
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic approaches and applications of sulfonimidates
Priscilla Mendonça
Matos
*ab and
Robert A.
Stockman
 *a
*a
aDepartment of Chemistry, University of Nottingham, Nottingham NG7 2RD, UK
bCAPES Foundation, Ministry of Education of Brazil, DF 70040-020, Brazil
First published on 3rd August 2020
Abstract
This review article explores the synthesis of the organosulfur(VI) species named sulfonimidates, focusing on their synthesis from sulfur(II), sulfur(IV) and sulfur(VI) reagents, and investigates their recent resurgeance in interest as intermediates to access other important organosulfur compounds. Sulfonimidates have been utilized as precursors for polymers, sulfoximine and sulfonimidamide drug candidates and as alkyl transfer reagents.
1. Introduction
The discovery of sulfonimidate synthesis over 50 years ago1 has arguably allowed the development of this class of organosulfur compounds to be intensively studied and flourish over time.2 In this review article, we describe specifically the synthesis of these important sulfur-containing compounds, and their subsequent transformations to other sulfur(VI) derivatives including sulfonimidamides3 and sulfoximines,4 the latter of which has increased prominence due to their medicinal chemistry properties (Fig. 1).5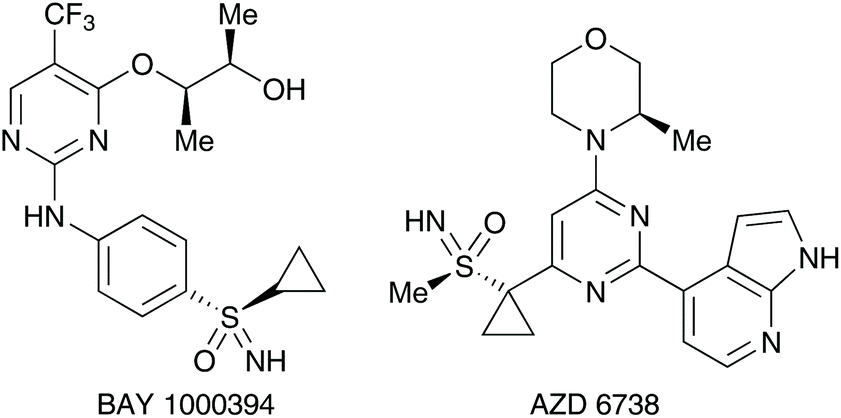 | ||
| Fig. 1 Drug candidates found with a sulfur(VI) centre. | ||
Sulfonimidates are a sulfur(VI) species bearing a tetrahedral sulfur centre, with four different groups attached (this review consistently represents them as S–O, S–C, S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and SO as shown in Fig. 2). The stereogenic sulfur centre of sulfonimidates can act as viable chiral templates that can be employed in asymmetric syntheses – an important application that is discussed in detail in the latter part of this review. One of the main advantages of such compounds is the possibility to modify up to three points of diversity; the O–R1 bond, the S–C(R2) bond and finally the nitrogen R3 substituent. The foregoing R1 and R2 substituents bear carbon containing alkyl or aryl moieties, whilst the broadest variation is found at the R3 substituent.6–16 Furthermore, sulfonimidates can be divided into two categories, acyclic and cyclic sulfonimidates. Cyclic variations of sulfonimidates arise when R1 and R3 are linked, usually through a short carbon chain that has been derived from chiral amino alcohols.
N and SO as shown in Fig. 2). The stereogenic sulfur centre of sulfonimidates can act as viable chiral templates that can be employed in asymmetric syntheses – an important application that is discussed in detail in the latter part of this review. One of the main advantages of such compounds is the possibility to modify up to three points of diversity; the O–R1 bond, the S–C(R2) bond and finally the nitrogen R3 substituent. The foregoing R1 and R2 substituents bear carbon containing alkyl or aryl moieties, whilst the broadest variation is found at the R3 substituent.6–16 Furthermore, sulfonimidates can be divided into two categories, acyclic and cyclic sulfonimidates. Cyclic variations of sulfonimidates arise when R1 and R3 are linked, usually through a short carbon chain that has been derived from chiral amino alcohols.
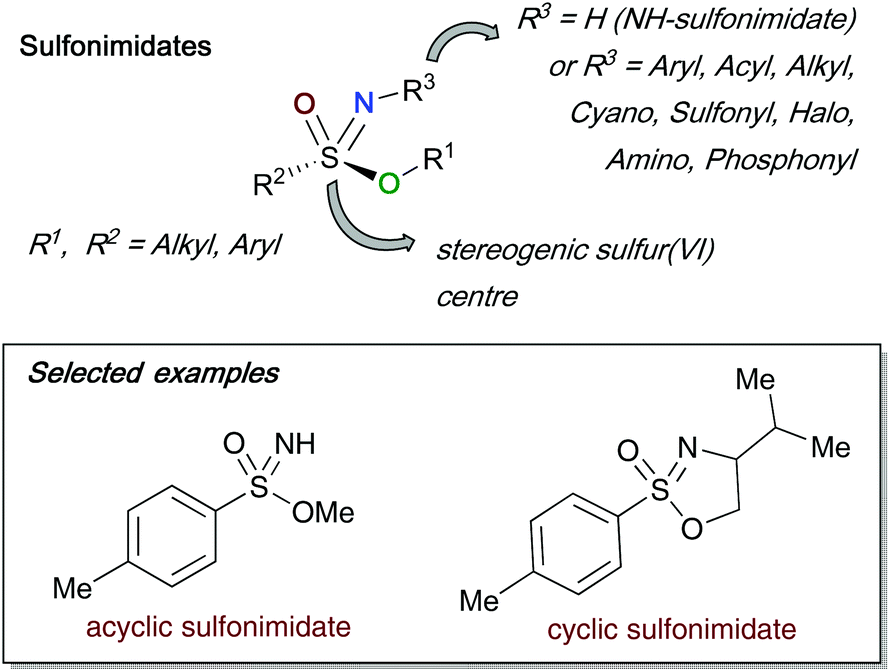 | ||
| Fig. 2 General overview of the sulfonimidate structure. | ||
Aside from their most prominent application as building blocks to access alternative sulfur(VI) compounds, sulfonimidates have found uses as alkyl transfer reagents to acids, alcohols and phenols,17 playing on the lability of sulfonimidates under acidic conditions (also noted by Levchenko in 1967).1 In addition to their acid sensitivity, they are also susceptible to elevated temperatures, being converted into sulfonamides over extended periods of time.18 The use of elevated temperatures can therefore limit applications of sulfonimidates – but, in the case of polymer synthesis, sulfonimidate decomposition at raised temperatures proved a novel way to access poly(oxothiazene) polymers8c and thionylphosphazene monomers and polymers.8d
The review is organised into two main sections – acyclic and cyclic sulfonimidates. The topics reported have focused on both seminal works as well as recent advancements on important organic chemistry features including syntheses and stereochemistry details where appropriate. Additionally, we wish to highlight to the readership that a complete overview focusing on a broad range of chiral sulfinyl compounds other than sulfonimidates has just been published this year.2b
2. Synthesis of acyclic sulfonimidates and their applications
2.1 Synthesis of acyclic sulfonimidates via sulfonimidoyl halides
Most of the important early contributions towards the synthesis of acyclic sulfonimidates were developed from the esterification of alcohols or phenols with racemic sulfonimidoyl chlorides. Levchenko and co-workers discovered that oxidation of arylsulfinyl chlorides 1 with sodium salts of N-chloroarylsulfonamides afforded sulfonimidoyl chloride 2 (Scheme 1a).19 Alternatively, sulfonimidoyl chlorides 3 bearing an N-alkyl moiety could also be accessed by the reaction of N,N-dichloroalkylamines on arene sulfinyl chlorides 1. Subsequent substitution of 3 with sodium phenoxides gave the desired sulfonimidates 4 (Scheme 1b).1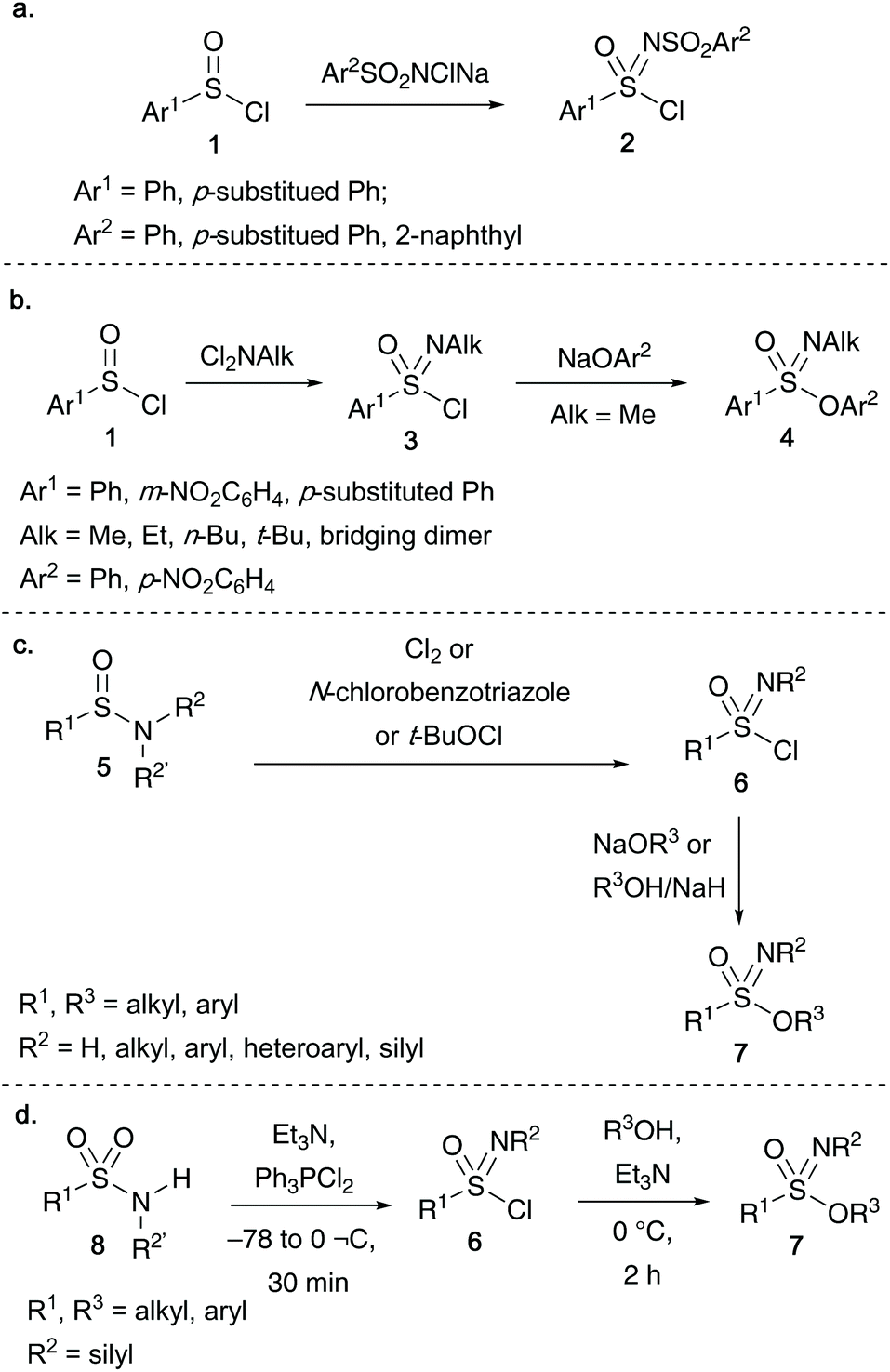 | ||
| Scheme 1 Important historical routes towards sulfonimidoyl chlorides and sulfonimidates. (a) Levchenko's synthesis of aryl-substituted sulfonimidoyl chlorides 2; (b) Levchenko's route to N-alkyl derived sulfonimidoyl chlorides 3 and conversion to sulfonimidates 4; (c) Johnson's oxidative chlorination of sulfinamides 5 to access sulfonimidoyl chlorides 6, and subsequent transformation into sulfonimidates 7; (d) Roy's synthesis of sulfonimidoyl halides 6 and sulfonimidates 7 from N-silylated sulfonamides 8 and dihalophosphoranes. | ||
In the 1970s, Johnson and co-workers identified an alternative route to access sulfonimidoyl chlorides 6 through the oxidation of sulfinamides 5 using either chlorine/N-chlorobenzotriazole,20a,b or tert-butyl hypochlorite20c as the oxidant (Scheme 1c). Reacting sulfonimidoyl chlorides 6 with either sodium alkoxides or sodium hydride-alcohol afforded sulfonimidates 7, furthermore, O-alkyoxy reagents gave slightly higher yields than the corresponding O-aryloxy derived reagents.20c This methodology was later utilized by Okuma and co-workers to access sulfonimidates (and subsequent transformation into amino(aryloxy)-oxosulfonium salts by reaction with Et3OBF4).21
Roy and co-workers disclosed that sulfonamides 8 react with bulky halophosphoranes to undergo a rearrangement reaction to access the key sulfonimidoyl chlorides 6 (Scheme 1d).8a The same group also disclosed that the bromo-derivative of 6, sulfonimidoyl bromide, could be isolated when Ph3PBr2 was used. The desired sulfonimidates 7 could be isolated in yields of up to 78% after reacting sulfonimidoyl chloride 6 with alcohol and triethylamine at 0 °C for 2 hours.8a Roy's N-silylsulfonimidates synthesized via this method were subsequently utilized in polymerization studies,8b,c investigating the reactivity of the Si–N bond under thermal polycondensation conditions (Turner and co-workers also utilized N-silylsulfonimidates to react with N-silylphosphoranimines, giving access to thionylphosphazene monomers and polymers).8d
In the 1970s, Johnson reported that access to optically active sulfonimidates 12 could be achieved when starting the synthesis from Andersen-type reagent (−)-menthyl (S)-benzenesulfinate 9 (Scheme 2). The starting reactant 9 has a known absolute configuration, and previous studies from Johnson's group showed that reaction of 9 with nucleophiles such as the lithium salt of methylamine or methyl magnesium bromide proceed with inversion of configuration.22 Moreover, studies by Montanari23 and Nudelman and Cram24 had previously highlighted that displacement of sulfinates by lithium amine salts occurs with inversion of configuration at sulfur, a similar feature that was found in Johnson's transformation of 9 to 10. Subsequent oxidation of 10 to 11 (retention of configuration), followed by reaction of sodium phenoxide with sulfonimidoyl chloride 11 (inversion of configuration) afforded sulfonimidate 12. The transformation of 10 to 12 was carried out without isolation of sulfonimidoyl chloride 11, which was found to racemise at high temperatures – maintaining 11 in an ethereal solution at −78 °C and transferring it to a solution of phenoxide in DMF at 0 °C proved fruitful, affording sulfonimidate 12 in 69% optical purity (97% on recrystallisation). Furthermore, during the oxidation step, pyridine was added as a hydrogen chloride scavenger to avoid racemisation.25
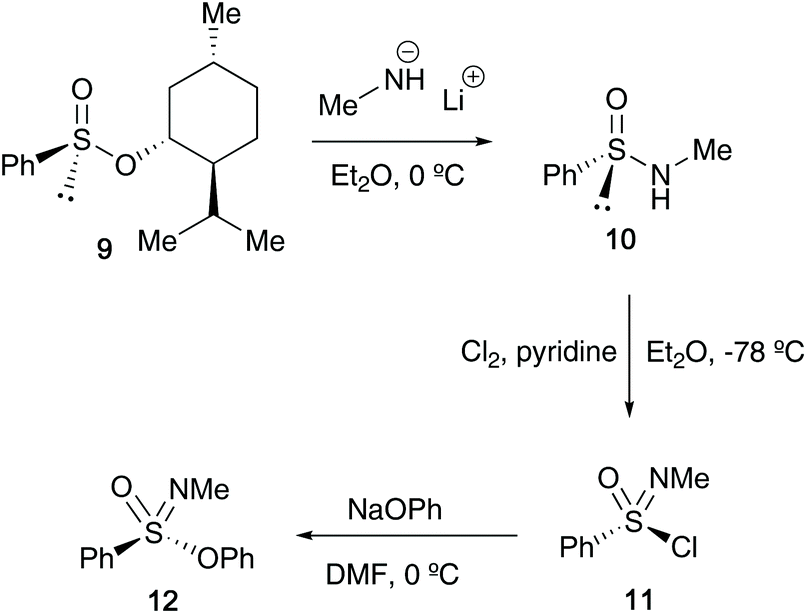 | ||
| Scheme 2 Johnson's synthesis of optically active sulfonimidates 12. | ||
The synthesis of sulfonimidates from sulfonimidoyl halides has received a resurgence in attention – this may be due to the fact that sulfonimidates are being identified as a useful intermediate towards the synthesis of other sulfur(VI) containing molecules found in drug discovery programmes, such as sulfoximines and sulfonimidamides.
Nobel prize winner Barry Sharpless has recently expanded his click-chemistry concept towards sulfur-fluoride exchange chemistry or ‘SuFEx’ chemistry, and in 2018, reported the synthesis of sulfonimidates 7 from sulfonimidoyl fluorides 13 (Scheme 3). The authors advocate the use of sulfonimidoyl fluorides over the analogous chlorides due to the latter's lability to hydrolysis. Sharpless showed that through DBU-promoted formation of the S–O bond in the sulfonimidate product, complex natural products containing a phenol motif including (+)-δ-tocopherol and capsaicin could successfully be incorporated in excellent yields.26
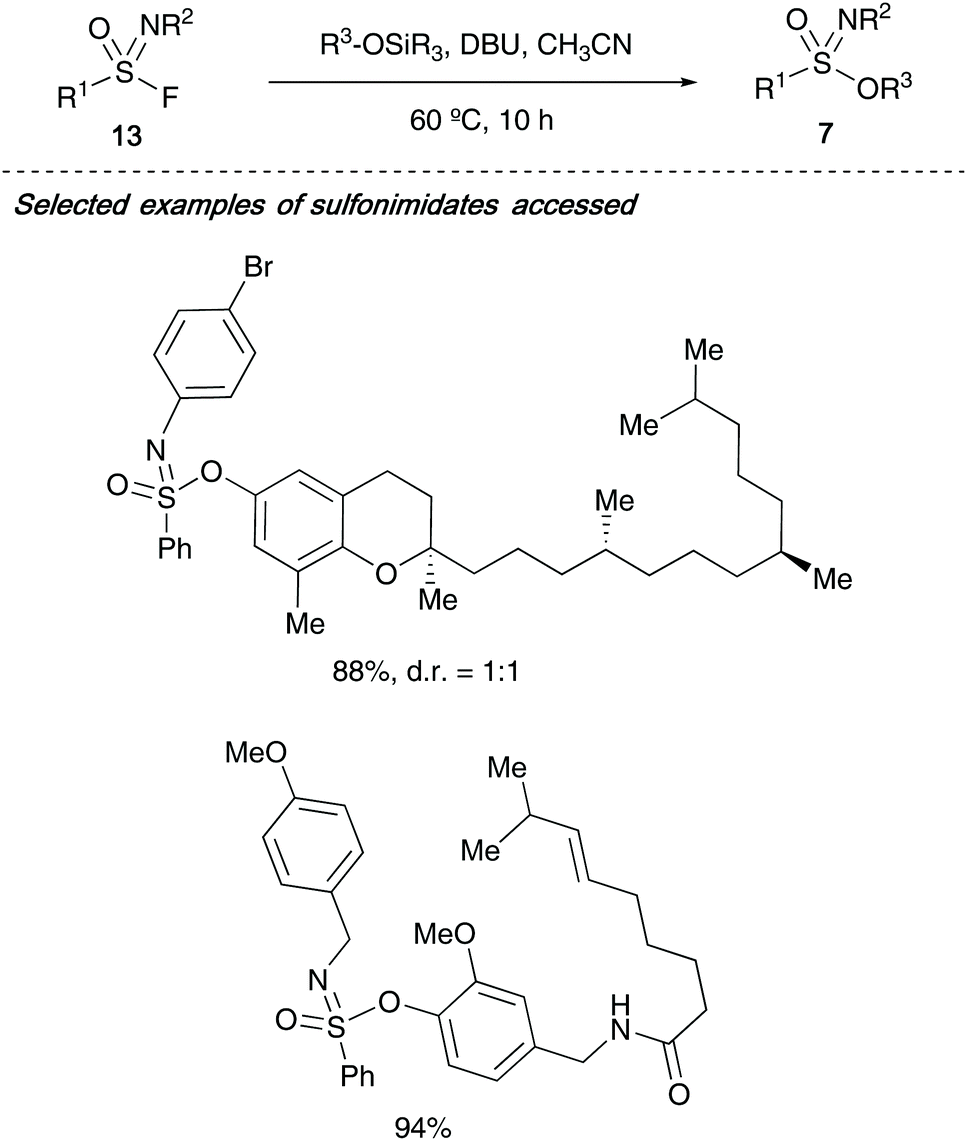 | ||
| Scheme 3 Sharpless's synthesis of sulfonimidates from sulfonimidoyl fluorides. | ||
Following on from Sharpless's use of sulfonimidoyl fluorides to access sulfonimidates, a joint research article from Wright, Oehlrich and co-workers also in 2018 led to the development of a bench stable sulfonimidate 15 that can be easily scaled for medicinal chemistry programmes looking to access CF3-derived sulfonimidamides 17/18 (Scheme 4).27a The synthesis of sulfonimidates was realised by reacting tetrafluorophenol and sulfonimidoyl fluoride 14 – a precursor that the same group previously reacted with amines directly to access sulfonimidamides.27b The authors show that their sulfonimidate 15 had a few key advantages over its predecessor (sulfonimidoyl fluorides) for the synthesis of sulfonimidamides; firstly, the sulfonimidate reagent was found to be more reactive towards amines than sulfonimidoyl fluorides. Secondly, during the sulfonimidamide formation reactions, the authors report less unwanted sulfonamide by-product was observed when the bench stable sulfonimidate was utilised. The authors have shown a single example in which two equivalents of methyl lithium were reacted with the sulfonimidate, affording the desired sulfoximine 16 in 60% yield (Scheme 4).
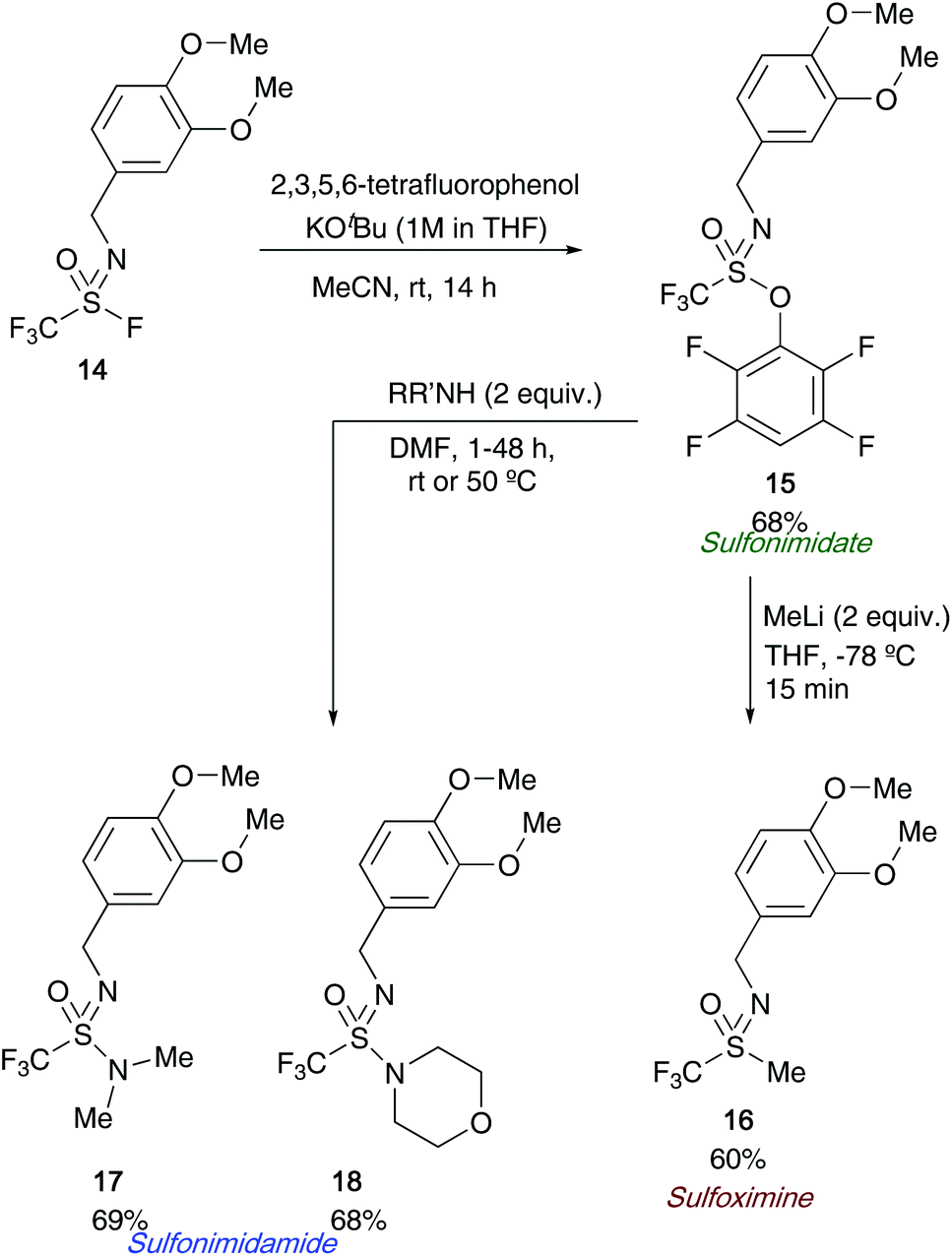 | ||
| Scheme 4 Wright and Oehlrich's bench stable sulfonimidate from sulfonimidoyl fluorides, and further applications of sulfonimidates. | ||
Building on the work of Sharpless's SuFEx chemistry, this year Zuilhof and co-workers reported the synthesis of sulfonimidates 20 from sulfonimidoyl fluorides 19 and phenols (a silicon-free SuFEx approach).28 The authors show that utilizing a stoichiometric amount of base (DBU), the formation of the target products can be achieved in excellent yields in short reaction times (as little as 2 minutes, see Scheme 5). Moreover, submitting an enantioenriched sulfonimidoyl fluoride 21 afforded sulfonimidates 22 in enantiomeric excesses up to 99% ee.
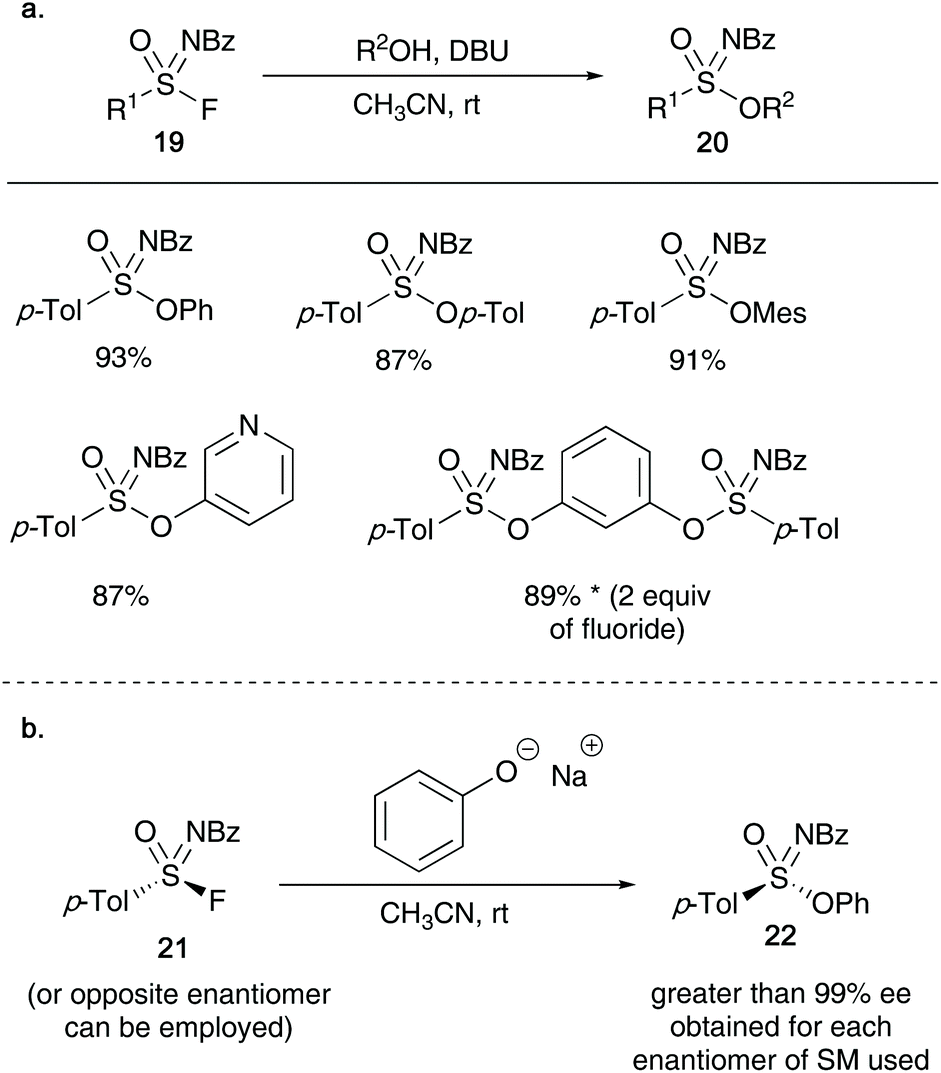 | ||
| Scheme 5 (a) Zuilhof's synthesis of sulfonimidates using a silicon free approach; (b) synthesis of optically active sulfonimidates. | ||
Three mechanistic routes were debated by the authors. Their investigations on the asymmetric synthesis of sulfonimidates ruled out an SN1 type intermediate, favoring a SN2 or addition–elimination mechanism – enantiomeric excesses were maintained when using an excess (10 equiv.) of good nucleophilic phenols, and, ee was lost when a poor, electron-deficient phenol was utilized. Furthermore, quantum chemical calculations conducted show a preference for the addition–elimination mechanism (potential energy surface analysis) when sodium phenolate was used as a nucleophile – interestingly, the calculations showed that DBU was not necessary to aid fluoride loss when sodium phenolate was used as a nucleophile source (Scheme 6).
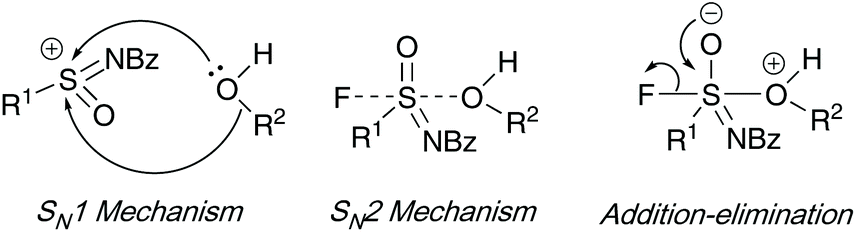 | ||
| Scheme 6 Zuilhof's proposed mechanistic routes to access sulfonimidates 20 and 22. | ||
In 2019, Ni and Hu reported the use of sulfonimidoyl fluorides as reagents to affect the conversion of alcohols 23/26 into alkyl fluorides 25/28 (Scheme 7).29 When 4-fluorophenethyl derived alcohol 23 was reacted with SulfoxFluor in the presence of DBU, 19F NMR spectroscopy indicated that a sulfonimidate species 24 had formed after 3 minutes. After a further 10 minutes, slow consumption of sulfonimidate 24 provided the desired alkyl fluoride product 25. The absence of DBU resulted in no reaction. In their proposed mechanism, DBU deprotonates the alcohol species 26 giving the alcoholate anion, that undergoes nucleophilic addition to SulfoxFluor to furnish a pentacoordinate intermediate. Protonated DBU promotes the loss of fluorine to yield sulfonimidate 27, that subsequently undergoes nucleophilic displacement by DBU-HF to produce the alkyl fluoride 28, along with a sulfonamide salt.
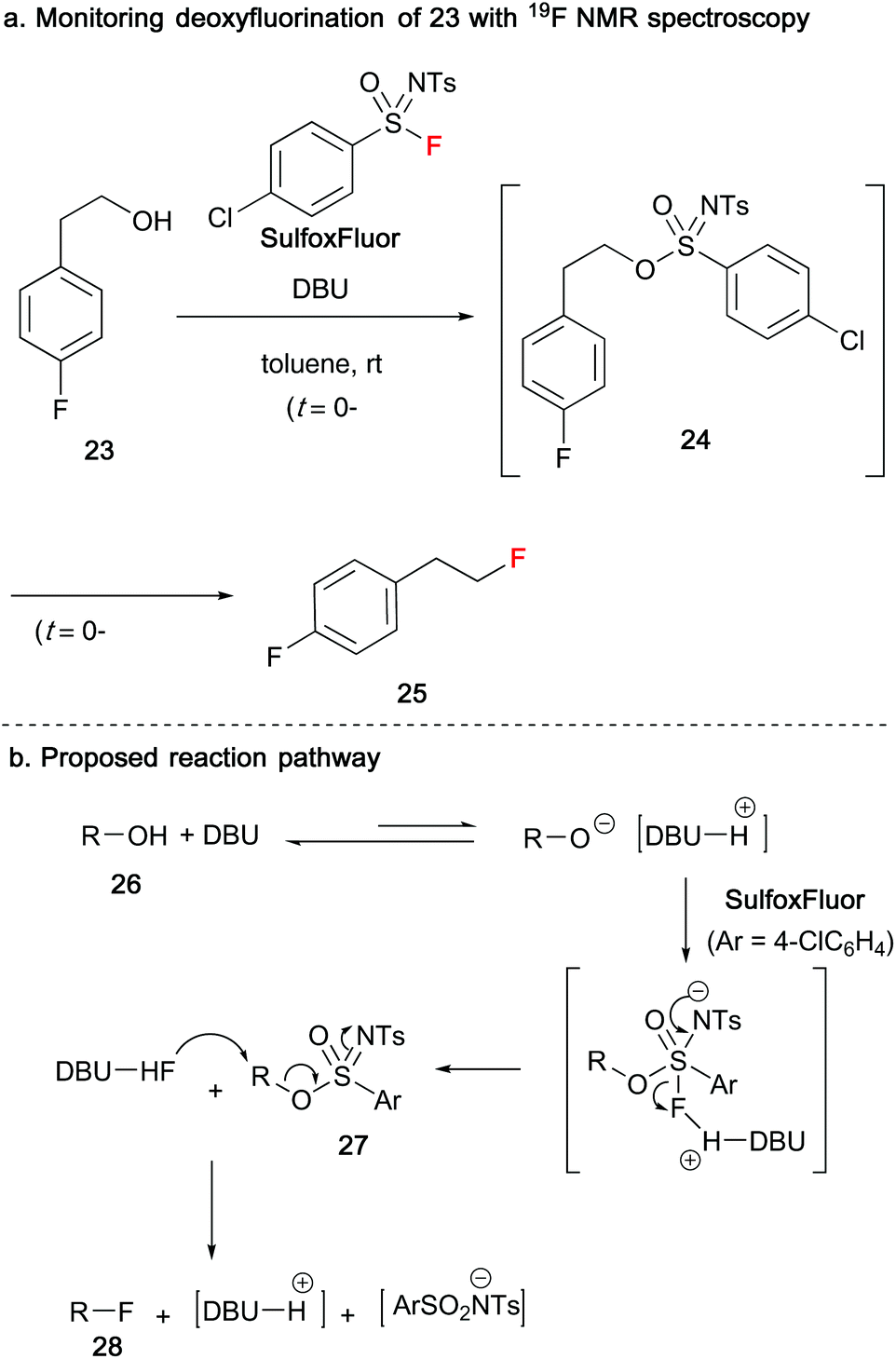 | ||
| Scheme 7 Ni and Hu's deoxyfluorination of alcohols. | ||
2.2 Synthesis of acyclic sulfonimidates from sulfinyl hydroxylamines and applications as alkyl transfer reagents
In 1973, Maricich et al. showed that sulfinyl hydroxylamines 29 undergo a rapid rearrangement to sulfonimidates 32/33, in which an alkoxy group migrates from the nitrogen to the sulfur atom.30 The sulfonimidates formed were unstable and were found to transfer their alkyl group to a range of alcohols when heated at 50 °C forming ethers 35 along with the corresponding sulfonamide 34 (Scheme 8).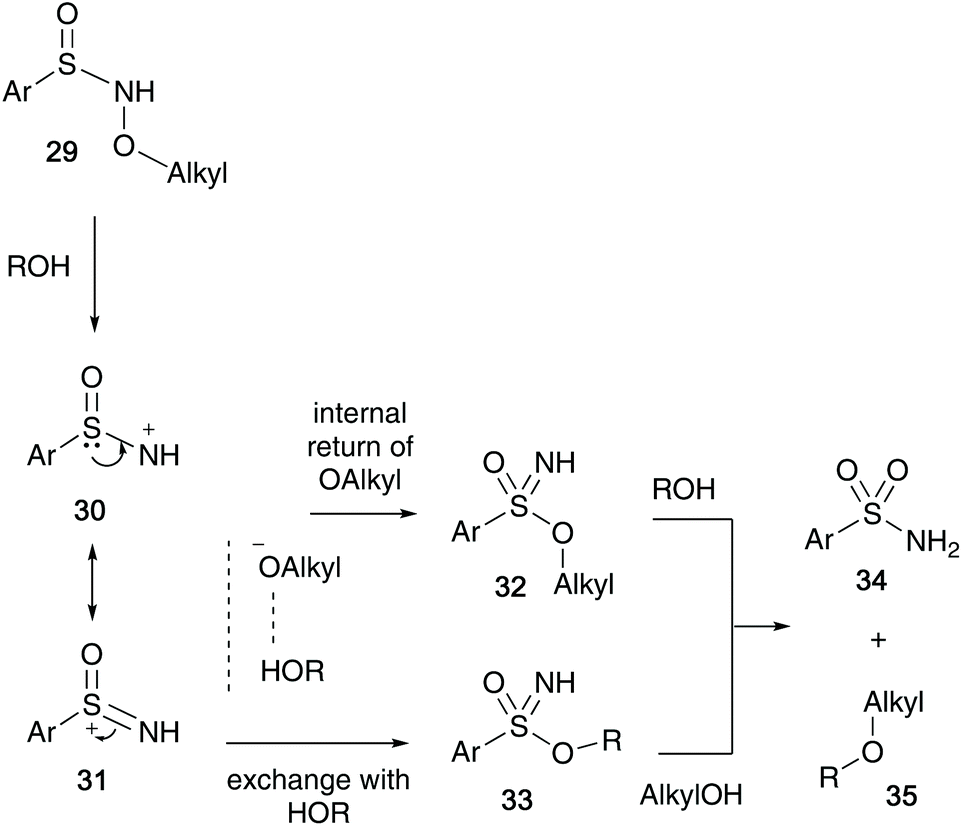 | ||
| Scheme 8 Maricich's proposed mechanism of alkyl transfer to alcohols. | ||
The reaction proceeds via a mechanism involving sulfonimidate species 32 or 33, that may have been formed through a dissociation process of the migrating alkoxy group and the solvent (ROH). Two possible sulfonimidates were postulated that could transfer the alkyl substituent to the alcohol. They also believe that a delocalised nitrenium species 30/31, stabilised by the sulfinyl electron pair is possible, and this was supported when cyclic N-acyl derivative 36 failed to rearrange after refluxing in ortho-dichlorobenzene for 20 hours, since the nitrenium ion from 36 would be destabilised by the N-acyl group (Scheme 9).
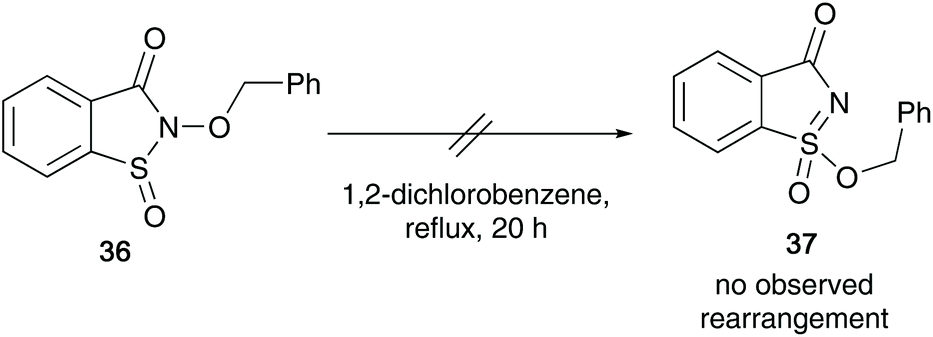 | ||
| Scheme 9 Maricich's attempted rearrangement of N-acyl derived N-alkoxybenzenesulfinamides 36. | ||
In 2013, the same author exploited the application of their sulfonimidate rearrangements, utilizing them as alkyl transfer reagents to a range of acids, alcohols and phenols.17 Utilizing nitro-containing sulfonimidate 38, the ethylation of acids to esters was achieved without a catalyst – however, the reaction required catalytic amounts of fluoroboric acid-dimethyl ether complex (10 mol%) for the conversion of alcohols and phenols to ethers (yields up to 94%, Scheme 10). For each reaction conducted, the side product isolated was the resulting sulfonamide 39 from the sulfonimidate reagent.
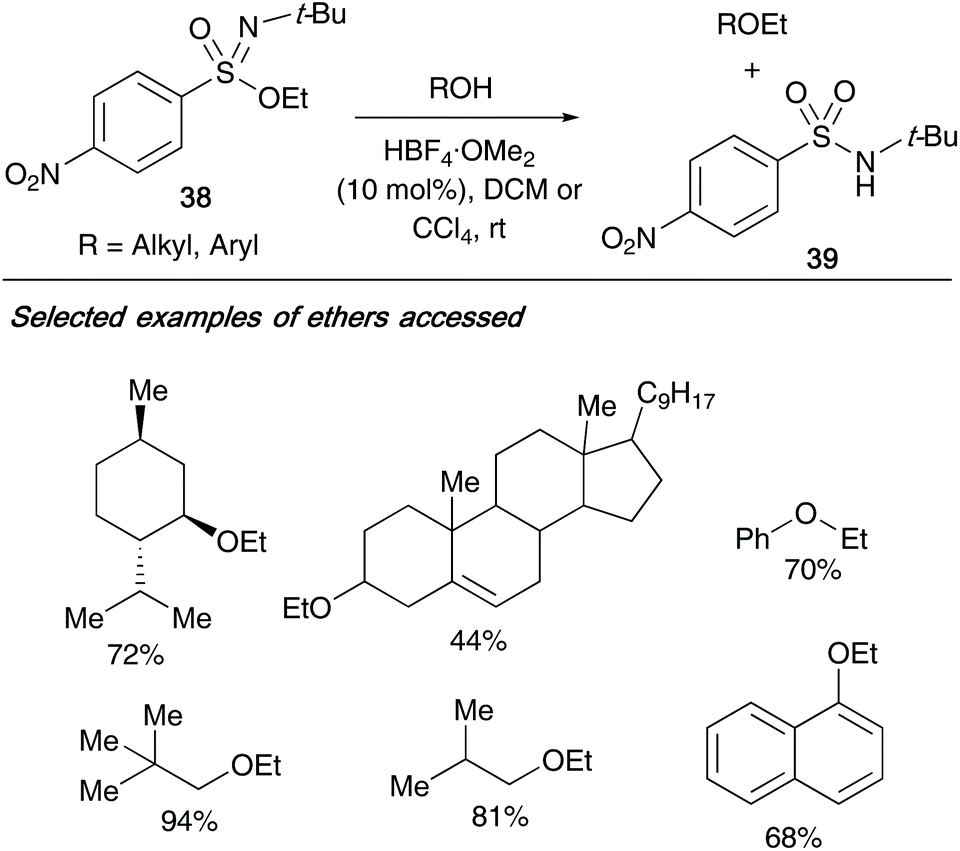 | ||
| Scheme 10 Maricich's sulfonimidate 38, and its application as an alkylation transfer reagent. | ||
2.3 Synthesis of acyclic sulfonimidates from sulfinamides utilising hypervalent iodine species & applications towards sulfoximine synthesis
Malacria and co-workers reported that sulfonimidates 41 could be formed from sulfinamides 40, iodosobenzene and alcohols in a one-pot procedure, utilising mild conditions, allowing access to a broad range of sulfonimidates in yields up to 95% (Scheme 11).6 The reaction tolerated a variety of primary alcohols (except benzyl alcohol), and further limitations were found on increasing the steric hindrance on the alcohol – iso-propyl alcohol required a longer reaction time, whilst reaction failure was observed when using tert-butanol. Furthermore, phenol only resulted in degradation, with no O-aryl sulfonimidate formed. | ||
| Scheme 11 Malacria's synthesis of sulfonimidates from sulfinamides and iodosobenzene. | ||
Malacria's procedure built upon the initial report of Maricich's N-alkoxybenzenesulfinamides,30 in which they devised a way of oxidising the sulfinamide to form the key N–O bond, and minimised the amounts of unwanted sulfonamide 42 that was known to occur when oxidants such as m-CPBA were employed.31 The use of a mild oxidising agent derived from iodine(III)32 allowed the necessary N–O bond formation to proceed smoothly (Scheme 11).
Their initial mechanistic proposal is highlighted in Scheme 12 – solvation of iodosylbenzene 43 in the desired alcohol, followed by attack on 44 by sulfinamide species 45 initiates the synthesis.6,33 This species can form a dissociated intermediate 47, that in the presence of the remaining alkoxide anion 48 can re-attack the nitrogen centre and expel the iodobenzene moiety to give the key N-alkoxybenzenesulfinamides intermediate 49, first identified by Maricich (Scheme 8). This can then rapidly rearrange to yield the key sulfonimidate product 33.
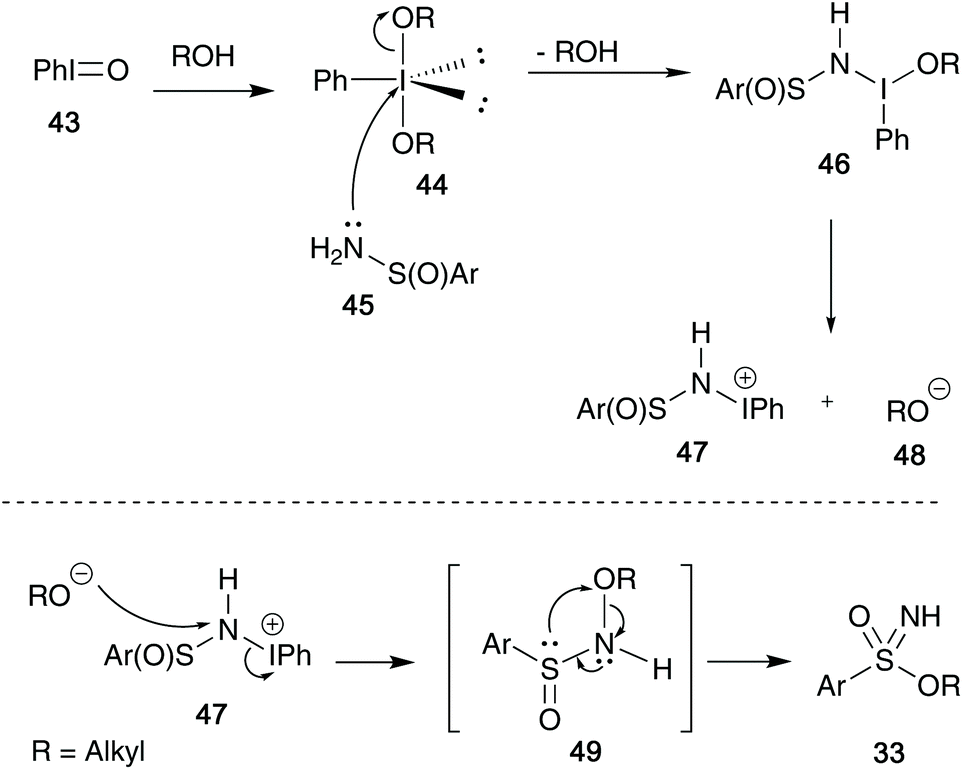 | ||
| Scheme 12 Malacria's initial mechanistic proposal for the formation of sulfonimidates 33. | ||
During the course of conducting an asymmetric version of the above transformation, the authors found that the reaction proceeded with retention of configuration at the sulfur centre in the product. As a result of these findings, Malacria proposed an alternative, more reasonable model (Scheme 13) – initial sulfinamide 51 attack on solvated iodosylbenzene 50 yields intermediate 52. A rearrangement of 53via a 6-electron transition state allowed access to sulfonimidates 54 with retention of configuration (Scheme 13a). However, from intermediate 53, an alternative route via an three-atom rearrangement (first reported by Reggelin and co-workers)34 would afford a sulfonimidoyl iodonium 55 – a species that could be responsible for loss in enantiomeric excess during the transformation, yielding sulfonimidate 56 (Scheme 13b).
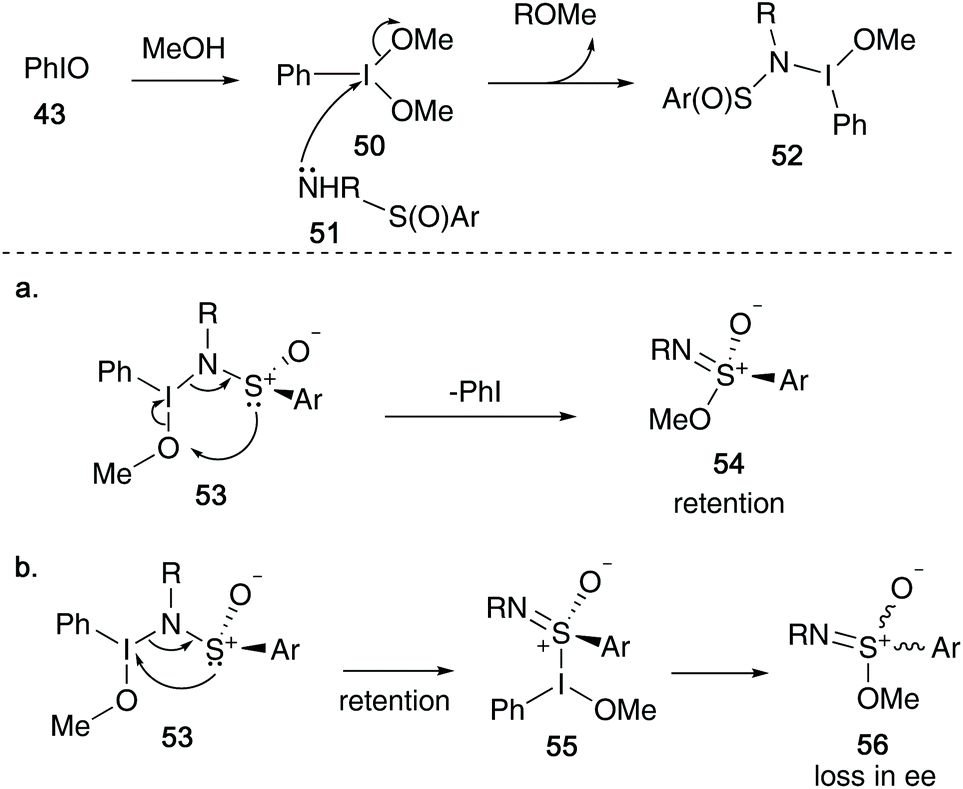 | ||
| Scheme 13 Malacria's revised mechanism for the preparation of sulfonimidates. | ||
In 2006, Malacria showed that N-aryl sulfinamides 57 could also be utilised in the formation of sulfonimidates 58 with their one-pot procedure utilising diacetoxyiodobenzene. With these examples, aryl derived sulfinamides required diacetoxyiodobenzene (DIB) and a mild base such as magnesium oxide to achieve high yields (Scheme 14).7
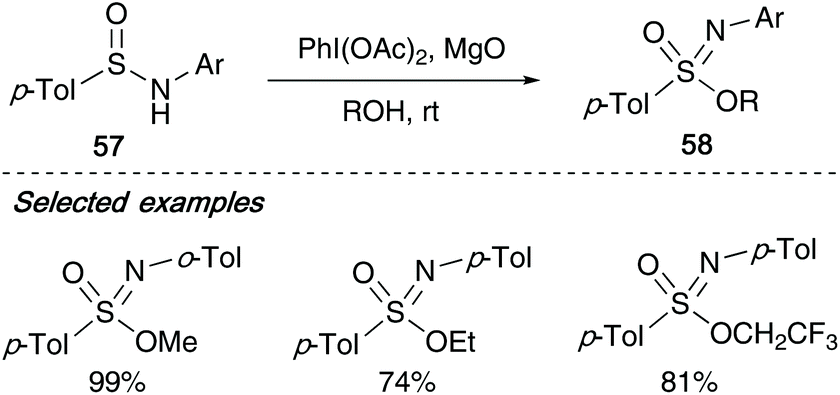 | ||
| Scheme 14 Malacria's synthesis of arylsulfonimidates. | ||
In 2018, Bull, Luisi and co-workers have most recently reported the conversion of aryl thiols 59 into sulfonimidates 60 utilising hypervalent iodine reagents and ammonium carbamate as nitrogen source (Scheme 15).16 Using a 4-equivalent loading of ammonium carbamate forced the reaction to produce exclusively sulfonimidate (at 1 equivalent of nitrogen source, sulfonamide was formed as the major reaction product). The reaction scope allowed a range of aryl thiols to participate in the reaction – substitution on the aromatic ring at ortho, meta and para with both electron withdrawing and donating substituents were well tolerated affording sulfonimidates in good yields. In one case, a cycloalkylthiol was submitted to the optimised conditions, and the desired sulfonimidate was isolated in moderate yield. Whilst the exact sequence of events to form the sulfonimidate 60/64 from thiol 59 is unclear, the mechanism of Bull and Luisi's method was based on a range of control experiments that allowed the identification of disulfides 61 and sulfinate esters 62 during the reaction (Scheme 15).
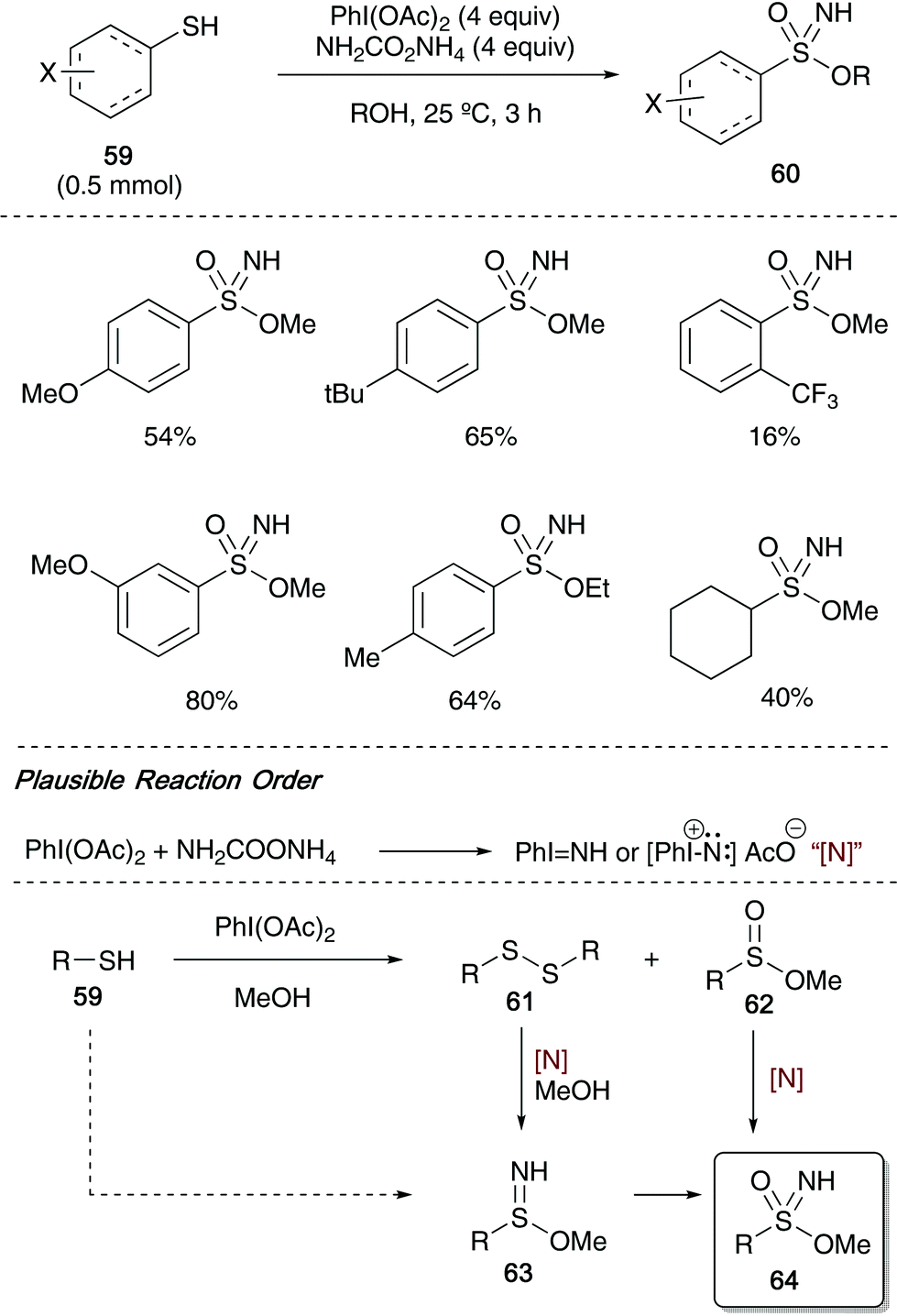 | ||
| Scheme 15 Bull and Luisi's sulfonimidate 60/64 synthesis from aryl thiols 59. | ||
In the same year, Stockman and co-workers utilized sulfonimidates 7 as versatile intermediates to access a broad library of sulfoximines 66.35 Building on the methodology developed by Malacria and co-workers, Stockman utilized iodosylbenzene to oxidise a range of sulfinamides 65 in an excess of alcohol as solvent to access a range of sulfonimidates that differ at three key positions (Scheme 16). During the oxidation reaction, modifying the R1 substituent to include allyl, cyclopropyl, methyl and phenyl moieties were all well tolerated – inclusion of a tert-butyl group however proved unsuccessful. Furthermore, a range of primary alcohols could be incorporated, as well as varying the N-moiety of the sulfinamide 65 (cycloalkyl, tert-butyl, substituted aryls) without loss of reactivity in the transformation. A broad library of sulfonimidates 7 (where OR3 was OEt) were then converted to sulfoximines 66 by Grignard displacement of the OR3 group.
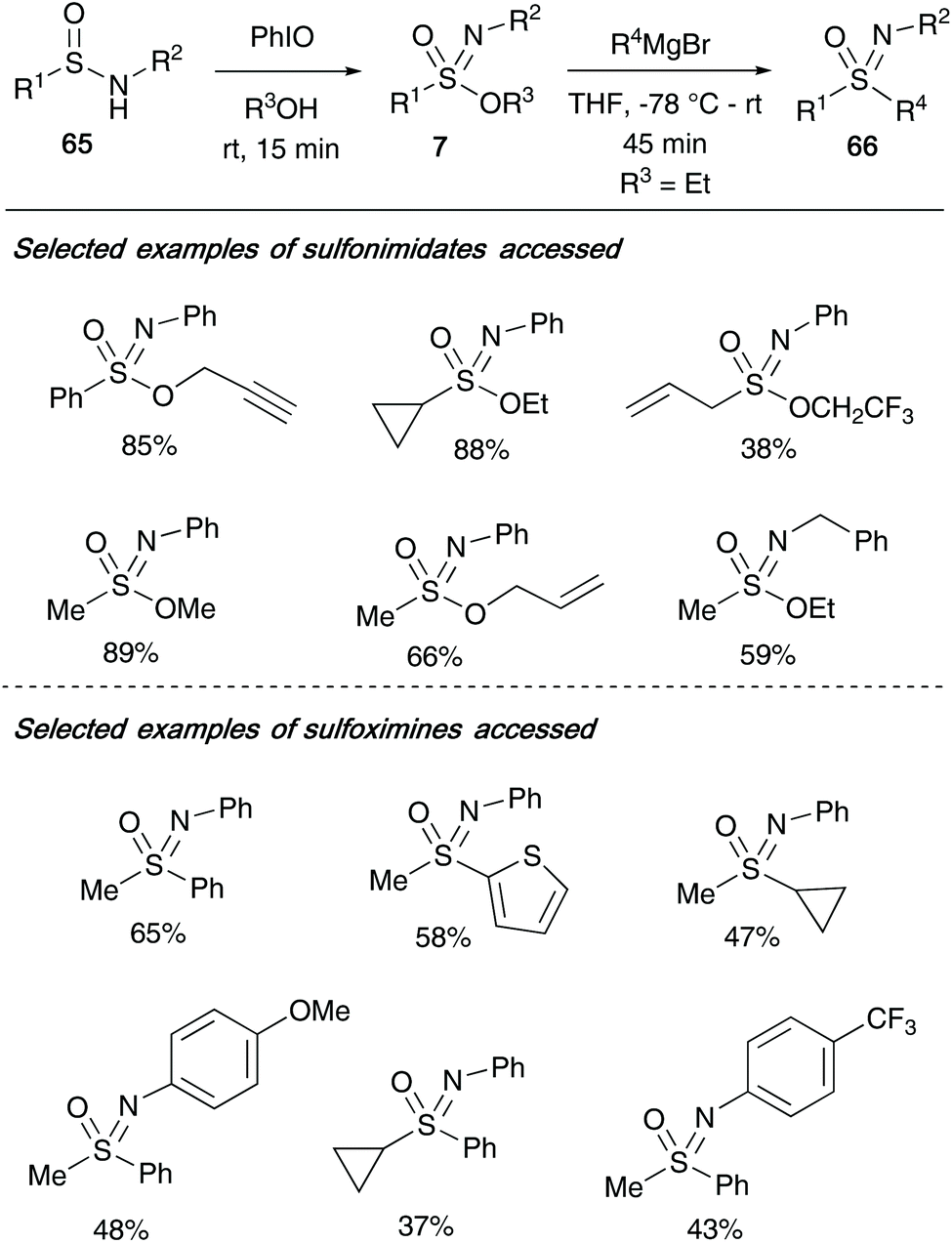 | ||
| Scheme 16 (a) Stockman's synthesis of sulfonimidates 7 and, (b) application of sulfonimidates to access a diverse library of sulfoximines 66. | ||
In 2019, Bolm and co-workers used O-benzotriazolyl sulfonimidates 69/70 as active intermediates in order to access sulfonimidamides 71 (Scheme 17).36 Their group reported that aromatic or heteroaromatic diazonium salts 68 could be reacted with N-tritylsulfinylamine 67![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 37 and 1-hydroxybenzotriazole hydrate with 1.1 equiv. of N-methyl-piperidine gives the desired sulfonimidates 69. The method utilizes dimethyl carbonate, a green solvent for synthesis, and benefits from fast reaction times. As shown in Scheme 17, a range of functional groups from the diazo-aryl/heteroaryl moiety were well tolerated under the reaction conditions, providing structural diversity. Control experiments with TEMPO as radical scavenger showed complete suppression of sulfonimidate formation, indicating a potential radical mechanism involving pre-organized aggregates of the reactants. Bolm also showed that sulfonimidates 70 could be converted into sulfonimidamides 71 through reaction with either primary or secondary amines (2.0 or 1.2 equiv., respectively) under basic conditions (Scheme 17b). To highlight just a few of the examples, aliphatic amines such as ethylamine, thiomorpholine and azepane reacted smoothly affording the target sulfonimidamides 71 in good yields.
37 and 1-hydroxybenzotriazole hydrate with 1.1 equiv. of N-methyl-piperidine gives the desired sulfonimidates 69. The method utilizes dimethyl carbonate, a green solvent for synthesis, and benefits from fast reaction times. As shown in Scheme 17, a range of functional groups from the diazo-aryl/heteroaryl moiety were well tolerated under the reaction conditions, providing structural diversity. Control experiments with TEMPO as radical scavenger showed complete suppression of sulfonimidate formation, indicating a potential radical mechanism involving pre-organized aggregates of the reactants. Bolm also showed that sulfonimidates 70 could be converted into sulfonimidamides 71 through reaction with either primary or secondary amines (2.0 or 1.2 equiv., respectively) under basic conditions (Scheme 17b). To highlight just a few of the examples, aliphatic amines such as ethylamine, thiomorpholine and azepane reacted smoothly affording the target sulfonimidamides 71 in good yields.
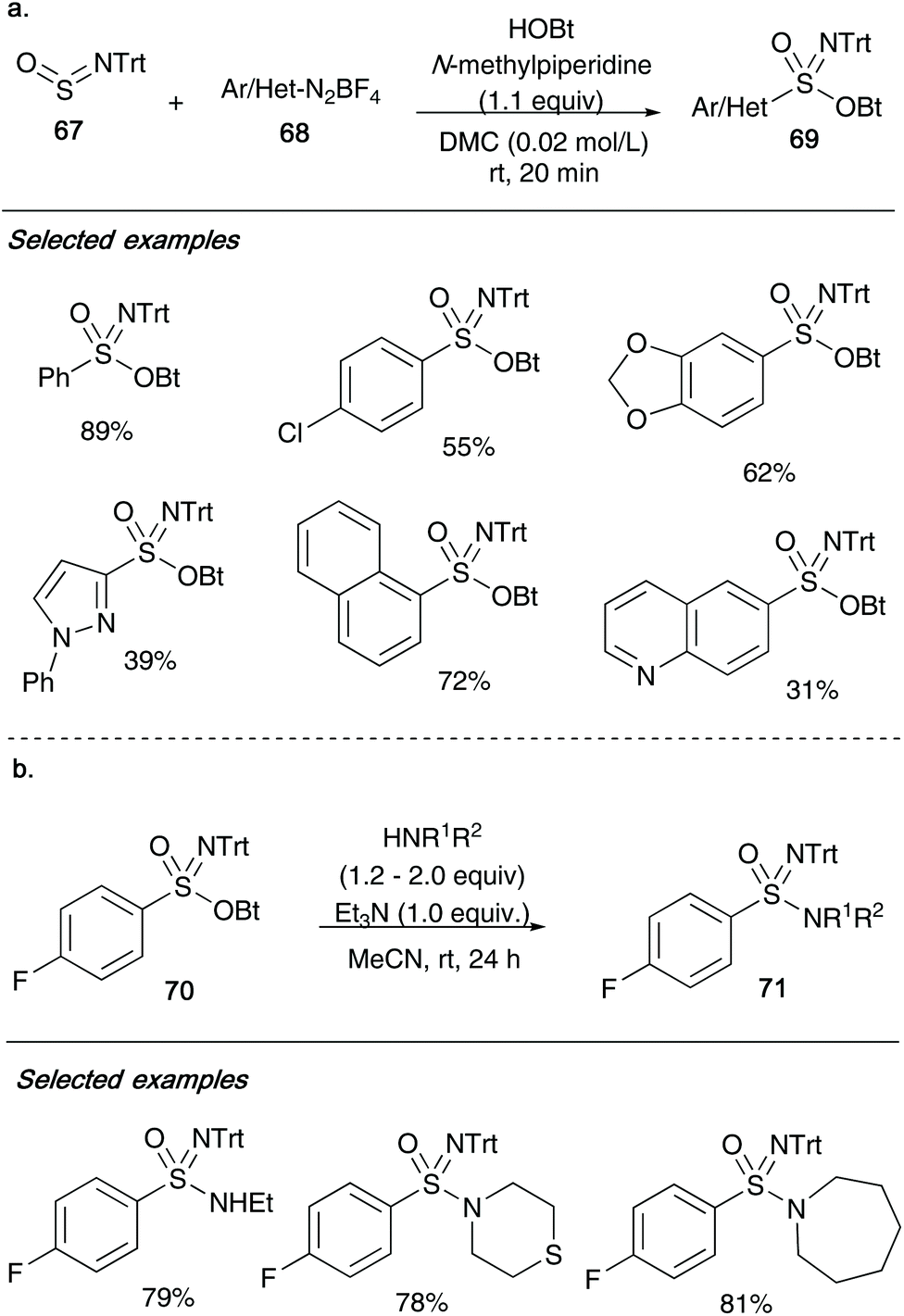 | ||
| Scheme 17 (a) Bolm's synthesis of sulfonimidates 69 from N-tritylsulfinylamine 67, heteroaromatic diazonium salts 68 and 1-hydroxybenzotriazole; (b) application to access sulfonimidamides 71 from sulfonimidates 70. | ||
3. Synthesis of cyclic sulfonimidates
3.1 Synthesis of cyclic sulfonimidates via sulfonimidoyl chlorides
The research reported by the group of Reggelin has dominated this particular field of sulfur(VI) chemistry over the last three decades. In Reggelin's seminal work on the synthesis of cyclic sulfonimidates in 1992, a one pot procedure in which (S)-O-trimethylsilyl valinol 72 and p-toluenesulfinyl chloride 73 were reacted accessing sulfinamide 74, followed by an oxidation affording the desired sulfonimidate products 75a and 75b without need of isolating reaction intermediates (Scheme 18).38 Reggelin utilised potassium fluoride to conduct the desilylation of the primary alcohol, thus allowing the cyclisation to occur. Furthermore, sulfonimidates 75a and 75b were reacted with various organometallic reagents affording sulfoximines in up to quantitative yields.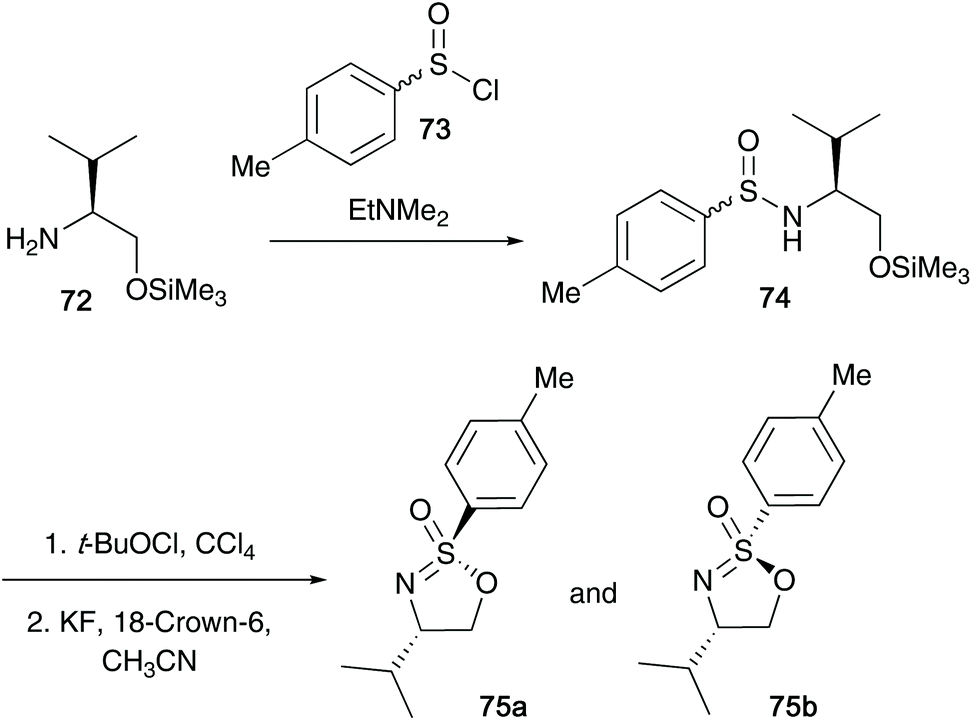 | ||
| Scheme 18 Reggelin's one-pot synthesis of cyclic sulfonimidates 75. | ||
In 1995, Reggelin followed up his seminal work by reporting an alternative route to access cyclic sulfonimidates 78 (Scheme 19).39 Sulfinamides 76 bearing chiral amino alcohols were identified as suitable starting materials that would be subjected to oxidative chlorination conditions to afford sulfonimidoyl chlorides 77. Previous reports by Johnson and Cram independently showed that sulfinamides are oxidised using tert-butyl hypochlorite at low temperatures to give sulfonimidoyl chlorides, proceeding with retention of configuration.25,40 However, sulfonimidoyl chlorides are not configurational stable at raised temperatures used in ensuing nucleophilic reactions.25 Reggelin showed that intramolecular attack of the internal alcohol at the sulfur centre, in the presence of DBU, was expeditious at −78 °C, and that the desired isomeric purity of the starting material is retained in the isolated product.
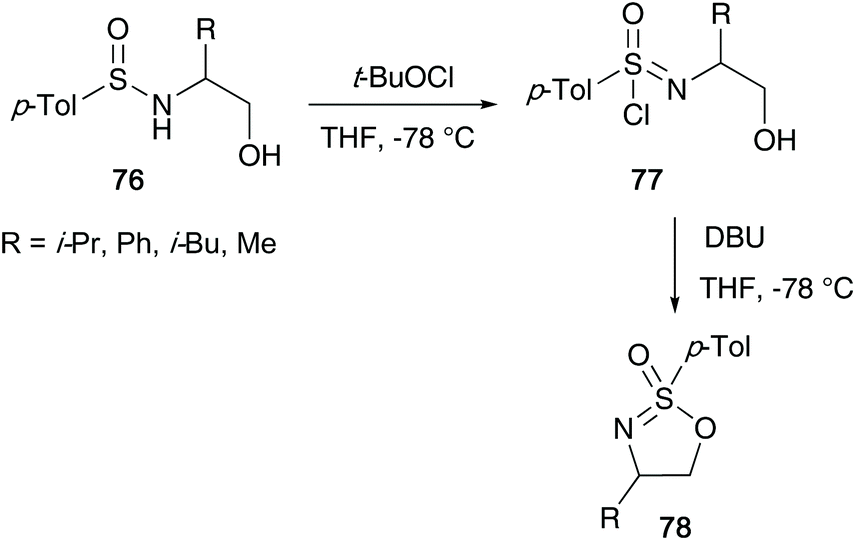 | ||
| Scheme 19 Reggelin's synthesis of cyclic sulfonimidates 78. | ||
Nearly two decades ago, Reggelin shed light on the mechanism believed to be in operation during the synthesis of cyclic sulfonimidates.34 He found through optimisation reactions that two particular pathways were in operation, depending on the base used in the reaction (Scheme 20). Oxidative cyclisation of an enantiomer of sulfinamide involved two key stereoselective steps; the first process is an oxidative chlorination reaction to afford sulfonimidoyl chloride intermediate 81, that proceeds with retention of configuration at sulfur. The mechanism of this chlorination was proposed to firstly involve chlorination of the nitrogen, followed by rearrangement to give the sulfonimidoyl chloride, which was supported through NMR control experiments (integration of meta-aromatic signals to NH signal changed from 2:1 to 3.58:1 during the course of the chlorination reaction). The second step is a base-induced cyclisation. If the base used is bulky, for example, DBU, and the reaction conducted at low temperatures, the chloride is displaced directly by the tethered alcohol in a SN2-like fashion, giving the product with inversion of configuration at sulfur (path a, Scheme 20). However, when dimethylethylamine was used as a smaller, more nucleophilic base (path b), sulfonimide ammonium salt 82 would be generated as an intermediate, which in turn could undergo base-catalysed cyclisation to form sulfonimidate 75b with retention of configuration due to a double displacement. A third pathway was identified (path c), which shows that if the base used is not strong enough, such as pyridine or 2,6-lutidine, then hydrolysis of the intermediate sulfonimidoyl chloride was found.
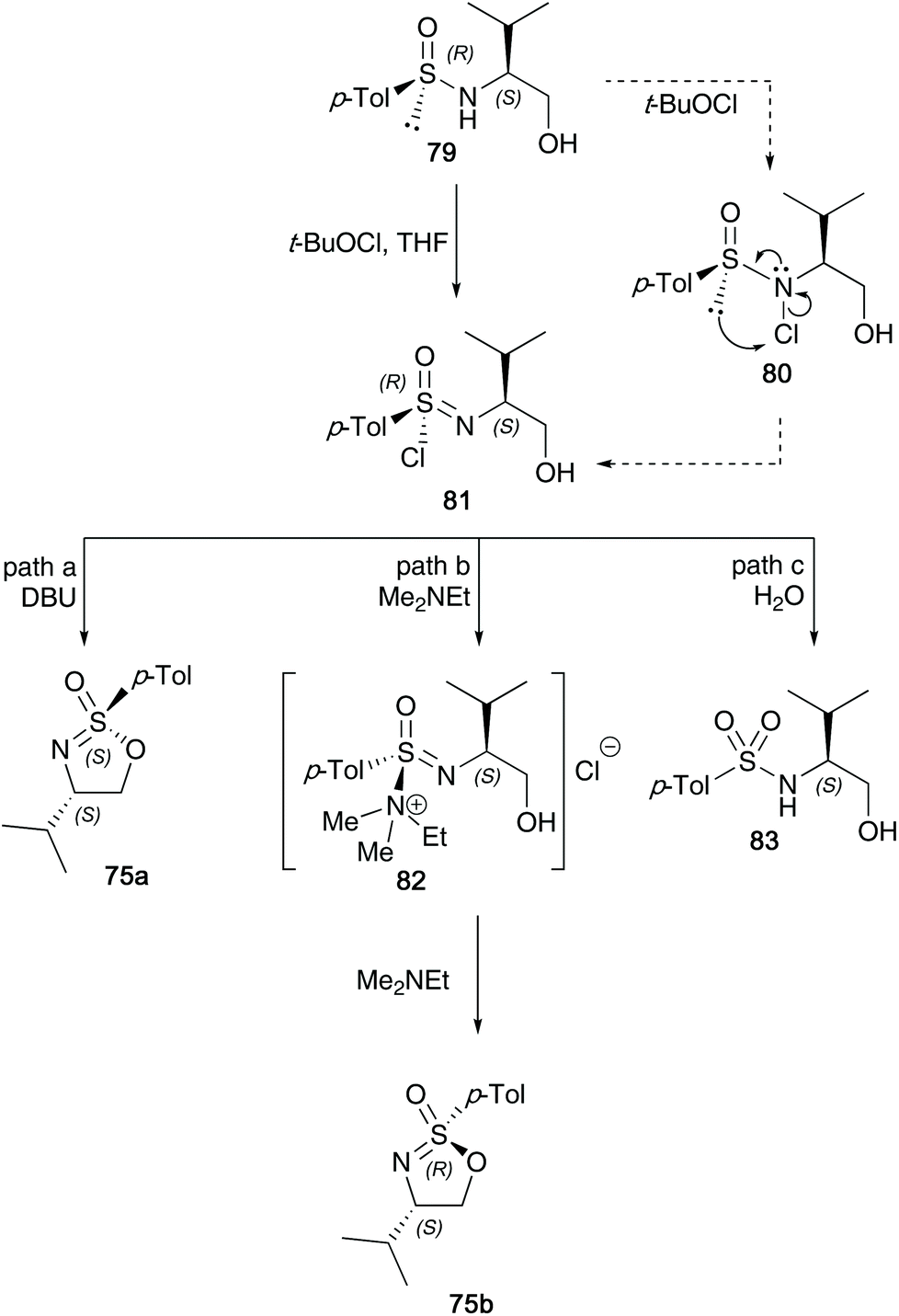 | ||
| Scheme 20 Oxidative chlorination of sulfinamides and cyclisation of the generated sulfonimidoyl chlorides to the target sulfonimidates 75a and 75b. | ||
This intricate procedure comes with a few drawbacks; firstly, the separation of diastereomeric sulfinamide starting materials is necessary (crystallisation and recrystallisation is required to purify the cyclic sulfonimidate products). During these recrystallizations, the authors disclose that for each sulfonimidate shown in Scheme 20, a maximum yield of around 35% was achieved for each epimer.
In the same article, Reggelin later described the conversion of the sulfonimidoyl chlorides 81 into sulfonimidoyl bromides 84 using 1.2 equivalents of KBr and 5 mol% 18-crown-6 (Scheme 21). These species were initially thought to be labile, albeit undetectable intermediates, yet NMR studies conducted at −20 °C showed <5% of a species that was not the chloride 81, but potentially the bromide 84. Through further NMR experiments at low temperatures, they observed that starting from either a 1:1, 18:1 or 1:5 diastereomeric mixture of sulfinamides 79/epi-79, an enriched ratio of 9:1 was observed in favour of the final sulfonimidate product 75b. The catalyst loading was found to be crucial in maintaining the stereoselectivity and found that at catalyst loadings of 0.5 mol%, the selectivity halved to 4.6:1 in favour of the target sulfonimidate 75b. The reaction was impressively shown to be reproducible on a large scale (checked up to a 300 g scale). The authors reported that the bromides react faster than chlorides. Furthermore, fast interconversion between the epimers is observed (dynamic epimer differentiation, in which epi-84 reacts 9–10 times faster than 84).
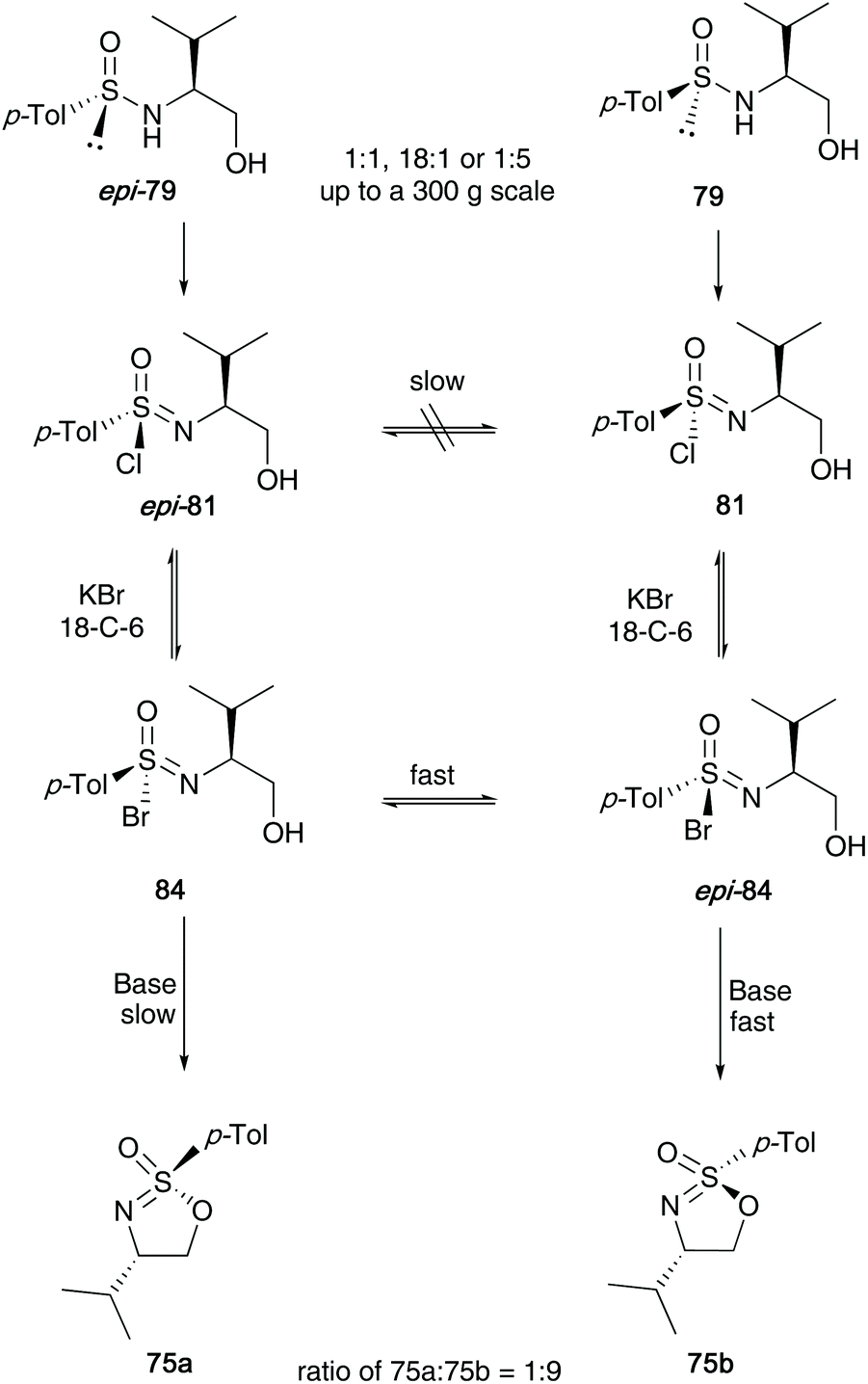 | ||
| Scheme 21 Formation of sulfonimidoyl bromides 84 and their epimerisation and interconversion. | ||
3.2 Cyclic sulfonimidates as chiral templates to access sulfoximines
In 1994, Reggelin and co-workers utilized their chiral sulfonimidate template 75a to access enantiomerically pure allyl-derived sulfoximines.41 After reaction of the sulfonimidate core with allyl lithium,38 the desired sulfoximines 85 were accessed (Scheme 22). Subsequently, the authors utilized a deprotonation and transmetallation step to obtain optically active titanated 2-alkenylsulfoximines 86 – such compounds could be reacted with aldehydes (2-methylpropanal shown below) under γ-hydroxyalkylation conditions to access chiral allyl alcohol substrates (product 87 was obtained in 86% yield starting from 85).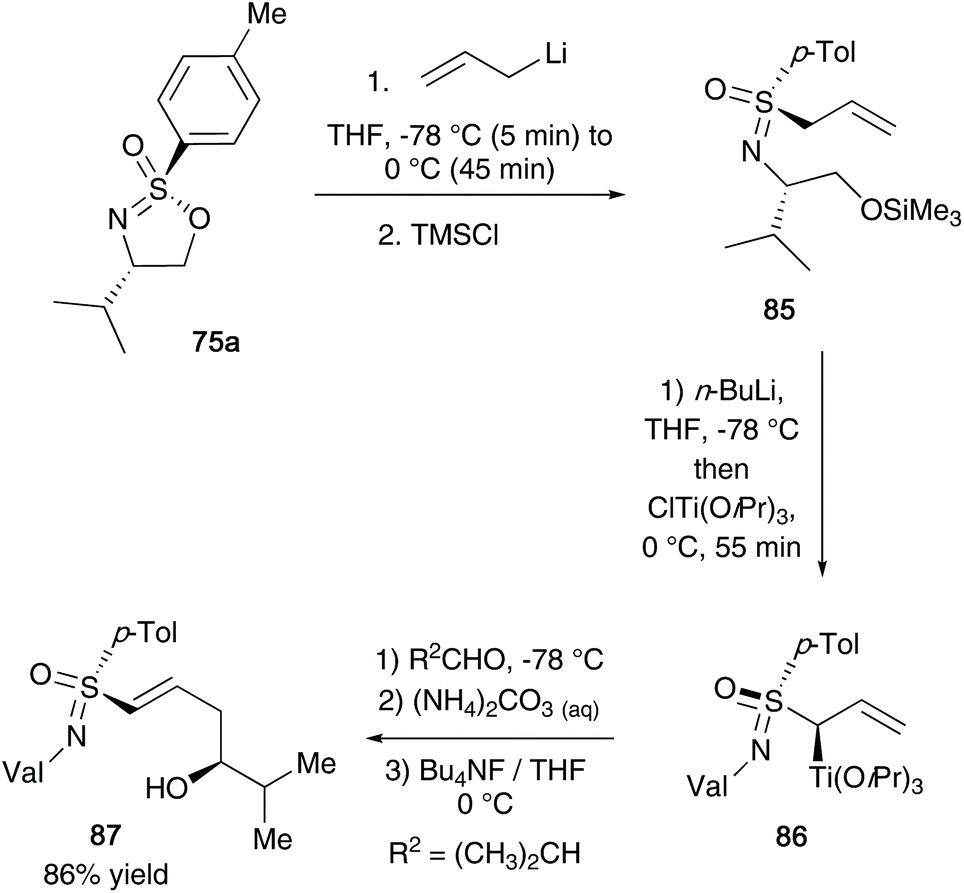 | ||
| Scheme 22 Reggelin's synthesis of alkenyl sulfoximines 85 and further modifications to 86 and 87. | ||
The synthesis of important oxygen and nitrogen containing heterocyclic compounds42 was realized using the common intermediate derived from alkenyl organometallic addition to optically active cyclic sulfonimidate 75. The diastereoselective γ-hydroxyalkylation of 2-alkenylsulfoximines 88 with enantiomeric lactaldehydes afforded tri- and tetrasubstituted tetrahydrofuran derivatives 89a/b (shown in Scheme 23).42a They were also able to access pyrrolidine derivatives 90 from the addition of α-aminoaldehydes to titanated 2-alkenylsulfoximines as the key synthetic step.42b Moreover, the oxabicyclic systems could be accessed from (2-cyclohexenylmethyl)sulfoximines.42c
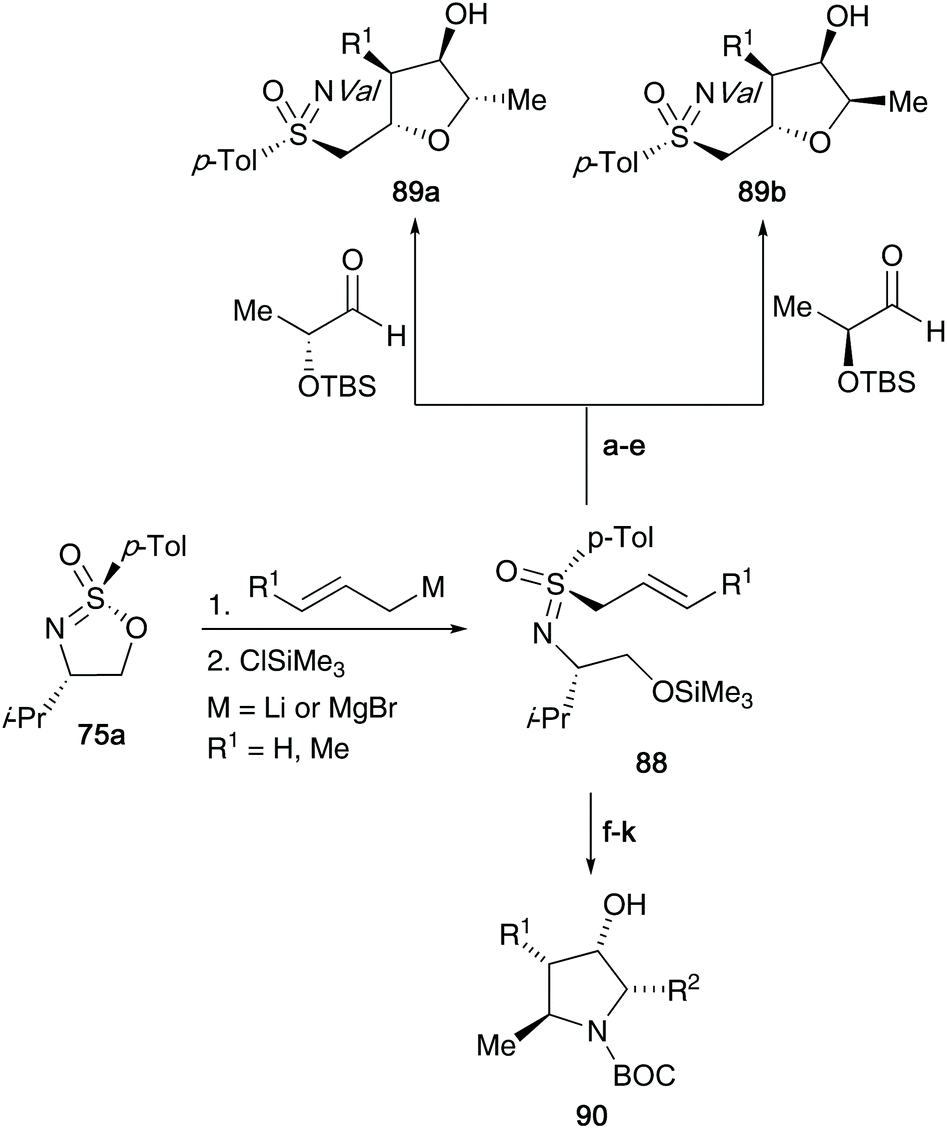 | ||
| Scheme 23 Application of 2-alkenylsulfoximines 88 towards the construction of important organic heterocycles 89 and 90. Reaction conditions: (a) nBuLi, THF, −78 °C; (b) ClTi(OiPr)3, 0 °C; (c) aldehyde, −78 °C; (d) (NH4)2CO3; (e) Bu4NF; (f) n-BuLi, toluene, −78 °C; (g) ClTi(OiPr)3, 0 °C; (h) FMOC-protected α-aminoaldehydes, −78 °C; (i) piperidine, 20 °C, (NH4)2CO3, NH4Cl; (j) K2CO3, MeOH; BOC2O, NaHCO3; (k) SmI2. | ||
Following on from their studies on using metalated 2-alkenylsulfoximines to access (poly)heterocyclic ring systems, Reggelin showed that reacting α- and β-aminoaldehydes with a range of titanated 2-alkenylsulfoximines allowed access to highly substituted aza(poly)cyclic compounds 94 (Scheme 24).42d Moreover, the authors employed 2-alkenylsulfoximines 91 bearing a range of chiral units (derived from cyclic sulfonimidates 75a/75b, allowing variation of the configuration at the sulfur atom and also at the amino alcohol carbon chiral centre) to control the stereochemical outcome in the final heterocycles obtained. Depending on the ring size of the heterocycle required, a range of preformed α- and β-aminoaldehydes could be reacted with titanated 2-alkenylsulfoximines, followed by nitrogen driven nucleophilic attack intramolecularly on the acceptor-substituted olefin bond in the vinyl sulfoximines to generate the desired heterocyclic core structures. Access to this key intramolecular nucleophilic attack is a result of piperidine driven cleavage of the nitrogen Fmoc group (the authors also use hydrazine when the nitrogen atom is protected with phthalic anhydride, a method that benefits from an easily separable phthalhydrazide byproduct). A final samarium iodide/lithium naphthalenide/RANEY®-nickel cleavage of the sulfonimidoyl moiety affords the nitrogen heterocycles.
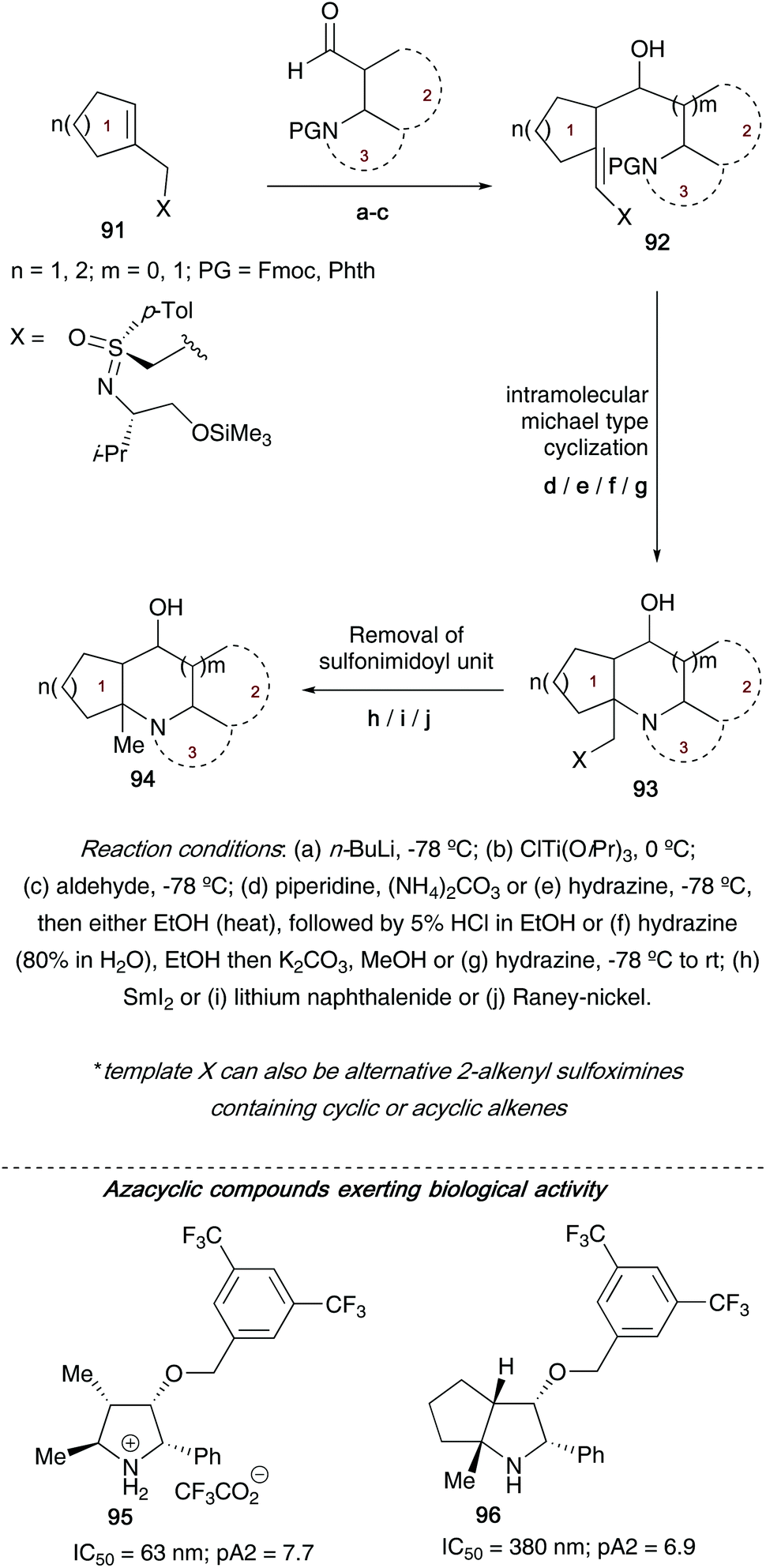 | ||
| Scheme 24 General overview of route to accessing (poly)heterocyclic ring systems 94, and some of the key compounds explored in biological assays. | ||
During the allyl transfer step, chirality at the carbon atoms in the newly formed bond was controlled by both the prochirality of the double bond, and sense of chirality in the sulfur auxiliary. Examples of nitrogen heterocycles accessed using this approach include pyrrolidines, piperidines, pyrrolizidine, benzopiperidines, 2-azabicycloalkanes and azapolycycles. A number of the molecules synthesized using this protocol were aiming to be type III peptidomimetics – an important group of pharmaceuticals targeting enzyme inhibition. Two heterocyclic ring systems were tested for bioactivity in NK1-functional assays; both pyrrolidine derivative 95 and azabicycle 96 showed high activities.
This work was further extended by exploring the synthesis of endocyclic 2-alkenyl sulfoximines (derived from sulfonimidate template 75b) bearing an additional heteroatom (nitrogen) or group (acetal, that could later be converted into the ketone), thereby installing an extra point of diversity into ring system 1 (see Scheme 24 for numbering of rings) on the bicycles generated. Some examples of the endocyclic 2-alkenyl sulfoximines 97–100 employed by Reggelin in this subsequent work are shown below in Fig. 3.42e
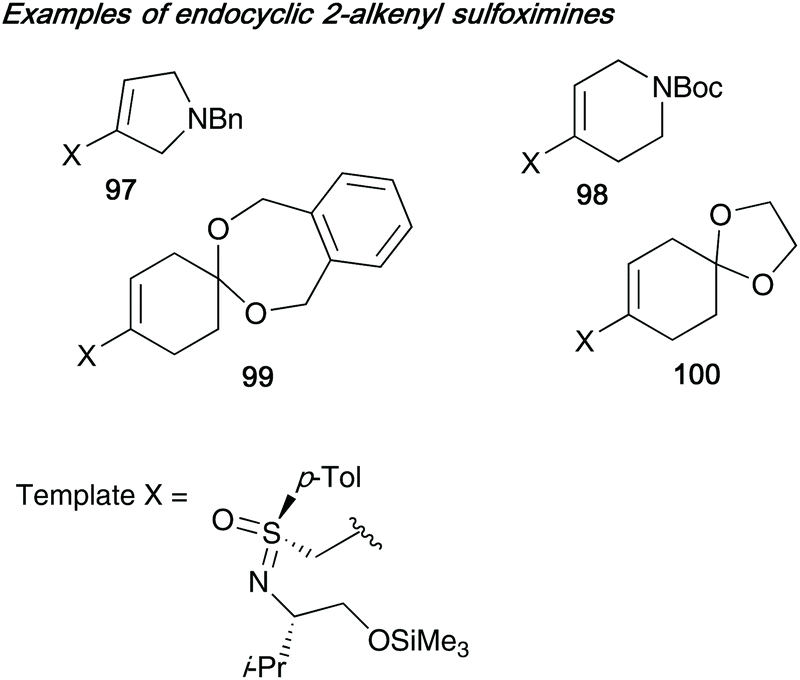 | ||
| Fig. 3 Endocyclic 2-alkenyl sulfoximines 97–100 employed to add a further point of diversity to bicyclic ring systems. | ||
One issue that was further addressed utilizing this type of methodology was the cleavage of the sulfonimidoyl auxiliary in tandem with C–C bond formation to give the desired azaheterocyclic systems.42f Whilst RANEY® Nickel, lithium naphthalenide and samarium iodide all facilitate the transformation, they result in a methyl group in the desired product which is unable to undergo further modifcations (see Scheme 24, 93 → 94). Utilizing a technique employed by Julia and co-workers for the olefinating desulfurization of sulfones using haloalkylmagnesium/lithium derived carbenoids,43 Reggelin was able to remove the sulfonimidoyl moiety in compound 101 employing the carbenoid iodomethylmagnesium iodide. The resulting olefin products 102 from β-elimination could be isolated in good to excellent yields (selected examples shown below, Scheme 25).
 | ||
| Scheme 25 Reggelin's C–C bond-forming desulfurizations of sulfoximines. | ||
These metalated sulfoximines proved integral towards the synthesis of important natural products, for example, euglobals G1 and G2 and arenaran A.42g 2-Oxabicyclo[n.3.0]alkane core structures were accessed using 2-cyclohexenylmethyl- and 2-cyclopentenylmethyl sulfoximines in an overall one-pot approach.
Cyclic sulfonimidates also proved to be important building blocks to access bis(sulfoximines).44 Subjecting one equivalent of the cyclic template 75b to MeLi gave the monosulfoximine product, but deprotonation with LHMDS and reaction with a further equivalent of template 75b gave the desired bis(sulfoximine) product 103 in 62% yield (one-pot) (Scheme 26). The designed bis(sulfoximines) were trialled as ligands in copper catalysed 1,4-additions to 2-cyclohexenone. Enantiomeric excesses for the isolated addition products reached up to 36% ee.
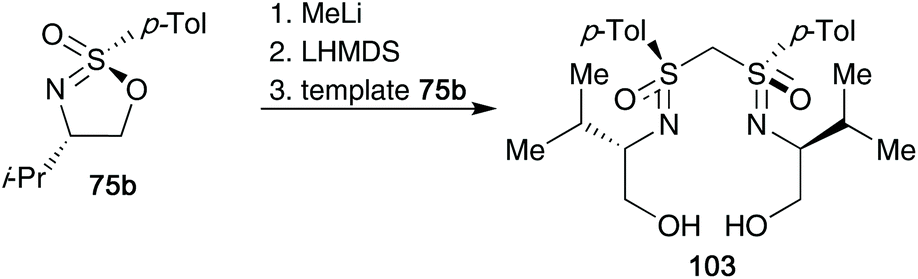 | ||
| Scheme 26 Utilization of template 75b to access bis(sulfoximines) 103. | ||
In 2012, Reggelin discussed methods to cleave the template from bis-sulfoximines (two main examples highlighted), affording free NH-bis sulfoximines. Firstly, a racemic para-methoxyphenyl derived template 104 was subjected to DDQ oxidation conditions – the desired racemic bis-NH sulfoximine 105 was obtained in up to 54% yield (Scheme 27a). For their aforementioned optically active valinol derived template 103, these conditions could not be employed. Instead a three-step protocol, including a mesylation, bromination and a final zinc induced β-elimination, afforded the enantiomerically pure NH-bis-sulfoximine 106 (see Scheme 27b).45
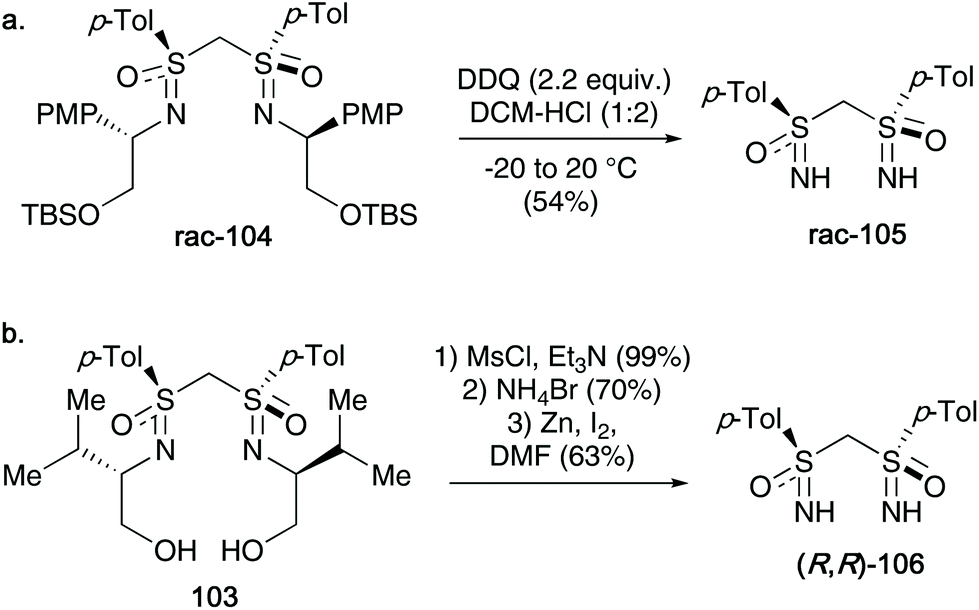 | ||
| Scheme 27 (a) DDQ promoted cleavage of the PMP group towards racemic bis-NH sulfoximines 105; (b) three-step procedure to access optically active bis-NH sulfoximines 106. | ||
The same group later applied these cyclic sulfonimidates to access optically active phosphanylated sulfoximines 107 – these compounds were subsequently employed as ligands in palladium-catalyzed allylic substitution reactions (Scheme 28).46 The ligands can be accessed through a short synthetic route over 4 steps, and ligand 107 was found to be highly enantioselective when applied in the palladium-catalyzed asymmetric allylic substitution of 1,3-diphenylallyl acetate and dimethylmalonate.
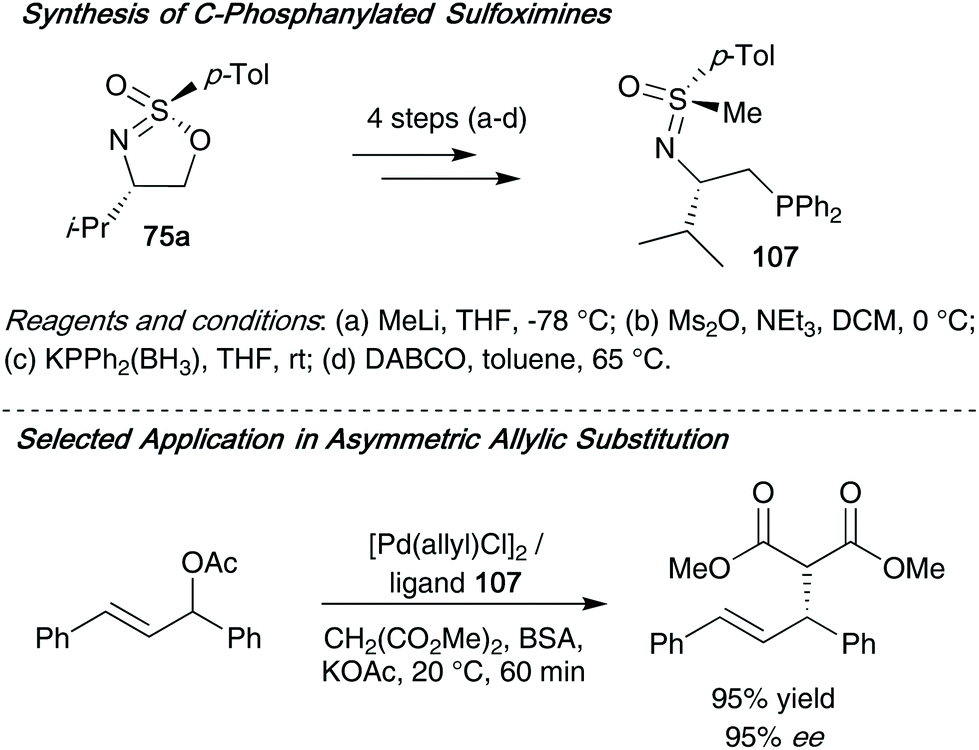 | ||
| Scheme 28 (a) Synthesis of ligand 107 from sulfonimidate 75a; (b) utilization of ligand 107 in allylic substitution reactions. | ||
Sulfonimidates such as 75b were also shown to participate as electrophiles in Barbier-type reactions with dihalomethanes and n-BuLi (Scheme 29). Reggelin showed that S-(chloromethyl)sulfoximines 108 from this reaction could then be reacted with potassium hydride in THF to access their desired oxathiazines 109.47 The protocol could also be conducted in a one-pot synthesis, providing a good yield of 66% for the desired oxathiazines 109.
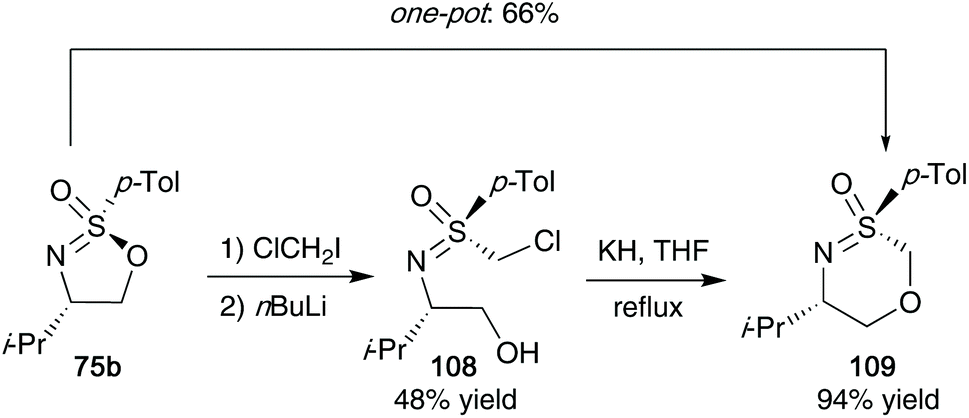 | ||
| Scheme 29 Reggelin's route to access oxathiazines 109 from cyclic sulfonimidates 75b. | ||
In 2020, the Stockman group reported that optically active cyclic sulfonimidate templates 111 could be employed to access enantioenriched NH-sulfoximines 113 (overall route shown in Scheme 30a).48 Building on the work reported by Reggelin and co-workers, amino acid derived phenyl glycinol provided a suitable auxiliary that could be carried through the oxidation of sulfinamides 110 using t-BuOCl/DBU to give cyclic sulfonimidates 111, followed by a Grignard addition step to yield sulfoximine products 112. Importantly, the auxiliary could be easily removed using molecular oxygen under basic conditions in MTBE to obtain the final NH-sulfoximines 113 whilst maintaining optical purity. For clarity, one epimer is shown in Scheme 30, however, the reaction proceeds smoothly for the other epimer as well under the reaction sequence, resulting in isolation of the opposite enantiomers of 113 shown (in similarly high enantiomeric excesses). Such optically active compounds synthesized mimic structures of chiral sulfoximines currently involved in drug discovery programmes, and as such may provide a useful tool to accessing necessary enantiomers for future drug molecule syntheses.
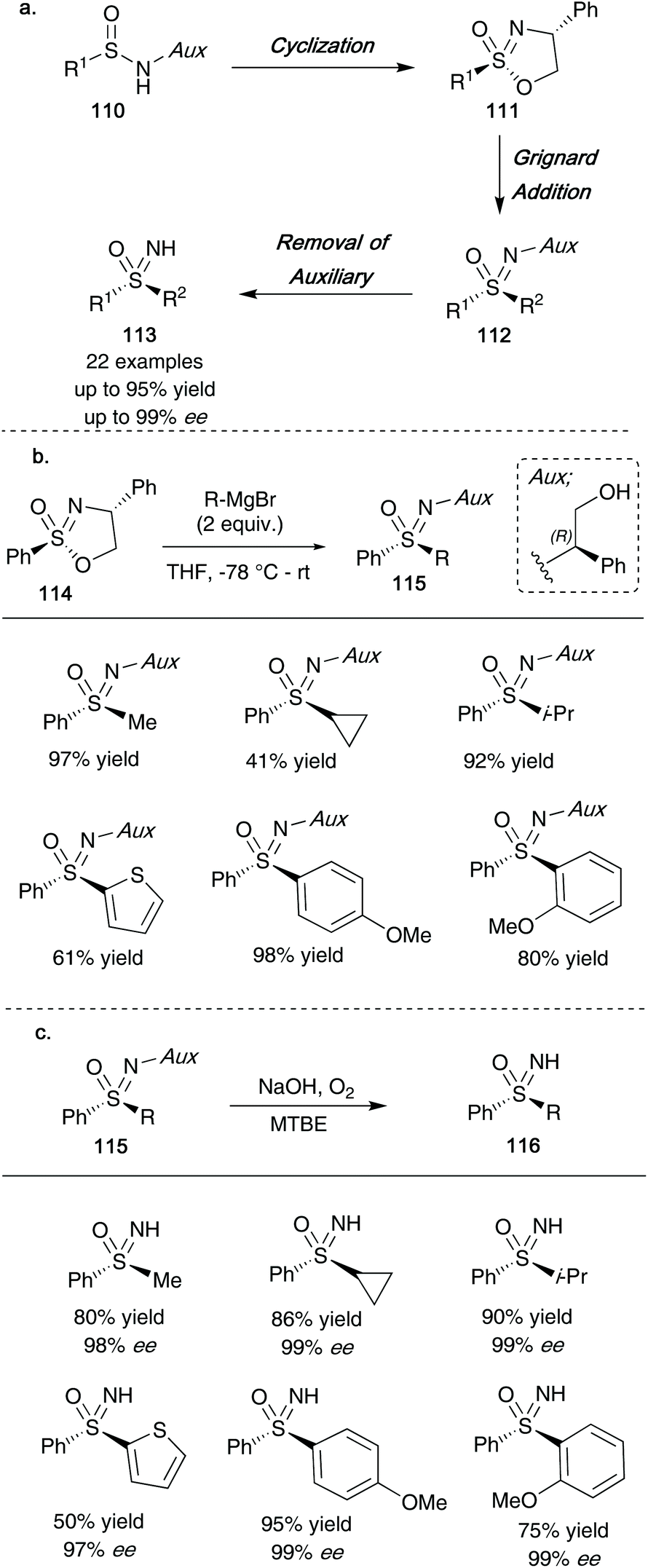 | ||
| Scheme 30 (a) Stockman's general route to enantioenriched NH-sulfoximines 113; (b) Grignard additions to sulfonimidate derived template 114; (c) oxidative debenzylation of sulfoximines 115 to give NH-sulfoximines 116. | ||
4. Conclusions
This review article has summarised the synthetic approaches to access sulfonimidates, and their various utilities and applications towards accessing high value compounds over the last half century. Since their discovery, a plethora of methods have been discovered to access these molecules. With the presence of a stereogenic tetrahedral sulfur centre, sulfonimidates have found applications as chiral templates to access enantioenriched sulfoximines – these in turn can subsequently be converted into other chiral molecules including highly substituted saturated heterocycles. In other cases, exploiting weaknesses of sulfonimidates, including acid sensitivity and heat lability, have led to important discoveries, such as acting as alkyl transfer agents or the synthesis of sulfur containing polymers. Due to the rising interest in other sulfur(VI) compounds including sulfonimidamides and sulfoximines as pharmaceutical leads in the medicinal chemistry sector, the need to develop fast, efficient routes using sulfonimidates as reactive (and potential optically active) intermediates is ever present. To address the growing need for routes to access these sought-after molecules, we envisage this field to continue to expand over the coming years. Sulfonimidate chemistry still offers many undiscovered leads and applications, which we anticipate to see in the near future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the CAPES foundation (P. M. M.) and the University of Nottingham for funding.References
- E. S. Levchenko, L. N. Markovskii and A. V. Kirsanov, Zh. Org. Khim., 1967, 3, 1273–1282 CAS.
- For reviews highlighting seminal works in sulfonimidate chemistry, please see: (a) S. G. Pyne, Sulfur Rep., 1999, 21, 281–334 CrossRef CAS; (b) E. Wojaczyńska and J. Wojaczyński, Chem. Rev., 2020, 120, 4578–4611 CrossRef PubMed.
- For selected reviews on sulfonimidamide chemistry, please see: (a) G. C. Nandi and P. I. Arvidsson, Adv. Synth. Catal., 2018, 360, 2976–3001 CrossRef CAS; (b) P. K. Chinthakindi, T. Naicker, N. Thota, T. Govender, H. G. Kruger and P. I. Arvidsson, Angew. Chem., Int. Ed., 2017, 56, 4100–4109 CrossRef CAS PubMed.
- For selected reviews on sulfoximine chemistry, please see: (a) H. Zhou and Z. Chen, Chin. J. Org. Chem., 2018, 38, 719–737 CrossRef CAS; (b) A. Hosseinian, L. Z. Fekri, A. Monfared, E. Vessally and M. Nikpassand, J. Sulfur. Chem., 2018, 39, 674–698 CrossRef CAS; (c) A.-L. Barthelemy and E. Magnier, C. R. Chim., 2018, 21, 711–722 CrossRef CAS; (d) M. Reggelin and C. Zur, Synthesis, 2000, 1–64 CrossRef CAS; (e) S. Wiezorek, P. Lamers and C. Bolm, Chem. Soc. Rev., 2019, 48, 5408–5423 RSC; (f) V. Bizet, R. Kowalczyk and C. Bolm, Chem. Soc. Rev., 2014, 43, 2426–2438 RSC; (g) U. Lücking, Angew. Chem., Int. Ed., 2013, 52, 9399–9408 CrossRef; (h) J. P. Colomer, M. Traverssi and G. Oksdath-Mansilla, J. Flow Chem., 2020, 10, 123–138 CAS; (i) J. A. Bull, L. Degennaro and R. Luisi, Synlett, 2017, 28, 2525–2538 CrossRef CAS; (j) V. Bizet, C. M. M. Hendriks and C. Bolm, Chem. Soc. Rev., 2015, 44, 3378–3390 RSC; (k) U. Lücking, Org. Chem. Front., 2019, 6, 1319–1324 RSC.
- For select references highlighting the use of sulfur(VI) as potential drug candidates, please see: (a) U. Lücking, M. Krüger, R. Jautelat and G. Siemeister, Sulfoximine-substituted pyrimidines for use as CDK and/or VEGF inhibitors, the production thereof and their use as drugs, WO2005/037800A1, 2004; (b) U. Lücking, R. Jautelat, M. Krüger, T. Brumby, P. Lienau, M. Schäfer, H. Briem, J. Schulze, A. Hillisch, A. Reichel, A. M. Wengner and G. Siemeister, ChemMedChem, 2013, 8, 1067–1085 CrossRef PubMed; (c) K. M. Foote, J. W. M. Nissink and P. Turner, Morpholino pyrimidines and their use in therapy, WO2011/154737A1, 2011.
- D. Leca, L. Fensterbank, E. Lacôte and M. Malacria, Org. Lett., 2002, 4, 4093–4095 CrossRef CAS.
- A. Felim, A. Toussaint, C. R. Phillips, D. Leca, A. Vagstad, L. Fensterbank, E. Lacôte and M. Malacria, Org. Lett., 2006, 8, 337–339 CrossRef CAS PubMed.
- (a) A. K. Roy, J. Am. Chem. Soc., 1993, 115, 2598–2603 CrossRef CAS; (b) A. K. Roy, G. T. Burns, G. C. Lie and S. Grigoras, J. Am. Chem. Soc., 1993, 115, 2604–2612 CrossRef CAS; (c) A. K. Roy, J. Am. Chem. Soc., 1992, 114, 1530–1531 CrossRef CAS; (d) V. Chunechom, T. E. Vidal, H. Adams and M. L. Turner, Angew. Chem., Int. Ed., 1998, 37, 1928–1930 CrossRef CAS.
- (a) J. Kümmerlen and A. Sebald, J. Am. Chem. Soc., 1993, 115, 1134–1142 CrossRef; (b) Y. Endo, K. Shudo and T. Okamoto, J. Am. Chem. Soc., 1982, 104, 6393–6397 CrossRef CAS.
- A. M. Pinchuk, L. N. Markovskii, E. S. Levchenko and V. I. Shevchenko, Zh. Obshch. Khim., 1967, 37, 852–855 CAS.
- E. S. Levchenko, I. É. Sheinkman and A. V. Kirsanov, Zh. Obshch. Khim., 1963, 33, 3315–3323 CAS.
- E. S. Levchenko, E. S. Kozlov and A. V. Kirsanov, Zh. Obshch. Khim., 1962, 32, 2585–2592 CAS.
- P. P. Kornuta and V. I. Shevchenko, Zh. Obshch. Khim., 1970, 40, 551–553 CAS.
- R. Koller, K. Stanek, D. Stolz, R. Aardoom, K. Niedermann and A. Togni, Angew. Chem., Int. Ed., 2009, 48, 4332–4336 CrossRef CAS.
- J. Iley, A. R. Bassindale and P. Patel, J. Chem. Soc., Perkin Trans. 2, 1984, 77–80 RSC.
- A. Tota, S. St John-Campbell, E. L. Briggs, G. O. Estévez, M. Afonso, L. Degennaro, R. Luisi and J. A. Bull, Org. Lett., 2018, 20, 2599–2602 CrossRef CAS PubMed.
- T. J. Maricich, M. J. Allan, B. S. Kislin, A. I.-T. Chen, F.-C. Meng, C. Bradford, N.-C. Kuan, J. Wood, O. Aisagbonhi, A. Poste, D. Wride, S. Kim, T. Santos, M. Fimbres, D. Choi, H. Elia, J. Kaladjian, A. Abou-Zahr and A. Mejia, Synthesis, 2013, 3361–3368 CrossRef CAS.
- B. C. Challis and J. N. Iley, J. Chem. Soc., Perkin Trans. 2, 1985, 699–703 RSC.
- E. S. Levchenko, N. Y. Derkach and A. V. Kirsanov, Zh. Obshch. Khim., 1960, 30, 1971–1975 CAS.
- (a) E. U. Jonsson, C. C. Bacon and C. R. Johnson, J. Am. Chem. Soc., 1971, 93, 5306–5308 CrossRef CAS; (b) C. R. Johnson, E. U. Jonsson and C. C. Bacon, J. Org. Chem., 1979, 44, 2055–2061 CrossRef CAS; (c) C. R. Johnson and A. Wambsgans, J. Org. Chem., 1979, 44, 2278–2280 CrossRef CAS.
- K. Okuma, K. Nakanishi and H. Ohta, J. Org. Chem., 1984, 49, 1402–1407 CrossRef CAS.
- E. U. Jonsson and C. R. Johnson, J. Am. Chem. Soc., 1971, 93, 5308–5309 CrossRef CAS.
- S. Colonna, R. Giovini and F. Montanari, Chem. Commun., 1968, 865–866 RSC.
- A. Nudelman and D. J. Cram, J. Am. Chem. Soc., 1968, 90, 3869–3870 CrossRef CAS.
- C. R. Johnson, E. U. Jonsson and A. Wambsgans, J. Org. Chem., 1979, 44, 2061–2065 CrossRef CAS.
- B. Gao, S. Li, P. Wu, J. E. Moses and K. B. Sharpless, Angew. Chem., Int. Ed., 2018, 57, 1939–1943 CrossRef CAS.
- (a) M. Wright, C. Martínez-Lamenca, J. E. Leenaerts, P. E. Brennan, A. A. Trabanco and D. Oehlrich, J. Org. Chem., 2018, 83, 9510–9516 CrossRef CAS; (b) C. S. Richards-Taylor, C. Martínez-Lamenca, J. E. Leenaerts, A. A. Trabanco and D. Oehlrich, J. Org. Chem., 2017, 82, 9898–9904 CrossRef CAS PubMed.
- D.-D. Liang, D. E. Streefkerk, D. Jordaan, J. Wagemakers, J. Baggerman and H. Zuilhof, Angew. Chem., 2020, 59, 7494–7500 CrossRef CAS PubMed.
- J. Guo, C. Kuang, J. Rong, L. Li, C. Ni and J. Hu, Chem. – Eur. J., 2019, 25, 7259–7264 CrossRef CAS PubMed.
- T. J. Maricich, R. A. Jourdenais and T. A. Albright, J. Am. Chem. Soc., 1973, 95, 5831–5832 CrossRef CAS.
- F. A. Davis, P. Zhou and G. V. Reddy, J. Org. Chem., 1994, 59, 3243–3245 CrossRef CAS.
- P. J. Stang and V. V. Zhdankin, Chem. Rev., 1996, 96, 1123–1178 CrossRef CAS PubMed.
- D. Leca, K. Song, M. Amatore, L. Fensterbank, E. Lacôte and M. Malacria, Chem. – Eur. J., 2004, 10, 906–916 CrossRef CAS PubMed.
- M. Reggelin and B. Junker, Chem. – Eur. J., 2001, 7, 1232–1239 CrossRef CAS.
- P. M. Matos, W. Lewis, J. C. Moore and R. A. Stockman, Org. Lett., 2018, 20, 3674–3677 CrossRef CAS PubMed.
- M. Bremerich, C. M. Conrads, T. Langletz and C. Bolm, Angew. Chem., Int. Ed., 2019, 58, 19014–19020 CrossRef CAS PubMed.
- For seminal work regarding the use of N-tritylsulfinylamine (TrNSO) to access sulfonimidamides, please see: T. Q. Davies, A. Hall and M. C. Willis, Angew. Chem., Int. Ed., 2017, 56, 14937–14941 CrossRef CAS PubMed.
- M. Reggelin and H. Weinberger, Tetrahedron Lett., 1992, 33, 6959–6962 CrossRef CAS.
- M. Reggelin and R. Welcker, Tetrahedron Lett., 1995, 36, 5885–5886 CrossRef CAS.
- M. R. Jones and D. J. Cram, J. Am. Chem. Soc., 1974, 96, 2183–2190 CrossRef CAS.
- M. Reggelin and H. Weinberger, Angew. Chem., Int. Ed. Engl., 1994, 33, 444–446 CrossRef.
- (a) M. Reggelin, H. Weinberger and T. Heinrich, Liebigs. Ann., 1997, 1881–1886 CrossRef CAS; (b) M. Reggelin and T. Heinrich, Angew. Chem., Int. Ed., 1998, 37, 2883–2886 CrossRef CAS; (c) M. Reggelin, H. Weinberger, M. Gerlach and R. Welcker, J. Am. Chem. Soc., 1996, 118, 4765–4777 CrossRef CAS; (d) M. Reggelin, B. Junker, T. Heinrich, S. Slavik and P. Bühle, J. Am. Chem. Soc., 2006, 128, 4023–4034 CrossRef CAS; (e) M. Reggelin, J. Kühl, J. P. Kaiser and P. Bühle, Synthesis, 2006, 2224–2232 CrossRef CAS; (f) M. Reggelin, S. Slavik and P. Bühle, Org. Lett., 2008, 10, 4081–4084 CrossRef CAS PubMed; (g) M. Reggelin, M. Gerlach and M. Vogt, Eur. J. Org. Chem., 1999, 1011–1031 CrossRef CAS.
- (a) C. De Lima, M. Julia and J.-N. Verpeaux, Synlett, 1992, 133–134 CrossRef CAS; (b) P. Charreau, M. Julia and J.-N. Verpeaux, J. Organomet. Chem., 1989, 379, 201–210 CrossRef CAS.
- M. Reggelin, H. Weinberger and V. Spohr, Adv. Synth. Catal., 2004, 346, 1295–1306 CrossRef CAS.
- M. Reggelin, C. Mehler and J. P. Kaiser, Synlett, 2012, 1095–1098 CrossRef CAS.
- V. Spohr, J. P. Kaiser and M. Reggelin, Tetrahedron: Asymmetry, 2006, 17, 500–503 CrossRef CAS.
- J. Kühl and M. Reggelin, Synthesis, 2017, 49, 403–408 Search PubMed.
- P. M. Matos, W. Lewis, S. P. Argent, J. C. Moore and R. A. Stockman, Org. Lett., 2020, 22, 2776–2780 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2020 |