd0(PPh3)4 in the presence of phenysilane and afforded the desired cyclopeptide 4b. Oligonucleotide 10 (5′Y-ACCCTAACCCT-X3′) was prepared by solid-phase oligonucleotide syntheses (SPOS) using phosphoramidite chemistry, on the commercially available 3′-PT-amino-modifier C3 CPG. The thiol function was introduced at the 5′-terminus by using the commercially available 5′-S-trityl modifier C6 phosphoramidite. The introduction of an azide function was achieved through the diazo transfer reaction as described above on the S-trityl protected thiol oligonucleotide followed by thiol deprotection in the presence of silver nitrate as reported.29With Route B, the first step consists of the use of the TC reaction which was carried out under the above-mentioned conditions, i.e. in the presence of tris(2-carboxyethyl)phosphine (TCEP), potassium iodide (KI) and DIEA. In this case, the addition of KCl for cation shielding is not required. It should be noted that due to the presence of an azide group on oligonucleotide 10, TCEP had to be added during the reaction and not prior to the addition of all other components in order to avoid partial reduction of the azide group. Desalting using size-exclusion chromatography (SEC) followed by RP-HPLC purification afforded pure conjugate 11 with an isolated yield of 53%. A first intramolecular CuAAC reaction was then performed in PBS buffer (pH 7.4) in the presence of CuSO4, THPTA ligand and sodium ascorbate at 37 °C for 2 h. As previously observed, the addition of KCl was required in order to neutralize unfavorable charge repulsions. After the addition of EDTA, desalting using size-exclusion chromatography (SEC) and RP-HPLC purification, conjugate 12 was obtained in 40% yield. The free amino function on the peptide moiety was then transformed into an azide by using a diazo transfer reaction which was carried out as described above to afford, in a quantitative yield, conjugate 13 after purification by size-exclusion chromatography. Conjugates 11–13 were characterized by ESI-MS analysis which showed a good agreement between the experimentally determined molecular weight and the calculated value (Fig. S26, S29 and S31†).
From intermediate 13, two pathways can be considered to access the expected product 1b. Indeed, a first OL could be performed to attach oligonucleotide 2 followed by a second CuAAC reaction or vice versa. As mentioned above, the major drawback of the CuAAC reaction is that there is no molecular weight difference between the starting material and the reaction product and the retention time difference is expected to analyze and purify the crude reaction mixture. Furthermore, the CuAAC reaction was found to be inefficient in the last step of the assembly of 1a. For these reasons, we envisioned to perform the CuAAC reaction first followed by the most reactive oxime reaction as the last step. The CuAAc reaction was carried out under the optimized conditions as described above, i.e. in the presence of 500 mM KCl for neutralizing the unfavorable charge repulsions between 13 and oligonucleotide 2. However, a low conversion rate was obtained at room temperature (around 20–30% according to LC-MS monitoring). Increasing the temperature to 60 °C was found to be beneficial for affording intermediate 14 with a satisfactory 48% yield after desalting using size-exclusion chromatography (SEC) and RP-HPLC purification.
The last step consisted of the one-pot deprotection of the aminooxy function and the oxime reaction. Treatment of conjugate 14 under mild acidic conditions (1% TFA) allowed the cleavage of the Eei protecting group and the simultaneous oxime bond formation. As anticipated, the monitoring of the reaction by LC-MS analysis showed the loss of the Eei protection as well as the loss of a water molecule thus demonstrating the formation of the oxime bond to afford the i-motif forming conjugate 1b (Fig. 3 and S35–S38†). It should be mentioned that the intermediate-free aminooxy compound was not observed which emphasizes the high reactivity of the oxime reaction. As expected, the efficiency of the oxime formation might have been promoted by the preorganization of the DNA strands into an i-motif structure at the acidic pH required to perform the oxime reaction.
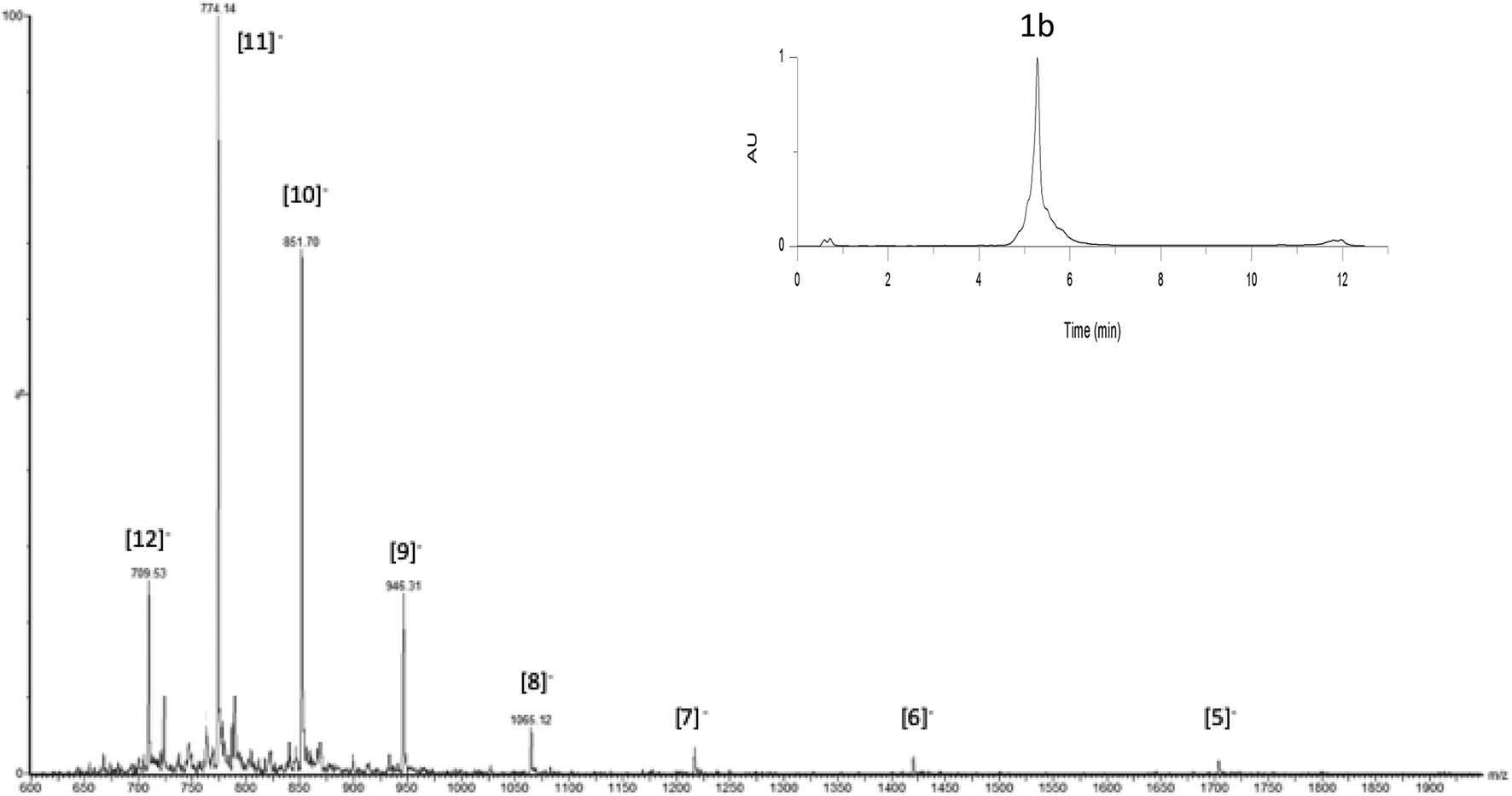 | ||
| Fig. 3 UPLC-MS monitoring of OL for the crude transformation of 14 to 1b corresponding to all peaks. Inset: UPLC profile of the crude mixture, column heated at 60 °C, detection at 260 nm. | ||
Characterization of i-motif formation
To confirm the formation of an i-motif structure within 1b and to gage its stability, CD studies were performed at different pH values at a concentration of 2 μM at 20 °C. Previous investigations have indeed shown that i-motif DNA can be associated with a specific CD signature which is markedly distinct for the CD signature of C-rich DNA fragments not forming the i-motif structure.30 The CD spectrum of the biomolecular assembly 1b recorded at pH 4 displayed a positive peak at 286 nm and a negative peak at 262 nm, characteristic of the formation of the i-motif. For comparison, the CD spectrum of the native telomeric sequence 15 d(5′(CCCCTAA)3CCCT3′) was recorded and showed similar signals (Fig. 4A shows the CD spectra of 1a and 15 at pH = 4) that confirms the formation of the i-motif structure in 1b.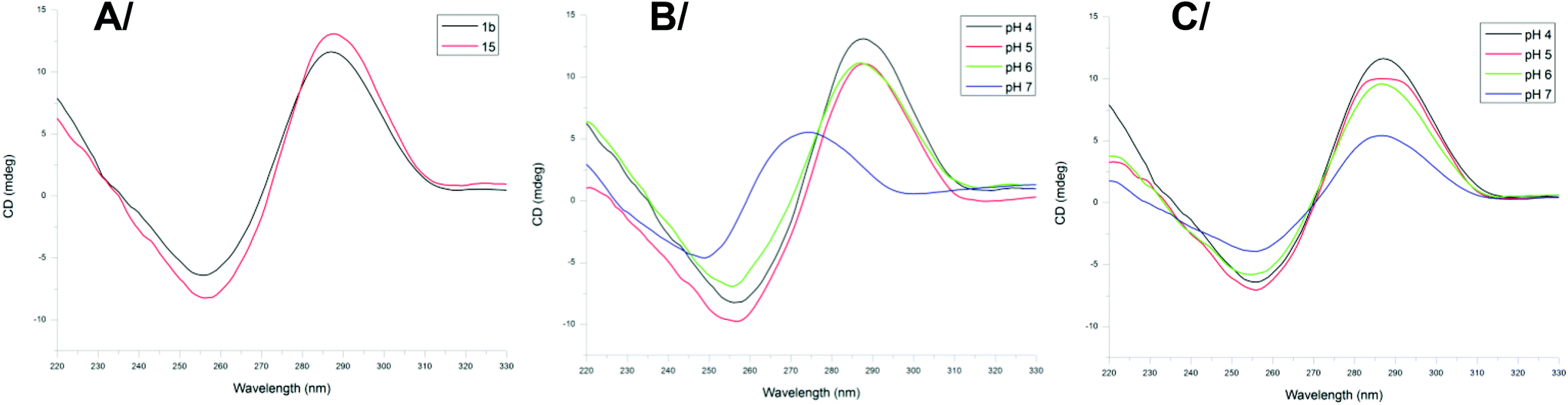 | ||
| Fig. 4 (A) CD spectra of biomolecular assembly 1b (black line) and control sequence 15 (red line) at pH = 4; (B) CD spectra of the native telomeric sequence 15 at different pH values; (C) CD spectra of biomolecular assembly 1b at different pH values. Measurements were carried out at 2 μM in 10 mM PBS buffer at 20 °C. | ||
The influence of pH on the formation of the i-motif structure was then investigated with both compounds 1b and oligonucleotide 15. By varying the pH from 4 to 7 the shape of the signal changed with the appearance of a positive peak at around 270 nm at pH = 7 (Fig. 4B) for the native telomeric sequence 15 which can be attributed to the “unstructured” DNA motif in agreement with reported studies.15a The intermediate conjugate 8 also shows the CD characteristics of unfolded C-rich DNA fragments at pH 7 (Fig. S39†), whereas for constrained system 1b, the characteristic signal of the i-motif structure is still observed at pH 7 (Fig. 4C). These data are of interest as they suggest that interaction studies with potential ligands as well as with biological components (e.g. proteins) might be performed at physiological pH with the mimic structure 1b.
The thermal stability of the i-motif structure formed within 1b was further evaluated by CD denaturation studies at pH 6 and at neutral pH 7 (Fig. S40 and S41†). At pH 6 the melting temperature was 40 °C, a temperature which is on par with the reported melting temperature of the native telomeric i-motif sequence at pH 5.5.15a On the other hand, we found that the telomeric i-motif structural mimic within 1b possesses a melting temperature of 31 °C at pH 7. This demonstrates a significant stabilization from the native structure, which was shown to unfold into a random coil at pH 6.5 with no determinable Tm at physiological pH,15a and further confirms that 1b might be exploited to study the interaction of i-motif-targeting ligands at neutral pH and room temperature.
Experimental section
General remarks
ESI mass spectra were recorded using an Esquire 3000 spectrometer from Bruker or an Acquity UPLC/MS system from Waters equipped with an SQ Detector 2 and a column heater set at 60 °C. MALDI-TOF mass spectra were recorded using an Autoflex Bruker with hydropiccolinic acid (HPA) (45 mg, ammonium citrate (4 mg) in 500 mL H2O/CH3CN) as the matrix. Peptides were analyzed in the positive mode and oligonucleotides and conjugates in the negative mode.Peptide synthesis
Oligonucleotide synthesis
Conjugate synthesis
:1, v/v) at 5.10−4 M. TCEP (2 equiv.), 500 mM KCl, DIEA (45 equiv.), and KI (60 equiv.) were added to this solution. The reaction mixture was stirred at room temperature for 3 h and the crude product was purified by RP-HPLC. Conjugate 7 was desalted using an NAP-25 column and freeze dried (74 nmol, 74% yield). ESI-MS (−): m/z calcd for C286H395N94O153P24S2: 8405.2, m/z found: 8403.5 [M − H]−.
Circular dichroism studies
Analyses were performed on a Jasco J-810 spectropolarimeter using a 1 cm length quartz cuvette. Spectra were recorded at 20 °C or every 5 °C in the range of 5 to 90 °C with a wavelength range of 220 to 330 nm. For each temperature, the spectrum was an average of three scans with a 0.5 s response time, a 1 nm data pitch, a 4 nm bandwidth and a 200 nm min−1 scanning speed. Melting temperatures were obtained using Boltzmann fit on Origin software. Each curve fit was only accepted with an rvalue > 0.99.Conclusion
In summary, we have designed and assembled a peptide–DNA conjugate capable of folding into a stable i-motif structure. The assembly was performed via the stepwise connection of two i-motif forming DNA strands through four consecutive orthogonal ligation reactions. The sequential synthetic pathway was optimized to provide full control in the assembly of such a sophisticated i-motif forming conjugate. This detailed optimization effort sheds some new light on the concrete hurdles associated with the assembly of such advanced molecular objects. For instance, we found that TC and CuACC reactions were less capable than the OL reaction to achieve the challenging formation of intramolecular covalent bonds within bulky molecular conjugates. We also further demonstrate that a mild acidic treatment can be used for the one-pot aminooxy deprotection and oxime bound formation in the presence of the acid-sensitive heteropolymer DNA sequence.The resulting conjugate was found by CD to fold, at room temperature, into an i-motif structure at acidic and neutral pH. This stabilization of the secondary structure was obtained without major modifications of a large portion of the native structure. The stabilized i-motif mimic may therefore be used to study, at physiologically relevant pH, the interaction of the i-motif with putative i-motif targeting ligands by using SPR. This unique molecular object might also be used to pull down and further characterize proteins that interact with i-motif structures. In both cases, one could take advantage of the biotin moiety built in the i-motif conjugate for surface immobilization. Finally, such a novel i-motif forming a conjugate with shifted pH sensitivity might be exploited to design original DNA nanodevices with new sets of characteristics and performances.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Agence Nationale de la Recherche (ANR-16-CE11-0006-01). The Nanobio-ICMG platform (FR2607) is acknowledged for providing facilities for the synthesis and purification of oligonucleotides (R. Lartia) as well as for mass spectrometry analyses (A. Durand, L. Fort and R. Guéret).References
- J. Spiegel, S. Adhikari and S. Balasubramanian, The Structure and Function of DNA G-Quadruplexes, Trends Chem., 2020, 2, 123–136 CrossRef.
- (a) K. Gehring, J.-L. Leroy and M. Guéron, A Tetrameric DNA Structure with Protonated Cytosine-Cytosine Base Pairs, Nature, 1993, 363(6429), 561–565 CrossRef CAS PubMed; (b) S. Benabou, A. Avino, R. Eritja, C. Gonzalez and R. Gargallo, Fundamental Aspects of the Nucleic Acid i-Motif Structures, RSC Adv., 2014, 4, 26956–26980 RSC.
- V. S. Chambers, G. Marsico, J. M. Boutell, M. Di Antonio, G. P. Smith and S. Balasubramanian, High-throughput Sequencing of DNA G-Quadruplex Structures in the Human Genome, Nat. Biotechnol., 2015, 33(8), 877–881 CrossRef PubMed.
- T. Tian, Y.-Q. Chen, S.-R. Wang and X. Zhou, G-Quadruplex: A Regulator of Gene Expression and Its Chemical Targeting, Chem, 2018, 4(6), 1314–1344 CAS.
- E. P. Wright, J. L. Huppert and Z. A. E. Waller, Identification of Multiple Genomic DNA Sequences Which Form i-Motif Structures at Neutral pH, Nucleic Acids Res., 2017, 45(6), 2951–2959 CrossRef CAS PubMed.
- (a) C. E. Kaiser, N. A. Van Ert, P. Agrawal, R. Chawla, D. Yang and L. H. Hurley, Insight into the Complexity of the i-Motif and G-Quadruplex DNA Structures Formed in the KRAS Promoter and Subsequent Drug-Induced Gene Repression, J. Am. Chem. Soc., 2017, 139(25), 8522–8536 CrossRef CAS PubMed; (b) S. Takahashi, J. A. Brazier and N. Sugimoto, Topological Impact of Noncanonical DNA Structures on Klenow Fragment of DNA Polymerase, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(36), 9605–9610 CrossRef CAS PubMed; (c) S. Kendrick, A. Muranyi, V. Gokhale, L. H. Hurley and L. M. Rimsza, Simultaneous Drug Targeting of the Promoter MYC G-Quadruplex and BCL2 i-Motif in Diffuse Large B-Cell Lymphoma Delays Tumor Growth, J. Med. Chem., 2017, 60(15), 6587–6597 CrossRef CAS PubMed.
- (a) Y. Chen, K. Qu, C. Zhao, L. Wu, J. Ren, J. Wang and X. Qu, Insights into the Biomedical Effects of Carboxylated Single-Wall Carbon Nanotubes on Telomerase and Telomeres, Nat. Commun., 2012, 3, 1074 CrossRef PubMed; (b) P. Skolakova, S. Foldynova-Trantirkova, K. Bednarova, R. Fiala, M. Vorlickova and L. Trantirek, Unique C. Elegans Telomeric Overhang Structures reveal the Evolutionarily Conserved Properties of Telomeric DNA, Nucleic Acids Res., 2015, 43(9), 4733–4745 CrossRef CAS PubMed.
- (a) B. Roy, P. Talukder, H. J. Kang, S. S. Tsuen, M. P. Alam, L. H. Hurley and S. M. Hecht, Interaction of Individual Structural Domains of hnRNP LL with the BCL2 Promoter i-Motif DNA, J. Am. Chem. Soc., 2016, 138(34), 10950–10962 CrossRef CAS PubMed; (b) L. Lannes, P. Young, C. Richter, N. Morgner and H. Schwalbe, Interaction of the N-Terminal Tandem Domains of hnRNP LL with the BCL2 Promoter i-Motif DNA Sequence, ChemBioChem, 2017, 18(20), 2033–2044 CrossRef CAS PubMed.
- S. Dzatko, M. Krafcikova, R. Hänsel-Hertsch, T. Fessl, R. Fiala, T. Loja, D. Krafcik, J.-L. Mergny, S. Foldynova-Trantirkova and L. Trantirek, Evaluation of the Stability of DNA i-Motifs in the Nuclei of Living Mammalian Cells, Angew. Chem., Int. Ed., 2018, 57(8), 2165–2169 CrossRef CAS PubMed.
- M. Zeraati, D. B. Langley, P. Schofield, A. L. Moye, R. Rouet, W. E. Hughes, T. M. Bryan, M. E. Dinger and D. Christ, i-Motif DNA Structures Are Formed in the Nuclei of Human Cells, Nat. Chem., 2018, 10(6), 631–637 CrossRef CAS PubMed.
- E. Ruggiero, S. Lago, P. Šket, M. Nadai, I. Frasson, J. Plavec and S. N. Richter, A Dynamic i-Motif with a Duplex Stem-Loop in the Long Terminal Repeat Promoter of the HIV-1 Proviral Genome Modulates Viral Transcription, Nucleic Acids Res., 2019, 47(21), 11057–11068 CrossRef CAS PubMed.
- S. S. Masoud and K. Nagasawa, i-Motif-Binding Ligands and Their Effects on the Structure and Biological Functions of i-Motif, Chem. Pharm. Bull., 2018, 66(12), 1091–1103 CrossRef CAS PubMed.
- J. Huang, Y. He, X. Yang, K. Wang, L. Ying, K. Quan, Y. Yang and B. Yin, i-motif-based Nano-Flares for Sensing pH Changes in Live Cells, Chem. Commun., 2014, 50, 15768–15771 RSC.
- W. Guo, C.-H. Lu, R. Orbach, F. Wang, X.-J. Qi, A. Cecconello, D. Seliktar and I. Willner, pH-Stimulated DNA Hydrogels Exhibiting Shape-Memory Properties, Adv. Mater., 2015, 27(1), 73–78 CrossRef CAS PubMed.
- (a) E. P. Wright, J. L. Huppert and Z. A. E. Waller, Identification of Multiple Genomic DNA Sequences Which Form i-Motif Structures at Neutral pH, Nucleic Acids Res., 2017, 45(22), 2951–2959 CrossRef CAS PubMed; (b) A. M. Fleming, Y. Ding, R. A. Rogers, J. Zhu, J. Zhu, A. D. Burton, C. B. Carlisle and C. J. Burrows, C. J. 4n–1 Is a “Sweet Spot” in DNA i-Motif Folding of 2′-Deoxycytidine Homopolymers, J. Am. Chem. Soc., 2017, 139(13), 4682–4689 CrossRef CAS PubMed.
- (a) L. Lacroix, J.-L. Mergny, J.-L. Leroy and C. Hélène, Inability of RNA To Form the i-Motif: Implications for Triplex Formation, Biochemistry, 1996, 35(26), 8715–8722 CrossRef CAS PubMed; (b) S. Robidoux and M. J. Damha, D-2-Deoxyribose and D-Arabinose, but Not D-Ribose, Stabilize the Cytosine Tetrad (i-DNA) Structure, J. Biomol. Struct. Dyn., 1997, 15(3), 529–535 CrossRef CAS PubMed; (c) H. A. Assi, R. El-Khoury, C. González and M. J. Damha, 2′-Fluoroarabinonucleic Acid Modification Traps G-quadruplex and i-Motif Structures in Human Telomeric DNA, Nucleic Acids Res., 2017, 45(20), 11535–11546 CrossRef PubMed; (d) A. Aviñó, M. Dellafiore, R. Gargallo, C. González, A. M. Iribarren, J. Montserrat and R. Eritja, Stabilization of Telomeric i-Motif Structures by (2′S)-2′-Deoxy-2′-C-Methylcytidine Residues, ChemBioChem, 2017, 18(12), 1123–1128 CrossRef PubMed.
- (a) V. B. Tsvetkov, T. S. Zatsepin, E. S. Belyaev, Y. I. Kostyukevich, G. V. Shpakovski, V. V. Podgorsky, G. E. Pozmogova, A. M. Varizhuk and A. V. Aralov, i-Clamp Phenoxazine for the Fine Tuning of DNA i-Motif Stability, Nucleic Acids Res., 2018, 46(6), 2751–2764 CrossRef CAS PubMed; (b) P. Školáková, D. Renčiuk, J. Palacký, D. Krafčík, Z. Dvořáková, I. Kejnovská, K. Bednářová and M. Vorlíčková, Systematic Investigation of Sequence Requirements for DNA i-Motif Formation, Nucleic Acids Res., 2019, 47(5), 2177–2189 CrossRef PubMed; (c) P. Perlíková, K. K. Karlsen, E. B. Pedersen and J. Wengel, Unlocked Nucleic Acids with a Pyrene-Modified Uracil: Synthesis, Hybridization Studies, Fluorescent Properties and i-Motif Stability, ChemBioChem, 2014, 15(1), 146–156 CrossRef PubMed; (d) B. Xu, G. Devi and F. Shao, Regulation of Telomeric i-Motif Stability by 5-Methylcytosine and 5-Hydroxymethylcytosine Modification, Org. Biomol. Chem., 2015, 13(20), 5646–5651 RSC; L. Lannes, S. Halder, Y. Krishnan and H. Schwalbe, Tuning the pH Response of i-Motif DNA Oligonucleotides, ChemBioChem, 2015, 16(11), 1647–1656 Search PubMed; (e) F. Seela, S. Budow and P. Leonard, Oligonucleotides Forming an i-Motif: the pH-Dependent Assembly of Individual Strands and Branched Structures Containing 2′-Deoxy-5-propynylcytidine, Org. Biomol. Chem., 2007, 5(12), 1858–1872 RSC; (f) B. Yang and M. T. Rodgers, Base-Pairing Energies of Proton-Bound Heterodimers of Cytosine and Modified Cytosines: Implications for the Stability of DNA i-Motif Conformations, J. Am. Chem. Soc., 2014, 136(1), 282–290 CrossRef CAS PubMed.
- (a) J.-L. Mergny and L. Lacroix, Kinetics and Thermodynamics of I-DNA Formation: Phosphodiester versus Modified Oligodeoxynucleotides, Nucleic Acids Res., 1998, 26(21), 4797–4803 CrossRef CAS PubMed; (b) J. A. Brazier, J. Fisher and R. Cosstick, Stabilization of the DNA i-Motif Structure by Incorporation of 3′-S-Phosphorothiolate Linkages, Angew. Chem., Int. Ed., 2006, 45(1), 114–117 CrossRef CAS PubMed.
- (a) P. Murat, D. Cressend, N. Spinelli, A. Van der Heyden, P. Labbe, P. Dumy and E. Defrancq, A Novel Conformationally Constrained Parallel G Quadruplex, ChemBioChem, 2008, 9(16), 2588–2591 CrossRef CAS PubMed; (b) V. Romanucci, M. Gaglione, A. Messere, N. Potenza, A. Zarrelli, S. Noppen, S. Liekens, J. Balzarini and G. Di Fabio, Hairpin Oligonucleotides forming G-Quadruplexes: New Aptamers with anti-HIV Activity, Eur. J. Med. Chem., 2015, 89, 51–58 CrossRef CAS PubMed; (c) M. Nikan, B. O. Patrick and J. C. Sherman, Crystal Structure of a Template-Assembled Synthetic G-Quadruplex, ChemBioChem, 2012, 13(10), 1413–1415 CrossRef CAS PubMed; (d) R. Ferreira, M. Alvira, A. Avino, I. Gomez-Pinto, C. Gonzalez, V. Gabelica and R. Eritja, Synthesis and Structural Characterization of Stable Branched DNA G-Quadruplexes using the Trebler Phosphoramidite, ChemistryOpen, 2012, 1, 106–114 CrossRef CAS PubMed; (e) G. Oliviero, N. Borbone, J. Amato, S. D'Errico, A. Galeone, G. Piccialli, M. Varra and L. Mayol, Synthesis of Quadruplex-Forming Tetra-end-Linked Oligonucleotides: Effects of the Linker Size on Quadruplex Topology and Stability, Biopolymers, 2009, 91(6), 466–477 CrossRef CAS PubMed.
- L. Bonnat, J. Dejeu, H. Bonnet, B. Gennaro, O. Jarjayes, F. Thomas, T. Lavergne and E. Defrancq, Templated Formation of Discrete RNA and DNA: RNA Hybrid G-Quadruplexes and Their Interactions with Targeting Ligands, Chem. – Eur. J., 2016, 22(9), 3139–3147 CrossRef CAS PubMed.
- R. Bonnet, T. Lavergne, B. Gennaro, N. Spinelli and E. Defrancq, Construction of anti-Parallel G-Quadruplexes through Sequential Templated Click, Chem. Commun., 2015, 51(23), 4850–4853 RSC.
- R. Bonnet, P. Murat, N. Spinelli and E. Defrancq, Click–click Chemistry on a Peptidic Scaffold for easy Access to Tetrameric DNA Structures, Chem. Commun., 2012, 48(48), 5992–5994 RSC.
- L. Bonnat, L. Bar, B. Gennaro, H. Bonnet, O. Jarjayes, F. Thomas, J. Dejeu, E. Defrancq and T. Lavergne, Template-Mediated Stabilization of a DNA G-Quadruplex formed in the HIV-1 Promoter and Comparative Binding Studies, Chem. – Eur. J., 2017, 23(23), 5602–5613 CrossRef CAS PubMed.
- (a) P. Murat, R. Bonnet, A. Van der Heyden, N. Spinelli, P. Labbe, D. Monchaud, M. P. Teulade-Fichou, P. Dumy and E. Defrancq, Template-Assembled Synthetic G-Quadruplex (TASQ): a Useful System for Investigating the Interactions of Ligands with Constrained Quadruplex Topologies, Chem. – Eur. J., 2010, 16(20), 6106–6114 CrossRef CAS PubMed; (b) G. Piraux, L. Bar, M. Abraham, T. Lavergne, H. Jamet, J. Dejeu, L. Marcelis, E. Defrancq and B. Elias, New Ruthenium-Based Probes for Selective G-Quadruplex Targeting, Chem. – Eur. J., 2017, 23(49), 11872–11880 CrossRef CAS PubMed; (c) L. Lecarme, E. Prado, A. De Rache, M. L. Nicolau-Travers, R. Bonnet, A. van Der Heyden, C. Philouze, D. Gomez, J. L. Mergny, H. Jamet, E. Defrancq, O. Jarjayes and F. Thomas, Interaction of Polycationic Ni(II)-Salophen Complexes with G-Quadruplex DNA, Inorg. Chem., 2014, 53(23), 12519–12531 CrossRef CAS PubMed.
- (a) O. P. Edupuganti, Y. Singh, E. Defrancq and P. Dumy, New Strategy for the Synthesis of 3′,5′-Bifunctionalized Oligonucleotide Conjugates through Sequential Formation of Chemoselective Oxime Bonds, Chem. – Eur. J., 2004, 10(23), 5988–5995 CrossRef CAS PubMed; (b) O. P. Edupuganti, O. Renaudet, E. Defrancq and P. Dumy, The Oxime Bond Formation as an Efficient Chemical Tool for the Preparation of 3′,5′-Bifunctionalised Oligodeoxyribonucleotides, Bioorg. Med. Chem. Lett., 2004, 14(11), 2839–2842 CrossRef CAS PubMed.
- (a) P. M. E. Gramlich, S. Warncke, J. Gierlich and T. Carell, Click-click-click: Single to Triple Modification of DNA, Angew. Chem., Int. Ed., 2008, 47(18), 3442–3444 CrossRef CAS PubMed; (b) S. De Tito, F. Morvan, A. Meyer, J. J. Vasseur, A. Cummaro, L. Petraccone, B. Pagano, E. Novellino, A. Randazzo, C. Giancola and D. Montesarchio, Fluorescence Enhancement upon G-Quadruplex Folding : Synthesis, Structure, and Biophysical Characterization of a Dansyl/Cyclodextrin-Tagged Thrombin Binding Aptamer, Bioconjugate Chem., 2013, 24(11), 1917–1927 CrossRef CAS PubMed.
- R. Lartia, P. Murat, P. Dumy and E. Defrancq, Versatile Introduction of Azido Moiety into Oligonucleotides through Diazo Transfer Reaction, Org. Lett., 2011, 13(20), 5672–5675 CrossRef CAS PubMed.
- M. Galibert, P. Dumy and D. Boturyn, One-Pot Approach to Well-Defined Biomolecular Assemblies by Orthogonal Chemoselective Ligations, Angew. Chem., Int. Ed., 2009, 48(14), 2576–2579 CrossRef CAS PubMed.
- B. A. Connolly and P. Rider, Chemical Synthesis of Oligonucleotides Containing a Free Sulphydryl Group and Subsequent Attachment of Thiol Specific Probes, Nucleic Acids Res., 1985, 13(12), 4485–4502 CrossRef CAS PubMed.
- J. Kypr, I. Kejnovska, D. Renciuk and M. Vorlickova, Circular Dichroism and Conformational Polymorphism of DNA, Nucleic Acids Res., 2009, 37(6), 1713–1725 CrossRef CAS PubMed.
- E. D. Goddard-Borger and R. V. Stick, An efficient, Inexpensive, and Shelf-Stable Diazotransfer Reagent: Imidazole-1-sulfonyl zide Hydrochloride, Org. Lett., 2007, 9, 3797–3800 CrossRef CAS PubMed.
- S. Foillard, M. O. Rasmussen, J. Razkin, D. Boturyn and P. Dumy, 1-Ethoxyethylidene, a New Group for the Stepwise SPPS of Aminooxyacetic Acid Containing Peptides, J. Org. Chem., 2008, 73(3), 983–991 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: RP-HPLC profiles of products and ESI-MS and MALDI TOF-MS spectra of purified ODNs, CD spectra. See DOI: 10.1039/d0ob01311k |
| This journal is © The Royal Society of Chemistry 2020 |