
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
E,Z-Selectivity in the reductive cross-coupling of two benzaldehydes to stilbenes under substrate control†
Nicolas
D'Imperio
,
Anna I.
Arkhypchuk
 and
Sascha
Ott
*
and
Sascha
Ott
*
Department of Chemistry, Ångström Laboratory, Uppsala University, Box 523, 75120 Uppsala, Sweden. E-mail: sascha.ott@kemi.uu.se
First published on 15th July 2020
Abstract
Unsymmetrical E- and Z-stilbenes can be synthesized from two differently substituted benzaldehydes in a MesP(TMS)Li-promoted reductive coupling sequence. Depending on the order of addition of the two coupling partners, the same olefin can be produced in either E- or Z-enriched form under identical reaction conditions. A systematic study of the correlation between the stereochemical outcome of the reaction and the substitution pattern at the two aldehydes is presented. The results can be used as guidelines to predict the product stereochemistry.
Introduction
The stereochemistry of carbon–carbon double bonds is of crucial importance for the chemical properties of alkenes1–4 and their function in nature5 and commodity chemicals.6,7 Thus, developing methods to selectively access either the E- or Z-isomer has been, and still is, at the heart of organic chemistry. A plethora of protocols for synthesizing olefins is available,8,9 among which the most widely used are the Wittig,10,11 Horner–Wadsworth–Emmons (HWE),12 Peterson,13 olefin metathesis14,15 and cross-coupling reactions.16,17 In classical carbonyl olefinations mediated by phosphorus compounds,9 namely the Wittig,18 Horner–Wittig19 and HWE20 reactions, the factors that influence the E- and Z-stereoselectivity are well understood and can be controlled, for example, by the nature of the phosphorus reagent, the base or the solvent.21–26 It should be pointed out that in almost all cases the stereochemical outcome of the reaction is determined by the reaction conditions or the type of olefinating reagent, and is usually not influenced by the nature of the aldehyde substrates. To the best of our knowledge, the only example in which a substituent at the aldehyde has been reported to influence the E–Z ratio of a Wittig olefination is in the case of benzaldehydes with heteroatoms such as halides or ethers in the ortho-position. This so-called “ortho-effect”27,28 can be exploited to synthesize enriched Z-stilbenes, as shown by Gilheany and co-workers.29–31 As shown in Scheme 1a, the reaction gives rise to higher proportions of Z-alkene when the aldehyde has an ortho-substituent. In contrast, an ortho-substituent on the phosphonium salt has no such effect, and the thermodynamically more stable E-stilbene is formed (Scheme 1b). From a mechanistic viewpoint, the Z-directing effect of the ortho-substituent arises from a secondary bonding interaction between the phosphorus and the ortho-heteroatom during the transition state.29,31 | ||
| Scheme 1 E- and Z-Stereoselective Wittig reactions of (a) ortho-substituted benzaldehydes, and (b) ortho-substituted phosphonium ylides. X = OMe, Br. | ||
In recent years, we have been interested in developing new phosphorus mediated cross-coupling reactions of carbonyl compounds to olefins.32–35 In one of our latest work, we have been able to couple two different benzaldehydes selectively to unsymmetrical 1,2-disubstituted stilbenes via phosphaalkene (2) and phosphinate (3) intermediates (Scheme 2).34 In this one-pot reaction, a first benzaldehyde A is converted to a phosphaalkene 2 which proceeds under Umpolung of the carbonyl-carbon. Subsequent activation of the phosphaalkene provides phosphinates 3 which react with the second aldehyde B to form an unsymmetrical stilbene. In contrast to the McMurry chemistry that is traditionally used for the reductive coupling of carbonyl compounds to alkenes, the reaction proceeds by an ionic mechanism, and allows the controlled preparation of unsymmetrical alkenes owing to the successive addition of the two aldehydes to the reaction. While developing this method, an interesting trend in the E–Z ratio of the formed products attracted our attention. Herein, we report on a systematic study of how the stereochemical outcome of the cross-coupling reaction can be altered simply by choosing the order of addition of the two benzaldehyde substrates. As will be shown by various examples, the reactions proceed to a large extent under substrate control, with electronic effects as well as the presence of ortho-substituents determining the product stereochemistry.
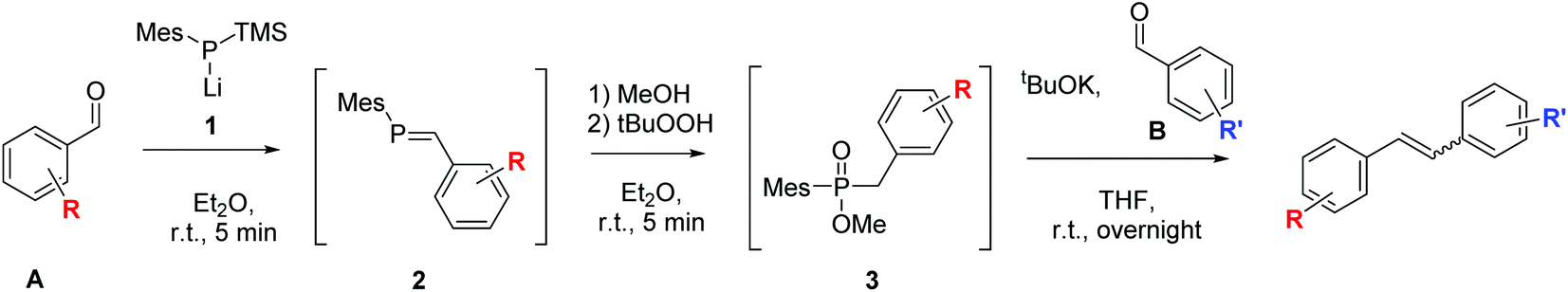 | ||
| Scheme 2 One-pot phosphorus mediated cross-coupling of two different benzaldehydes to E- and Z-olefins.34 | ||
Results and discussion
The influence of the aldehydes’ substituents on the stereochemical outcome of the reaction was tested and the existence of two separate effects in the coupling procedure was noticed. A first effect is of electronic nature, and best studied when the aldehydes are substituted in the para-position; a second one can be observed in the reaction of ortho-substituted benzaldehydes. Both effects will be discussed separately first, and then in combination. To understand the role that the electronic nature of the two benzaldehyde substrates has on the E–Z ratio of the newly formed double bond, a series of reactions between para-substituted benzaldehydes was investigated. The results of the study are summarized in Table 1.|
|
||||
|---|---|---|---|---|
| Entry | Aldehyde A | Aldehyde B | Isolated yield, % | E–Z ratio |
| 1 | H | Br | 46 | 70/30 |
| 2 | H | Me | 52 | 75/25 |
| 3 | H | OMe | 75 | 87/13 |
| 4 | Br | H | 45 | 100/0 |
| 5 | Me | H | 39 | 52/48 |
| 6 | OMe | H | 57 | 38/62 |
| 7 | Br | Br | 58 | 100/0 |
| 8 | H | H | 52 | 75/25 |
| 9 | Me | Me | 44 | 69/31 |
| 10 | OMe | OMe | 40 | 50/50 |
| 11 | Br | Me | 32 | 100/0 |
| 12 | Me | Br | 74 | 49/51 |
| 13 | Br | OMe | 35 | 100/0 |
| 14 | OMe | Br | 48 | 30/70 |
As shown in Table 1, entries 1–3, the electronic nature of the para-substituent on aldehyde B does not influence the outcome of the reaction to a great extent, as all three reactions predominantly form E-enriched products. A different picture emerges from a comparison between entries 4–6. With an electron deficient aldehyde A (entry 4, X = Br), the reaction forms exclusively the E-isomer. This selectivity is however compromised by moving to more and more electron-rich aldehydes A. Changing the X group from the electron-withdrawing (EWG) bromide to the electron-donating (EDG) methyl moiety (entry 5) leads to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of E- and Z-stilbenes, and in the case of the most electron-rich para-methoxy-substituted aldehyde A (entry 6), the Z-isomer is observed in a roughly 2:1 ratio. Entries 7–10 describe the results of homo-coupling experiments which in essence further corroborate the trends observed in entries 1–6. When X = Y = Br (entry 7), the reaction is, as expected, E-selective as a result of the EWG in aldehyde A. Alternative EWGs on aldehyde A such as a nitro-group have previously been shown to also give exclusively the E-isomer.34 However, with increasing electron-donating character of the para-substituent in aldehyde A, higher percentages of Z-isomers are formed.
:1 mixture of E- and Z-stilbenes, and in the case of the most electron-rich para-methoxy-substituted aldehyde A (entry 6), the Z-isomer is observed in a roughly 2:1 ratio. Entries 7–10 describe the results of homo-coupling experiments which in essence further corroborate the trends observed in entries 1–6. When X = Y = Br (entry 7), the reaction is, as expected, E-selective as a result of the EWG in aldehyde A. Alternative EWGs on aldehyde A such as a nitro-group have previously been shown to also give exclusively the E-isomer.34 However, with increasing electron-donating character of the para-substituent in aldehyde A, higher percentages of Z-isomers are formed.
Entries 11–14 illustrate the significance of the effects that are described herein, and ways to exploit them for the preferential preparation of E- or Z-isomers. Reactions 11, 12, 13, 14 employ the same starting materials under identical conditions, but exhibit a dramatic difference in E–Z selectivity. When aldehyde A carries an EWG (X = Br, entries 11 and 13), only the E-olefins are formed. Changing the order of addition turns the substrates with an EDG (X = Me (entry 12) and X = OMe (entry 14)) into aldehyde A, leading to the opposite preferential stereoselectivity with the Z-isomers becoming the predominant forms.
Summarizing the results from Table 1, it is clear that the electronic nature of aldehyde A has a great influence on the alkene stereochemistry, while that of aldehyde B is negligible. E-Alkenes are exclusively formed when aldehyde A is electron-deficient, while Z-alkenes become the major product for electron-rich aldehydes A. This trend can be observed in a study where aldehyde B is kept constant (for this study a para-Br) and aldehyde A varied. In entry 7, only the E-isomer is formed due to the use of an EWG substituent on aldehyde A. With electron-neutral benzaldehyde as the first coupling partner (entry 1), 30% of Z-isomer is generated. When a fairly good EDG like p-CH3 (entry 12) is used, the reaction forms E- and Z-isomers in an almost 1:1 ratio. Ultimately, with the stronger EDG p-OMe (entry 14), the E/Z ratio is 30/70. Plotting the proportion of Z-selectivity versus the Hammett parameter of the substituents on aldehyde A yields a straight line (see ESI, Fig. S33†), suggesting that it may be possible to predict the product stereochemistry for new combinations of reactants in the future.
The results presented in Table 1 nicely illustrate the strength of this procedure: the same olefin can be synthesized in higher E- or Z-form only depending on the order of addition of the two aldehydes during the sequence. The opportunities that the method offers are greater the higher the electronic difference between the two benzaldehydes.
Following the study on electronic effects, and inspired by the Z-directing ortho-effect described by Gilheany and co-workers,29,31 a series of experiments with different ortho-substituted benzaldehydes were performed (Table 2).
|
|
||||
|---|---|---|---|---|
| Entry | Aaldehyde A | Aldehyde B | Isolated yield, % | E–Z ratio |
| 1 | H | Br | 51 | 33/67 |
| 2 | H | Me | 38 | 34/66 |
| 3 | H | OMe | 67 | 41/59 |
| 4 | Br | H | 43 | 100/0 |
| 5 | Me | H | 76 | 71/29 |
| 6 | OMe | H | 57 | 75/25 |
| 7 | Br | Br | 36 | 62/38 |
| 8 | Me | Me | 33 | 37/63 |
| 9 | OMe | OMe | 57 | 36/64 |
Entries 1–3 describe reactions where the ortho-substitution is on aldehyde B. In all such cases, Z-olefins are formed as the major products. No influence from the electronic nature of the Y group on aldehyde B is noticeable, and the E–Z ratio is similar in all three cases. Entries 4–6 show examples in which the ortho-substitution is on aldehyde A. The stereochemical outcome is opposite compared to that in entries 1–3, and the E-isomer is the predominant form. When X = Br (entry 4), the reaction is 100% E-selective while with EDG moieties (entries 5 and 6), a certain percentage of Z-isomer is formed. These trends thus mirror the findings from Table 1, and no ortho-effect can be established for aldehydes A.
Entries 7–9 show examples of homocoupling reactions between ortho-substituted benzaldehydes. When X = Y = Br (entry 7), the reaction forms preferentially the E-isomer. This result has to be viewed in context of entries 1 and 4, in which the ortho-bromide is Z-directing for aldehyde B and E-directing for aldehyde A. The two effects are thus working in opposite directions, explaining the observed reaction outcome. With EDGs (entries 8 and 9), the situation changes, and the ortho-substituent in aldehyde B as well as the electronic effect of the EDG substituent in aldehyde A are both Z-directing. Consequently, both reactions give rise to predominantly the Z-isomer.
The above data show that the ortho-effect29 is an important aspect to consider when coupling two benzaldehydes. ortho-Substituents on aldehyde B exhibit a Z-directing effect, while those on aldehyde A are largely of electronic nature, and thus follow the trends from Table 1. As a result, it is possible to also direct the synthesis of a specific stilbene with ortho-substituents towards one or the other isomer by choosing the right order of addition of the two aldehydes. For example, when comparing entries 1 and 4, ortho-bromo-stilbene can intentionally be produced either with 100% E-selectivity, or in the Z-enriched form as a 1:2 isomeric mixture.
Summarizing the findings above, it is the electronic nature of aldehyde A and the ortho-effect of aldehyde B that determine the isomeric preference of the reaction. Depending on the exact structure of the starting aldehydes, the two effects can oppose or support each other, and can be used to control and predict the product stereochemistry. In certain combinations, changing the order of addition can have striking effects. One such example is illustrated in the coupling of p-methoxybenzaldehyde with o-bromobenzaldehyde (Scheme 3). If the former is used as aldehyde A and the latter as aldehyde B, the Z-alkene is formed predominantly due to the ortho-effect of aldehyde B and the EDG on aldehyde A, both of which are Z-directing (Scheme 3a).
 | ||
| Scheme 3 Example of how the electronic nature of aldehyde A and the ortho-effect on aldehyde B can be used to direct product isomer distribution. | ||
By reversing the order of addition, the ortho-bromide substituent now only exerts its electronic EWG effect and is E-directing, while the methoxy group in the para-position in aldehyde B has no effect. Consequently, the reaction is 100% E-selective (Scheme 3b). The examples in Scheme 3c and d follow the same rationale. In the coupling of electron-rich aldehyde A with a morpholino group in the para-position and o-bromobenzaldehyde as aldehyde B, both substituents are Z-directing. Conversely, with a reversed order of addition of the two coupling partners, the opposite isomer is predominately formed (Scheme 3d).
Conclusions
E- and Z-Enriched stilbenes can be synthesized by the coupling of two differently substituted benzaldehydes. Owing to the sequential addition of the two coupling partners at different stages of the sequence, different E- and Z-directing effects on the two aldehydes can be exploited. Chart 1 represents a practical tool to predict which combination of substituents can be used to direct the synthesis towards E- or Z-enriched stilbenes. In the left column, substrate combinations that give rise to Z-enriched stilbenes are depicted. These contain examples where aldehyde A has an EDG, or where aldehyde B features an ortho-substituent. Conversely, as summarized in the right column, E-stilbenes are formed from electron-deficient aldehydes A, irrespective of the structure of aldehyde B. None of these opportunities are available with related carbonyl–carbonyl cross-couplings such as the McMurry reaction, or also more recent reports on the topic including transition-metal catalyzed variants.36 One limitation of the procedure, though, is the incompatibility of the procedure for the coupling of aldehydes with acidic protons due to acid–base chemistry with phosphorus based reagents 1 and 3. The isolated yields presented herein range from 32 to 75%, depending on the exact nature of the substituents and the reactivity they impose. While being admittedly modest, it is important to remember that these yields are isolated yields of one-pot coupling reactions. As such, they compare favorably with the overall yields of 2–3 step synthetic procedures that are typical in the Wittig-type olefinations.9,37 We therefore believe that the results presented herein can be useful in the preparation of pharmaceutically relevant compounds, which is the subject of on-going efforts in our group.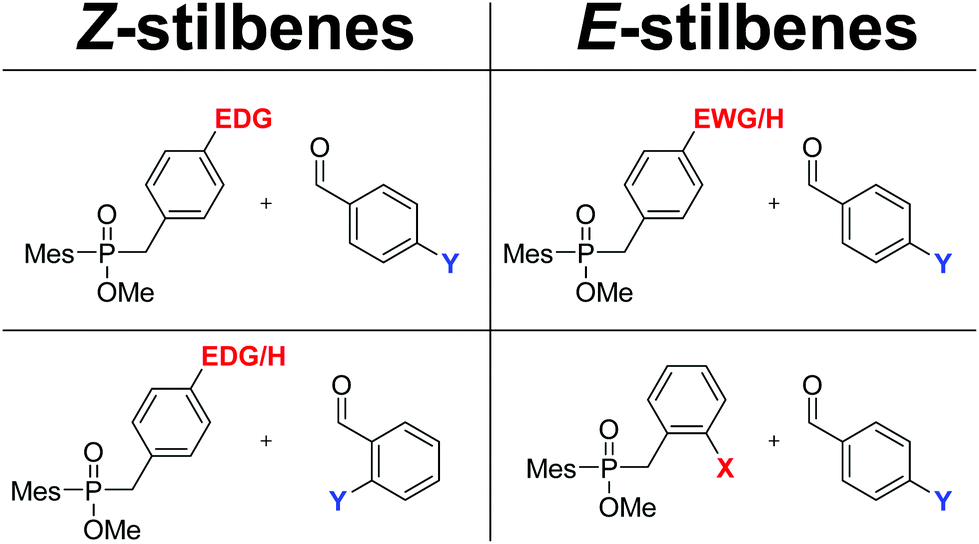 | ||
| Chart 1 X=EDG, EWG, H; substitution on the first aldehyde. Y = EDG, EWG, H; substitution on the second coupling partner. | ||
Experimental section
Materials and methods
The first half of the reaction up to the addition of MeOH was carried out in a glove box, while the second part can be conducted using regular Schlenk techniques. Glassware was flame-dried, and aldehydes dried/distilled prior to use. THF and Et2O were freshly distilled over Na/benzophenone under nitrogen. MesP(TMS)2 was synthesized according to literature procedures.38tBuOOH was dried with an azeotrope distillation of water and benzene from a commercial water solution of the peroxide. LiOEt and tBuOK were both used as a commercially available 1 M solution in THF. All the carbonyl compounds are commercially available. NMR spectra were recorded on a JEOL (400YH magnet) Resonance 400 MHz spectrometer. Chemical shifts (δ) are reported in ppm and coupling constants J in Hz. 1H NMR and 13C NMR chemical shifts are referenced to the residual protic solvent signal and 31P NMR spectra externally to 85% H3PO4(aq.). High-resolution mass spectra (HR-MS) were recorded on a Bruker QTOF spectrometer.Preparation of MesP(Li)TMS
To a solution of MesP(TMS)2 (1 eq., 1.60 g, 5.4 mmol) in 40 mL of dry THF was added a 1 M THF solution of LiOEt (1 eq.) at room temperature. The reaction mixture was stirred at ambient temperature for 4 hours until full conversion of the starting material to MesP(Li)TMS 1 was achieved, as judged from its typical P NMR shift (−187 ppm). The solvent was removed under reduced pressure to afford MesP(Li)TMS 1 as an orange oil without further purification. Dry Et2O was added and the yellow solution was stored in a glove box until further use for up to one month without visible changes.Detailed general procedure for the coupling of two aldehydes to stilbenes (Scheme 2)
To a yellow solution of 1 (1 eq., 351 mg, 1.52 mmol) in 15 mL dry Et2O were added 0.9 eq. of a first aldehyde (RCHO, R = Ph, Table 1, entry 8; 0.9 eq., 145 mg, 1.37 mmol) at ambient temperature; upon this addition, the reaction mixture changes immediately from bright yellow to a pale limpid yellow solution. The formation of the phosphaalkene 2 was monitored by 31P NMR using an external C6D6 standard. After complete formation, achieved within a few minutes, 0.2 mL of MeOH was added at room temperature with stirring. Upon addition of MeOH, the reaction mixture becomes immediately turbid. After 5 minutes, 1.2 eq. of tBuOOH were added to the reaction mixture at room temperature; no changes are visible after addition of the oxidant. The reaction was monitored by 31P NMR. Complete conversion to the desired phosphinate intermediate 3 is usually achieved within 5 minutes. The solvent was removed under vacuum and freshly distilled THF (15 mL) was added under an argon flow; the color of the reaction mixture at this stage is pale yellow. 0.9 eq. of a second aldehyde (RCHO, R = Ph, Table 1, entry 8; 0.9 eq., 145 mg, 1.37 mmol) were added to the reaction mixture, followed by 1.5 eq. of a 1 M THF solution of tBuOK; upon addition of the base, the mixture changes to a deep and limpid orange color. The reaction mixture was stirred at room temperature until phosphinate 3 is fully consumed. After completion of the reaction, water was added to the mixture. The aqueous phase was extracted with Et2O. The organic phase was dried over MgSO4, and the solvent was removed under reduced pressure to afford the crude product. The olefinic product was purified via silica gel column chromatography with a mixture of 5% of EtOAc in heptane. Isolated yield: 130 mg, 52%.Upscaling the reaction to gram scale (Scheme 2)
To a yellow solution of 1 (1 eq., 932 mg, 4.05 mmol) in 25 mL dry Et2O were added 0.9 eq. of a first aldehyde (RCHO, R = p-CH3-Ph, Table 1, entry 12; 0.9 eq., 437 mg, 3.64 mmol) at ambient temperature; upon this addition, the reaction mixture changes immediately from bright yellow to a pale limpid yellow solution. After stirring at ambient temperature for 5 minutes, 1 mL of MeOH was added, upon which the reaction mixture turns turbid immediately. After 5 minutes, 1.2 eq. of tBuOOH were added at room temperature, resulting in an exothermic reaction. The reaction was monitored by 31P NMR. Complete conversion to the desired phosphinate intermediate 3 is achieved within 5 minutes. The solvent was removed under vacuum and freshly distilled THF (25 mL) was added under an argon flow; the color of the reaction mixture at this stage is pale yellow. 0.9 eq. of a second aldehyde (RCHO, R = p-Br-Ph, Table 1, entry 12; 0.9 eq., 637 mg, 3.63 mmol) were added to the reaction mixture, followed by 1.5 eq. of a 1 M THF solution of tBuOK; upon addition of the base, the mixture changes to a deep and limpid orange color. Also in this case, the reaction is slightly exothermic. The reaction mixture was stirred at room temperature until full consumption of phosphinate 3. After completion of the reaction, water was added to the mixture. The aqueous phase was extracted with Et2O. The organic phase was dried over MgSO4, and the solvent was removed under reduced pressure to afford the crude product. The olefinic product was purified via silica gel column chromatography with a mixture of 5% of EtOAc in heptane. Isolated yield: 558 mg, 56%. E/Z ratio: 60/40.NMR spectroscopic data and the list of olefins
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for the work was provided by the Swedish Research Council and Uppsala University.Notes and references
- I. N. Ioffe and A. A. Granovsky, J. Chem. Theory Comput., 2013, 9, 4973–4990 CrossRef CAS PubMed.
- J. Jiang, C. Zheng, K. Zhu, J. Liu, N. Sun, C. Wang, H. Jiang, J. Zhu, C. Luo and Y. Zhou, J. Med. Chem., 2015, 58, 2538–2546 CrossRef CAS PubMed.
- M. Cushman, D. Nagarathnam, D. Gopal, A. K. Chakraborti, C. M. Lin and E. Hamel, J. Med. Chem., 1991, 34, 2579–2588 CrossRef CAS PubMed.
- M. Cushman, D. Nagarathnam, D. Gopal, H. M. He, C. M. Lin and E. Hamel, J. Med. Chem., 1992, 35, 2293–2306 CrossRef CAS PubMed.
- C. Rivière, A. D. Pawlus and J.-M. Mérillon, Nat. Prod. Rep., 2012, 29, 1317–1333 RSC.
- S. R. Marder, D. N. Beratan and L.-T. Cheng, Science, 1991, 252, 103–106 CrossRef CAS PubMed.
- B. De Filippis, A. Ammazzalorso, M. Fantacuzzi, L. Giampietro, C. Maccallini and R. Amoroso, ChemMedChem, 2017, 12, 558–570 CrossRef CAS PubMed.
- J. Wang, Stereoselective Alkene Synthesis, Springer-Verlag, Berlin Heidelberg, 2012 Search PubMed.
- T. Takeda, Modern Carbonyl Olefination, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2004 Search PubMed.
- G. Wittig and G. Geissler, Liebigs Ann. Chem., 1953, 580, 44–57 CrossRef CAS.
- G. Wittig, Science, 1980, 210, 600–604 CrossRef CAS PubMed.
- J. Boutagy and R. Thomas, Chem. Rev., 1974, 74, 87–99 CrossRef CAS.
- L. F. v. Staden, D. Gravestock and D. J. Ager, Chem. Soc. Rev., 2002, 31, 195–200 RSC.
- O. M. Ogba, N. C. Warner, D. J. O'Leary and R. H. Grubbs, Chem. Soc. Rev., 2018, 47, 4510–4544 RSC.
- D. Astruc, New J. Chem., 2005, 29, 42–56 RSC.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS.
- D. Roy and Y. Uozumi, Adv. Synth. Catal., 2018, 360, 602–625 CrossRef CAS.
- P. A. Byrne and D. G. Gilheany, Chem. Soc. Rev., 2013, 42, 6670–6696 RSC.
- J. Clayden and S. Warren, Angew. Chem., Int. Ed. Engl., 1996, 35, 241–270 CrossRef CAS.
- W. S. Wadsworth and W. D. Emmons, J. Am. Chem. Soc., 1961, 83, 1733–1738 CrossRef CAS.
- A. B. Juan and R. O. Liliana, Curr. Org. Chem., 2015, 19, 744–775 CrossRef.
- P. A. Byrne, in Investigation of Reactions Involving Pentacoordinate Intermediates, Springer, Berlin, Heidelberg, 2012, ch. 2, DOI: DOI:10.1007/978-3-642-32045-3_2.
- W. C. Still and C. Gennari, Tetrahedron Lett., 1983, 24, 4405–4408 CrossRef CAS.
- E. J. Corey and G. T. Kwiatkowski, J. Am. Chem. Soc., 1966, 88, 5653–5654 CrossRef CAS.
- K. Ando, Tetrahedron Lett., 1995, 36, 4105–4108 CrossRef CAS.
- K. Ando, J. Org. Chem., 1997, 62, 1934–1939 CrossRef CAS PubMed.
- H. Yamataka, K. Nagareda, K. Ando and T. Hanafusa, J. Org. Chem., 1992, 57, 2865–2869 CrossRef CAS.
- G. L. Keldsen and W. E. McEwen, J. Am. Chem. Soc., 1978, 100, 7312–7317 CrossRef CAS.
- E. C. Dunne, É. J. Coyne, P. B. Crowley and D. G. Gilheany, Tetrahedron Lett., 2002, 43, 2449–2453 CrossRef CAS.
- P. A. Byrne and D. G. Gilheany, J. Am. Chem. Soc., 2012, 134, 9225–9239 CrossRef CAS PubMed.
- P. A. Byrne, L. J. Higham, P. McGovern and D. G. Gilheany, Tetrahedron Lett., 2012, 53, 6701–6704 CrossRef CAS.
- K. Esfandiarfard, J. Mai and S. Ott, J. Am. Chem. Soc., 2017, 139, 2940–2943 CrossRef CAS PubMed.
- J. Mai, A. I. Arkhypchuk, A. K. Gupta and S. Ott, Chem. Commun., 2018, 54, 7163–7166 RSC.
- A. I. Arkhypchuk, N. D'Imperio and S. Ott, Org. Lett., 2018, 20, 5086–5089 CrossRef CAS PubMed.
- A. I. Arkhypchuk, N. D'Imperio and S. Ott, Chem. Commun., 2019, 55, 6030–6033 RSC.
- D. Ainembabazi, C. Reid, A. Chen, N. An, J. Kostal and A. Voutchkova-Kostal, J. Am. Chem. Soc., 2020, 142, 696–699 CrossRef CAS PubMed.
- D. C. Harrowven, I. L. Guy, M. Howell and G. Packham, Synlett, 2006, 2977–2980 CrossRef CAS.
- Y. Takeda, T. Nishida and S. Minakata, Chem. – Eur. J., 2014, 20, 10266–10270 CrossRef CAS PubMed.
- K. Song, P. Liu, J. Wang, L. Pang, J. Chen, I. Hussain, B. Tan and T. Li, Dalton Trans., 2015, 44, 13906–13913 RSC.
- V. G. Landge, J. Pitchaimani, S. P. Midya, M. Subaramanian, V. Madhu and E. Balaraman, Catal. Sci. Technol., 2018, 8, 428–433 RSC.
- S. Fu, N.-Y. Chen, X. Liu, Z. Shao, S.-P. Luo and Q. Liu, J. Am. Chem. Soc., 2016, 138, 8588–8594 CrossRef CAS PubMed.
- C. E. Janßen and N. Krause, Eur. J. Org. Chem., 2005, 2322–2329 CrossRef.
- S. Inomata, H. Hiroki, T. Terashima, K. Ogata and S.-i. Fukuzawa, Tetrahedron, 2011, 67, 7263–7267 CrossRef CAS.
- Y. Lu, X. Feng, B. S. Takale, Y. Yamamoto, W. Zhang and M. Bao, ACS Catal., 2017, 7, 8296–8303 CrossRef CAS.
- M. B. Andrus, C. Song and J. Zhang, Org. Lett., 2002, 4, 2079–2082 CrossRef CAS PubMed.
- V. G. Landge, J. Pitchaimani, S. P. Midya, M. Subaramanian, V. Madhu and E. Balaraman, Catal. Sci. Technol., 2018, 8, 428–433 RSC.
- J. Yang, C. Wang, Y. Sun, X. Man, J. Li and F. Sun, Chem. Commun., 2019, 55, 1903–1906 RSC.
- M. Das and D. F. O'Shea, Org. Lett., 2016, 18, 336–339 CrossRef CAS.
- A. L. Krasovskiy, S. Haley, K. Voigtritter and B. H. Lipshutz, Org. Lett., 2014, 16, 4066–4069 CrossRef CAS PubMed.
- S. Ceyhan, Y. Cetinkaya, A. Akdag and M. Balci, Tetrahedron, 2016, 72, 6815–6824 CrossRef CAS.
- T. Huang, T. Chen and L.-B. Han, J. Org. Chem., 2018, 83, 2959–2965 CrossRef CAS PubMed.
- S. Tanaka, K. Itami, K. Sunahara, G. Tatsuta and A. Mori, Chem. Commun., 2015, 51, 1949–1952 RSC.
- Y. Liu, L. Hu, H. Chen and H. Du, Chem. – Eur. J., 2015, 21, 3495–3501 CrossRef CAS PubMed.
- J. N. Ngwendson, W. N. Atemnkeng, C. M. Schultze and A. Banerjee, Org. Lett., 2006, 8, 4085–4088 CrossRef CAS PubMed.
- T. Yoshida and K. Mori, Chem. Commun., 2017, 53, 4319–4322 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of all isolated olefinic products. See DOI: 10.1039/d0ob01139h |
| This journal is © The Royal Society of Chemistry 2020 |