
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
General dual functionalisation of biomacromolecules via a cysteine bridging strategy†
Stephen J.
Walsh‡
 ab,
Jessica
Iegre‡
a,
Hikaru
Seki‡
a,
Jonathan D.
Bargh
a,
Hannah F.
Sore
a,
Jeremy S.
Parker
c,
Jason S.
Carroll
b and
David R.
Spring
*a
ab,
Jessica
Iegre‡
a,
Hikaru
Seki‡
a,
Jonathan D.
Bargh
a,
Hannah F.
Sore
a,
Jeremy S.
Parker
c,
Jason S.
Carroll
b and
David R.
Spring
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: spring@ch.cam.ac.uk
bCancer Research UK Cambridge Institute, University of Cambridge, Robinson Way, Cambridge, CB2 0RE, UK
cEarly Chemical Development, Pharmaceutical Development, R&D, AstraZeneca, Macclesfield, UK
First published on 14th May 2020
Abstract
Site-selective modification of peptides and proteins has resulted in the development of a host of novel tools for the study of cellular systems or the synthesis of enhanced biotherapeutics. There is a need for useful methodologies that enable site-selective modification of native peptides or proteins, which is even more prevalent when modification of the biomolecule with multiple payloads is desired. Herein, we report the development of a novel dual functional divinylpyrimidine (dfDVP) platform that enables robust and modular modification of peptides, antibody fragments and antibodies. These biomacromolecules could be easily functionalised with a range of functional payloads (e.g. fluorescent dyes, cytotoxic warheads or cell-penetrating tags). Importantly, the dual functionalised peptides and antibodies demonstrated exquisite bioactivity in a range of in vitro cellular assays, showcasing the enhanced utility of these bioactive conjugates.
Introduction
The modification of proteins can be leveraged for a variety of tasks such as the study of cellular processes or the generation of bioconjugate therapeutics.1,2 To an even greater extent, dual modification of peptides or proteins can provide valuable tools to aid in these investigations.3,4 Incorporation of multiple payloads can take on many formats; such as attachment of Förster resonance energy transfer (FRET) pairs, half-life extension strategies of protein/peptide-drug conjugates, increased cellular permeability of peptide-based drugs/imaging agents or macromolecular theranostics. Dual modification can typically be achieved in two ways: (1) modification of two different amino acids with separate payloads, or (2) use of a single linker with orthogonal reactive handles to introduce the distinct functionality.5 While this can be relatively straightforward in short peptides, site-selective dual modification of large proteins can be challenging.6Peptide drugs are attractive candidates for the perturbation of cellular protein–protein interactions (PPIs) due to their large binding surface area. However, linear peptides typically suffer from poor circulatory stability and cell permeability, thus limiting their therapeutic potential.7,8 The use of a linker to macrocyclise the peptide can improve these characteristics and the overall pharmacological profile of the peptide drug. Furthermore, the linker can be used to modify the physical properties of the peptide (e.g. solubility, permeability) or can be used to attach functional payloads (e.g. drugs, fluorescent dyes).9,10 Peptide macrocyclisation linkers that facilitate dual modification with these tags would enable further development of peptide drugs and probes.
Similarly, antibody–drug conjugates (ADCs) are a class of targeted bioconjugate therapeutics that have witnessed enormous interest in recent years due to their ability to selectively deliver highly potent cytotoxic agents to specific cell types.11 Eight ADCs have now obtained approval by the Food and Drug Administration (FDA) for the treatment of a range of solid and haematological cancers.12 While three of the approved ADCs with high drug loading are (nearly) homogeneous, the straightforward synthesis of homogeneous ADCs with low drug-to-antibody ratio (DAR), especially with uneven numbers, is more challenging.13 The synthesis of homogeneous ADCs is desirable as they typically display more robust and reliable pharmacokinetic profiles than the corresponding heterogeneous ADC mixtures.14,15 Furthermore, conjugation strategies that generate homogenous ADCs should also produce a conjugate with high levels of plasma stability to ensure that premature release of the cytotoxic payload in circulation does not occur and cause off-target side effects.16
A number of strategies that enable site-selective and stable dual modification of peptides or antibodies with different payloads have been reported.17–23 Alabi and co-workers have developed elegant strategies that use both antibody engineering and enzymatic modification strategies to introduce two different payloads onto antibodies.24,25 These efforts have enabled the masking of the inherent hydrophobicity of cytotoxic warheads with polyethylene glycol (PEG) chains or the incorporation of FRET pairs for studying trafficking and metabolism of the bioconjugates. In another approach, Levengood and co-workers used orthogonal cysteine protecting groups to generate ADCs containing two different auristatin cytotoxins.26 Similarly, Kumar et al. developed a heterotrifunctional maleimide reagent that enabled the modification of antibodies containing engineered cysteine residues with both auristatin and pyrrolobenzodiazepine dimer payloads.27
Cysteine bridging technologies have emerged as a highly useful strategy, both in the synthesis of macrocyclic peptides and ADCs.28–31 These methodologies focus on the use of a bis-reactive linker that selectively cross-links the free thiols of cysteine side chains. Exemplary work by Caddick, Chudasama and co-workers using pyridazinedione reagents has demonstrated the potential of this approach to effectively generate homogeneous and stable dual functional antibody bioconjugates.32,33 Therefore, the development of technologies that allow the simple construction of complex dual functional antibodies is an area of immense interest.
We have previously reported the used of divinylpyrimidine (DVP) reagents for the site-selective synthesis of exceptionally stable ADCs, and divinyltriazine reagents to synthesise functionalised macrocyclic peptides.34,35 Herein, we report the development of a novel dual functional DVP (dfDVP) reagent that can be universally applied to the modification of peptides or antibodies/antibody fragments (Fig. 1). The dfDVP reagent can be easily synthesised using a combination of solid- and solution-phase chemistry. Cross-linking of cysteines in a range of biomolecules was then achieved and several functional moieties were conjugated in a controlled and site-selective fashion. Furthermore, the dual functional bioconjugates exhibited remarkable biological activity in numerous in vitro assays.
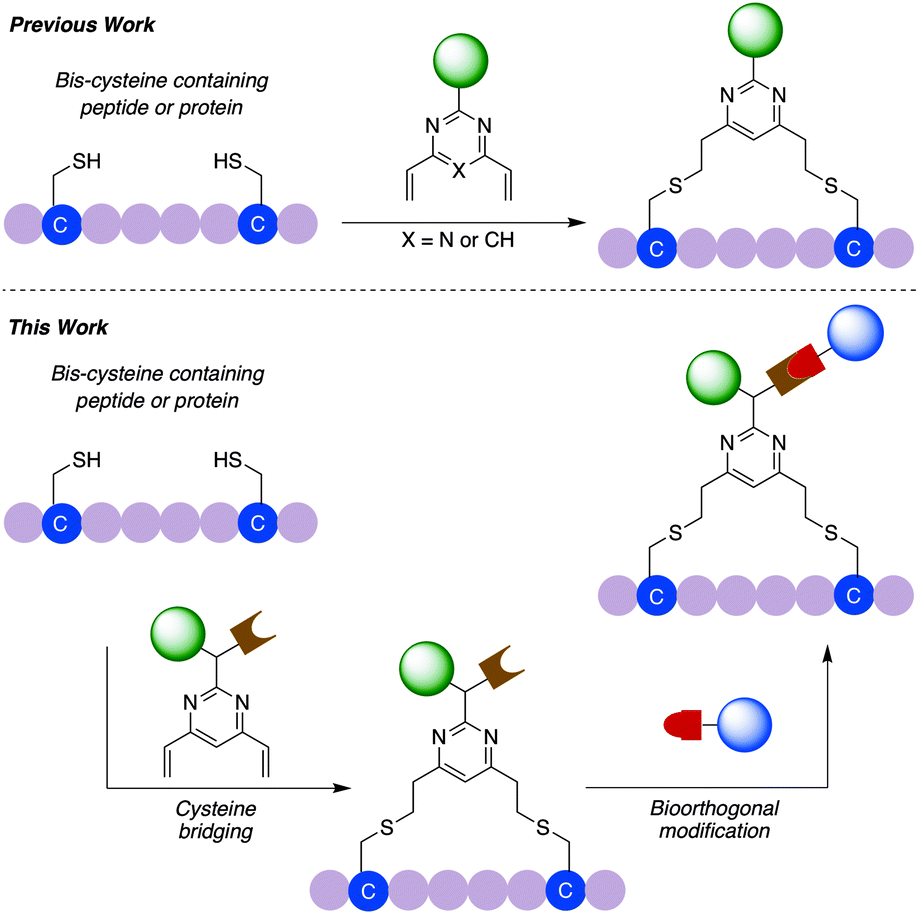 | ||
| Fig. 1 Schematic overview of previous work using divinylpyrimidine or divinyltriazine reagents for peptide or protein modification and the dual functional divinylpyrimidine (dfDVP) technology described in this work. In the first step, the dfDVP reagent cross-links two proteinogenic cysteine residues to selectively conjugate one payload, which is followed by a bioorthogonal click reaction to attach the second payload. | ||
Results and discussion
Linker design and synthesis
In commencing this work, we envisaged a general reagent that would facilitate modular modification of any peptide or protein containing neighbouring cysteine residues. As such dfDVP 1 was designed, hypothesising that a strategy resembling solid phase peptide synthesis could be used to easily assemble this scaffold (Fig. 2). This strategy would minimise purification and handling of complex and hydrophilic molecules in organic solvent. The dfDVP reagent was designed to contain a fluorescent dye (fluorescein), from reaction with fluorescein isothiocyanate (FITC) and an alkyne-containing amino acid (propargyl glycine), both of which could be inserted during on-resin synthesis. The alkyne could subsequently be used to conjugate a range of azide-functionalised payloads via click chemistry.36 Accordingly, fluorescein-containing amine 2 was synthesised on-resin following a standard Fmoc-protecting group strategy, and a condensation between the amine side chain of lysine and the isothiocyanate of FITC (ESI Scheme S1†). Upon cleavage from the Rink amide resin, amide coupling between DVP 3 (see ESI† for synthetic details) and amine 2 was achieved in good yield to generate the desired dfDVP linker 1. Furthermore, although fluorescein was attached in this case, it would also be feasible to attach other payloads on-resin using this strategy.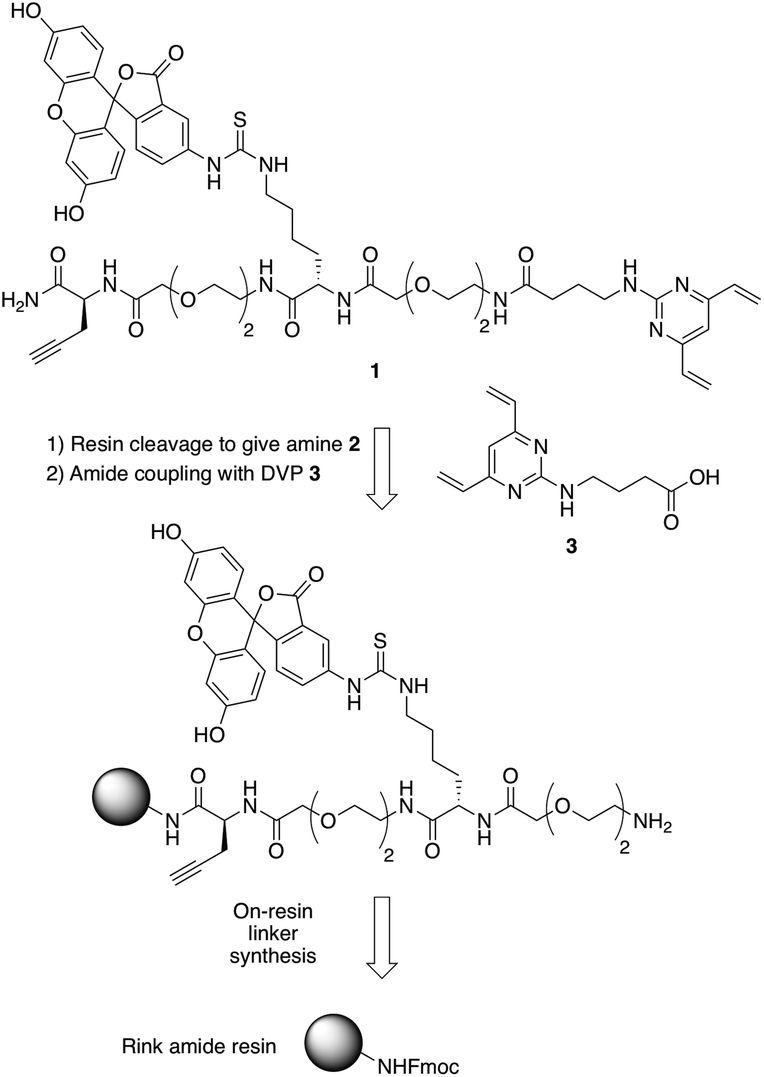 | ||
| Fig. 2 Retrosynthetic analysis of the solid- and solution-phase strategy for the synthesis of the dfDVP reagent 1. | ||
Peptide modification and biological evaluation
With dfDVP 1 in hand, investigations into its cysteine bridging and dual modification capabilities commenced. First, with the aim of generating a peptide modality with enhanced biological properties, dfDVP modification of a short oligopeptide was undertaken. The p53-targeting peptide Pep-p53 has previously been reported35 and as such, was synthesised and reacted with dfDVP 1 in a mixture of sodium phosphate buffer (NaPi, 95% v/v) and DMF (5% v/v) for 1 hour at room temperature (Fig. 3a). Macrocyclisation proceeded efficiently to generate the stapled peptide Pep-FITC. Pleasingly, the stapled peptide was the only reaction product observed by HPLC and LCMS analysis, with none of the bis-alkylated species observed. Next, to aid the cell permeability of the fluorescent peptide, a copper-catalysed azide–alkyne cycloaddition (CuAAC) was used to attach the azide-modified tri-arginine cell-penetrating tag N3-RRR to the stapled peptide.37 The CuAAC reaction proceeded smoothly and dual functionalised peptide Pep-FITC-RRR was obtained following HPLC purification. Having synthesised peptides with or without a cell-penetrating tag, we next wanted to investigate the permeability of the different peptides. The incorporation of the fluorescein moiety onto both peptides enabled the use of confocal microscopy to conduct this analysis. Accordingly, human osteosarcoma cells (U2OS) were treated with Pep-FITC (25 μM) and Pep-FITC-RRR at 37 °C. Cells were fixed after 0.5, 1 and 3 hours incubation and imaged via confocal microscopy. Pleasingly, we observed uptake of the dual functionalised stapled peptide, Pep-FITC-RRR at all time points, whereas no internalisation was detected for the mono functionalised stapled peptide Pep-FITC (Fig. 3b and ESI Fig. S6†). These results demonstrate the potential of our dfDVP methodology to simultaneously label and impart cell-permeability to otherwise non cell-permeable peptides.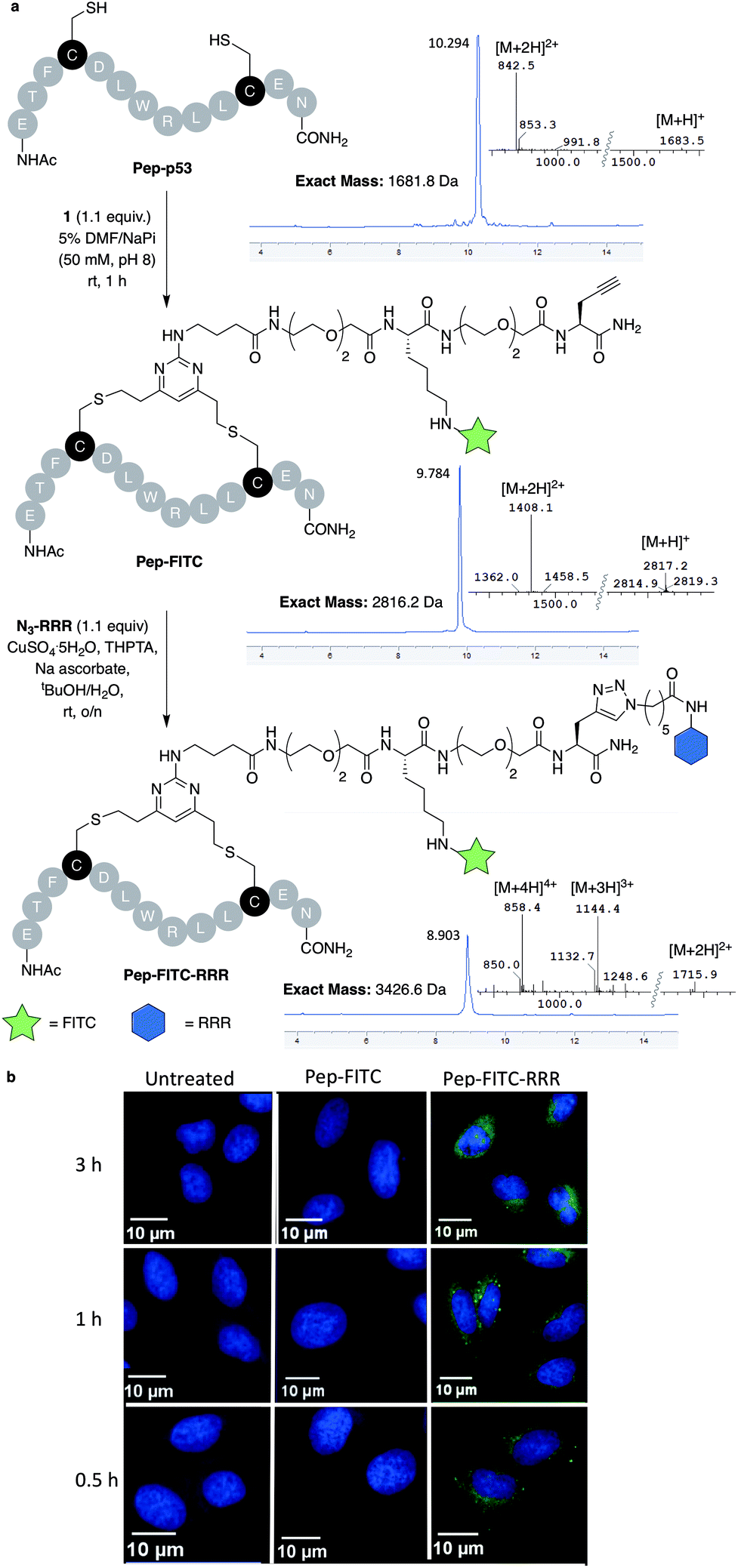 | ||
| Fig. 3 (a) Macrocyclisation of the Pep-p53 with dfDVP 1, followed by CuAAC to conjugate the cell-penetrating tag N3-RRR. HPLC and MS traces for the reaction products are shown in addition to the mass-to-charge ratio ions observed. (b) Confocal microscopy images obtained after treatment of U2OS cells with Pep-FITC and Pep-FITC-RRR. Additional confocal microscopy images with Pep-FITC-RRR can be found in Fig. S6.† | ||
Antibody fragment modification
We next aimed to demonstrate the dual modification potential on larger polypeptides. The fragment, antigen-binding (Fab) arm of an antibody contains a single disulfide that covalently links the two polypeptide chains. Trastuzumab is an anti-HER2 IgG1 antibody that is clinically used to treat HER2-positive breast cancer and comprises the antibody component in marketed ADCs, Kadcyla® and Enhertu®. The Fab of trastuzumab has also been studied in its own right as both a therapeutic agent and a delivery molecule in targeted therapeutics.32,38 It was proposed that modification of this Fab with both a fluorescent dye and a cytotoxic warhead would generate a valuable theranostic agent.39 Trastuzumab Fab was generated by sequential digestions with pepsin and papain and subsequently treated with tris(2-carboxyethyl)phosphine hydrochloride (TCEP) to reduce the single interchain disulfide (Fig. 4a, c, f and ESI Fig. S1†). The reduced protein was then reacted with dfDVP 1 (10 equiv.) for 1 hour at 37 °C. LCMS, SDS-PAGE and UV-vis analysis revealed >95% conversion to afford the desired conjugate, Fab-FITC (Fig. 4d, f and ESI Fig. S2†). To attach a drug molecule to the alkyne-functionalised antibody fragment, an azido-monomethyl auristatin E (MMAE) molecule was required. MMAE is a commonly used anti-mitotic drug in ADC therapeutics due to its sub-nanomolar cytotoxicity against a range of cancer types.34 Due to the complexity of the dual functional linker, it was hypothesised that cleavage of the drug from the linker would be required for cytotoxicity.40 We have recently reported the utility of sulfatase-cleavable linkers in MMAE-containing ADCs, and deemed this to be an appropriate cleavable group.41 Accordingly, azide-sulfate-MMAE 4 was prepared (Fig. 4b, see ESI† for synthetic details) and reacted with Fab-FITC in the presence of CuSO4·5H2O, THPTA and sodium ascorbate. Analysis of the reaction via LCMS suggested excellent conversion to the bis-functionalised antibody fragment, Fab-FITC-MMAE (Fig. 4e, f and ESI Fig. S3†). This cysteine bridging and subsequent CuAAC reaction demonstrated the utility of the dfDVP platform to modify complex biomolecules, with both modification steps proceeding efficiently.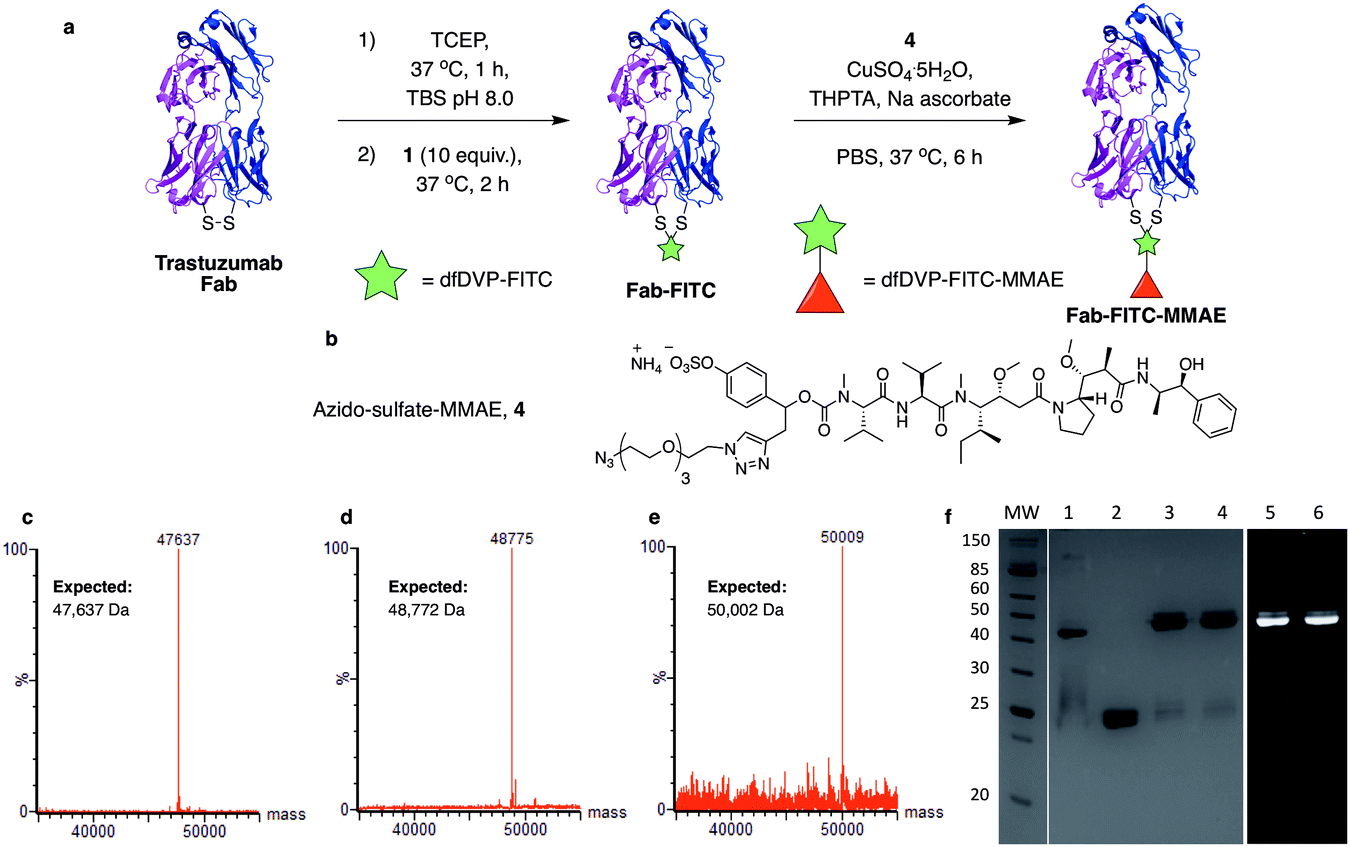 | ||
| Fig. 4 Dual modification of trastuzumab Fab. (a) Reaction of reduced trastuzumab Fab with dfDVP 1, followed by click attachment of (b) azido-sulfate-MMAE 4. LCMS analysis of (c) trastuzumab Fab, (d) Fab-FITC and (e) Fab-FITC-MMAE. (f) SDS-PAGE analysis of the dual modification. MW: molecular weight ladder, 1: non-reduced Fab, 2: reduced Fab, 3: Fab-FITC Coomassie stain, 4: Fab-FITC-MMAE Coomassie stain, 5: Fab-FITC fluorescence, 6: Fab-FITC-MMAE fluorescence. Lanes 3–6 were run under reducing conditions. TBS = Tris buffered saline (Tris HCl 25 mM pH 8, NaCl 25 mM, EDTA 0.5 mM pH 8). | ||
Antibody modification and biological evaluation
We next sought to modify an IgG antibody using this method, to obtain a fluorescent ADC bioconjugate with potential utility for simultaneous imaging and treatment of malignancies. IgG1 antibodies contain four interchain disulfides that can be easily reduced to produce four sites for the cross-linking of cysteine residues. As such, trastuzumab was reduced with TCEP and subsequently treated with dfDVP 1 (10 equiv. per disulfide) for 2 hours at 37 °C (Fig. 5a). Pleasingly, 90–95% conversion from the unreacted heavy and light chains to the bridged species Fab-FITC was observed by LCMS, SDS-PAGE and UV-vis analysis (Fig. 5b, c and ESI Fig. S4†). Similar to our previous observations with mono functional DVP reagents, the half-antibody species, formed through intrachain cross-linking of the cysteines in the hinge region of the heavy chains was observed to be the major reaction product.34 Despite this non-native rebridging, the full antibody remains intact due to non-covalent interactions and such ADCs remain biologically active.31,34,42 The fluorescent antibody Tras-FITC was then reacted with azido-sulfate-MMAE 4 in the presence of CuSO4·5H2O, THPTA and sodium ascorbate for 6 hours at 37 °C. Analysis of the reaction via LCMS revealed conversion to the fluorescent ADC Tras-FITC-MMAE with a DAR of approximately 3.0 (Fig. 5b, c and ESI Fig. S5†). Despite screening several reaction conditions, obtaining a DAR >3 could not be achieved. We postulate that the size of the linkers being attached allows attachment of one drug molecule in the hinge region of each antibody; however, conjugation of a second cytotoxin is prohibited due to the large size of the linker-payloads already conjugated. However, under all of the conditions tested, a controlled DAR of 3 was consistently achieved and we postulated that this drug loading would be sufficient to achieve potent and selective cytotoxicity.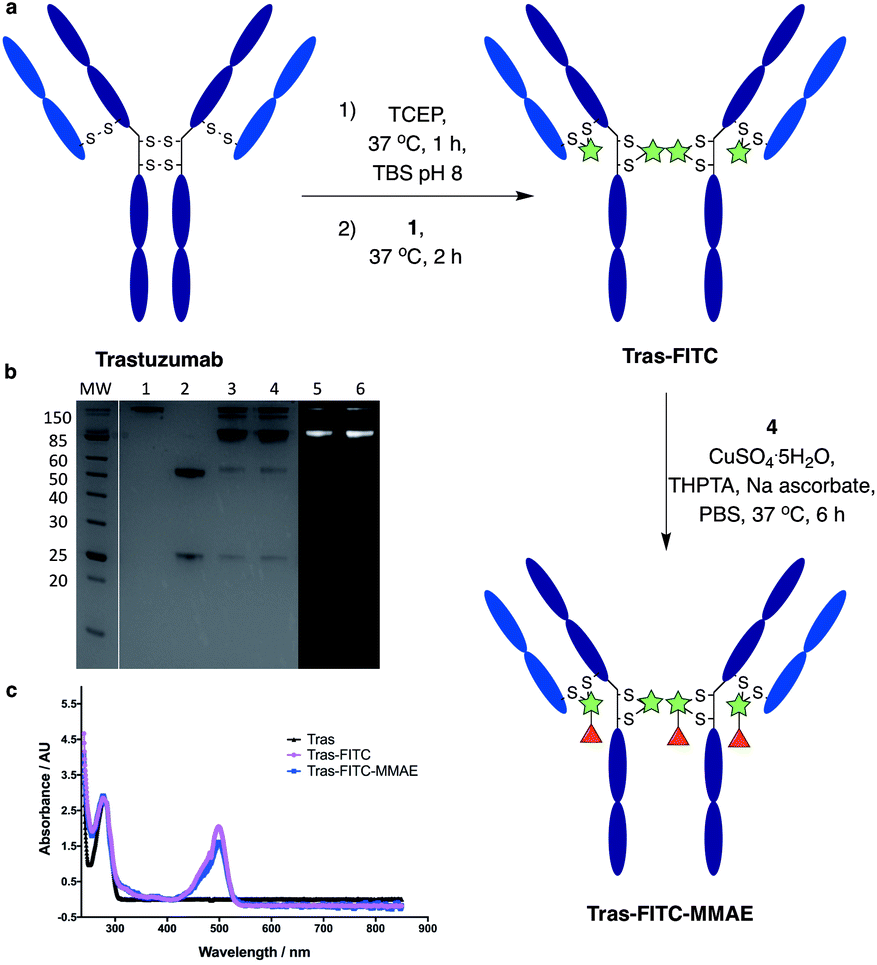 | ||
| Fig. 5 Dual modification of trastuzumab. (a) Reaction of reduced trastuzumab with dfDVP 1, followed by click attachment of azido-sulfate-MMAE 4. (b) SDS-PAGE analysis of the dual modification. MW: molecular weight ladder, 1: non-reduced trastuzumab, 2: reduced trastuzumab, 3: Tras-FITC Coomassie stain, 4: Tras-FITC-MMAE Coomassie stain, 5: Tras-FITC fluorescence, 6: Tras-FITC-MMAE fluorescence. Lanes 3–6 were run under reducing conditions. (c) UV-vis spectrum of trastuzumab, Tras-FITC and Tras-FITC-MMAE. TBS = Tris buffered saline. | ||
To investigate the biological activity of the trastuzumab conjugates, their cytotoxicity was initially evaluated. Both trastuzumab and Tras-FITC showed little effect on the viability of SKBR3 (HER2-positive) or MCF7 (HER2-negative) cell lines (Fig. 6a and b). In contrast, Tras-FITC-MMAE demonstrated exquisite dose-dependent cytotoxicity against SKBR3 cells, while having little effect on MCF7 cells (Fig. 6a and b). MMAE has previously been shown to have sub-nanomolar toxicity against both cell lines.34 This result highlights that modification of the antibody with a fluorescent dye does not cause a cytotoxic effect and that the cytotoxicity observed for Tras-FITC-MMAE was generated by the conjugated MMAE. Furthermore, this suggests that attachment of the fluorescent dye does not impede cleavage of MMAE from the antibody. Finally, to demonstrate the imaging capabilities of our bis-functionalised antibody, a microscopy-based cell selectivity and internalisation assay was conducted. First, SKBR3 and MCF7 cells were treated with Tras-FITC or Tras-FITC-MMAE for 1 hour at 4 °C. Incubation at this temperature allows receptor binding but not internalisation.32 The cells were then extensively washed to remove any unbound antibody and subsequently imaged using wide-field microscopy to ascertain the binding selectivity of the different antibody conjugates in both cell lines. Pleasingly, labelling was observed on the surface of SKBR3 cells for both Tras-FITC and Tras-FITC-MMAE (Fig. 6c). In contrast, no labelling of MCF7 cells was detected under the same treatment conditions (Fig. 6e). Finally, after incubation for 1 hour at 4 °C and washing to remove unbound antibodies, SKBR3 cells were incubated at 37 °C for 1 hour to enable receptor-antibody internalisation. Imaging of the cells via wide-field microscopy again suggested that internalisation had begun after this incubation period (Fig. 6d). This collective data demonstrates the exquisite cell-selective cytotoxicity and cell imaging potential of the dfDVP bis-functionalised trastuzumab conjugates. Moreover, the dfDVP platform enables the attachment of multiple payloads to antibodies without causing a negative effect on the activity of the antibody or either payload.
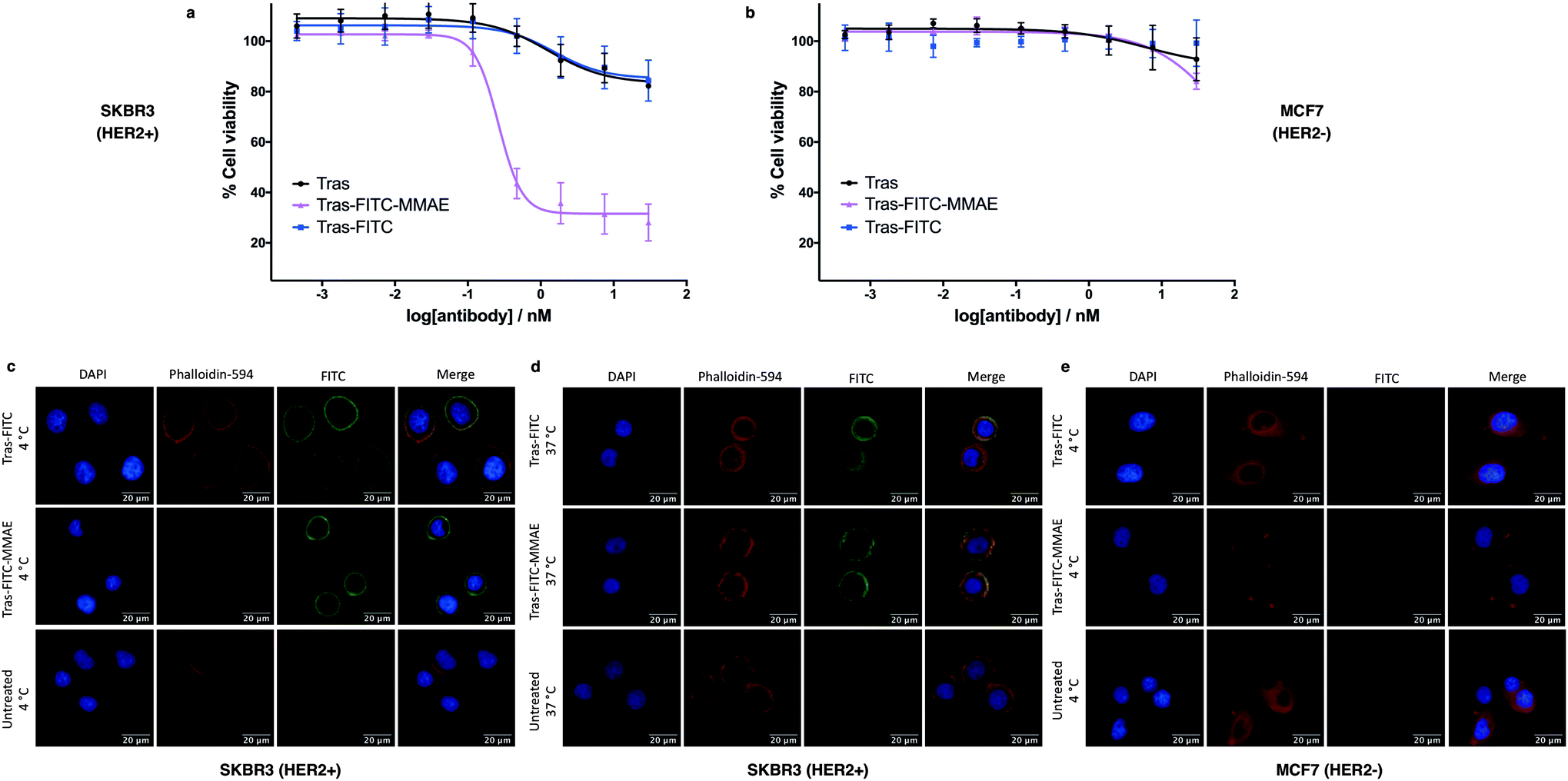 | ||
| Fig. 6 Biological evaluation of dfDVP modified trastuzumab. Cytotoxicity of the trastuzumab conjugates in (a) SKBR3 cells and (b) MCF7 cells. Widefield microscopy analysis of the antibody conjugates (c) in SKBR3 cells after 1 hour at 4 °C and (d) after a subsequent hour at 37 °C, and (e) in MCF7 cells for 1 hour at 4 °C. DAPI was used to stain nuclei and phalloidin-594 was used to stain actin. | ||
Conclusions
The primary objective of this work was to obtain a universal methodology to enable the dual functionalisation of a range of bis-cysteine-containing biomolecules. The dfDVP platform incorporates a tractable method of introducing complex functionality onto a biomolecule through two efficient and selective bioconjugation reactions. The dfDVP reagent 1 utilised in this work was routinely synthesised through an elegant combination of solid- and solution-phase chemistry and could be used to robustly modify oligopeptides, antibody fragments and IgGs with several different payloads. Furthermore, the synthesised bioconjugates demonstrated excellent bioactivity, as determined through a series of in vitro biological assays. This technology represents a novel technique that enables rapid construction of highly complex bioconjugates for the study or perturbation of biological systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Huw Naylor from the Microscopy Core at the CRUK Cambridge Institute for help with wide-field microscopy. We would like to thank Dr David Baker from AstraZeneca, Cambridge for help with confocal microscopy. SJW acknowledges support from AstraZeneca, The Cambridge Trusts and the EPSRC. JI acknowledges the EPSRC for funding. HS acknowledges Trinity College Cambridge for studentship support. JDB acknowledges an iCASE studentship from GlaxoSmithKline/EPSRC. DRS acknowledges support from the EPSRC (EP/P020291/1) and Royal Society (Wolfson Research Merit Award). The Spring lab acknowledges general lab support from the EPSRC, BBSRC, MRC and Royal Society.References
- E. A. Hoyt, P. M. S. D. Cal, B. L. Oliveira and G. J. L. Bernardes, Nat. Rev. Chem., 2019, 3, 147–171 CrossRef CAS.
- T. Tamura and I. Hamachi, J. Am. Chem. Soc., 2019, 141, 2782–2799 CrossRef CAS PubMed.
- A. Maruani, D. A. Richards and V. Chudasama, Org. Biomol. Chem., 2016, 14, 6165–6178 RSC.
- J. Iegre, P. Brear, D. J. Baker, Y. S. Tan, E. L. Atkinson, H. F. Sore, D. H. O'Donovan, C. S. Verma, M. Hyvönen and D. R. Spring, Chem. Sci., 2019, 10, 5056–5063 RSC.
- E. Gil de Montes, E. Jiménez-Moreno, B. L. Oliveira, C. D. Navo, P. M. S. D. Cal, G. Jiménez-Osés, I. Robina, A. J. Moreno-Vargas and G. J. L. Bernardes, Chem. Sci., 2019, 10, 4515–4522 RSC.
- F. F. Schumacher, J. P. M. Nunes, A. Maruani, V. Chudasama, M. E. B. Smith, K. A. Chester, J. R. Baker and S. Caddick, Org. Biomol. Chem., 2014, 12, 7261–7269 RSC.
- J. Iegre, J. S. Gaynord, N. S. Robertson, H. F. Sore, M. Hyvönen and D. R. Spring, Adv. Ther., 2018, 1, 1800052 CrossRef.
- T. Uhlig, T. Kyprianou, F. G. Martinelli, C. A. Oppici, D. Heiligers, D. Hills, X. R. Calvo and P. Verhaert, EuPA Open Proteom., 2014, 4, 58–69 CrossRef CAS.
- A. J. Rojas, C. Zhang, E. V. Vinogradova, N. H. Buchwald, J. Reilly, B. L. Pentelute and S. L. Buchwald, Chem. Sci., 2017, 8, 4257–4263 RSC.
- C. M. Grison, G. M. Burslem, J. A. Miles, L. K. A. Pilsl, D. J. Yeo, Z. Imani, S. L. Warriner, M. E. Webb and A. J. Wilson, Chem. Sci., 2017, 8, 5166–5171 RSC.
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaïa, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef CAS PubMed.
- H. Kaplon and J. M. Reichert, MAbs, 2019, 11, 219–238 CrossRef CAS PubMed.
- V. Chudasama, A. Maruani and S. Caddick, Nat. Chem., 2016, 8, 114–119 CrossRef CAS PubMed.
- B. Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S. F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saad, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, T. Nguyen, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawli, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS PubMed.
- P. Strop, S.-H. Liu, M. Dorywalska, K. Delaria, R. G. Dushin, T.-T. Tran, W.-H. Ho, S. Farias, M. G. Casas, Y. Abdiche, D. Zhou, R. Chandrasekaran, C. Samain, C. Loo, A. Rossi, M. Rickert, S. Krimm, T. Wong, S. M. Chin, J. Yu, J. Dilley, J. Chaparro-Riggers, G. F. Filzen, C. J. O'Donnell, F. Wang, J. S. Myers, J. Pons, D. L. Shelton and A. Rajpal, Chem. Biol., 2013, 20, 161–167 CrossRef CAS PubMed.
- S. C. Alley, D. R. Benjamin, S. C. Jeffrey, N. M. Okeley, D. L. Meyer, R. J. Sanderson and P. D. Senter, Bioconjugate Chem., 2008, 19, 759–765 CrossRef CAS PubMed.
- P. J. Burke, J. Z. Hamilton, S. C. Jeffrey, J. H. Hunter, S. O. Doronina, N. M. Okeley, J. B. Miyamoto, M. E. Anderson, I. J. Stone, M. L. Ulrich, J. K. Simmons, E. E. McKinney, P. D. Senter and R. P. Lyon, Mol. Cancer Ther., 2017, 16, 116–123 CrossRef CAS PubMed.
- M. R. Karver, R. Weissleder and S. A. Hilderbrand, Angew. Chem., Int. Ed., 2012, 51, 920–922 CrossRef CAS PubMed.
- M. T. Taylor, J. E. Nelson, M. G. Suero and M. J. Gaunt, Nature, 2018, 562, 563–568 CrossRef CAS PubMed.
- Y. Zhang, C. Zang, G. An, M. Shang, Z. Cui, G. Chen, Z. Xi and C. Zhou, Nat. Commun., 2020, 11, 1–10 CrossRef PubMed.
- R. Tessier, J. Ceballos, N. Guidotti, R. Simonet-Davin, B. Fierz and J. Waser, Chem, 2019, 5, 2243–2263 CAS.
- N. Nilchan, X. Li, L. Pedzisa, A. R. Nanna, W. R. Roush and C. Rader, Antibiot. Ther., 2019, 2, 71–78 Search PubMed.
- X. Li, J. T. Patterson, M. Sarkar, L. Pedzisa, T. Kodadek, W. R. Roush and C. Rader, Bioconjugate Chem., 2015, 26, 2243–2248 CrossRef CAS PubMed.
- D. N. Thornlow, E. C. Cox, J. A. Walker, M. Sorkin, J. B. Plesset, M. P. Delisa and C. A. Alabi, Bioconjugate Chem., 2019, 30, 1702–1710 CrossRef CAS PubMed.
- J. A. Walker, J. J. Bohn, F. Ledesma, M. R. Sorkin, S. R. Kabaria, D. N. Thornlow and C. A. Alabi, Bioconjugate Chem., 2019, 30, 2452–2457 CrossRef CAS PubMed.
- M. R. Levengood, X. Zhang, J. H. Hunter, K. K. Emmerton, J. B. Miyamoto, T. S. Lewis and P. D. Senter, Angew. Chem., Int. Ed., 2017, 56, 733–737 CrossRef CAS PubMed.
- A. Kumar, K. Kinneer, L. Masterson, E. Ezeadi, P. Howard, H. Wu, C. Gao and N. Dimasi, Bioorg. Med. Chem. Lett., 2018, 28, 3617–3621 CrossRef CAS PubMed.
- J. P. M. Nunes, M. Morais, V. Vassileva, E. Robinson, V. S. Rajkumar, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, Chem. Commun., 2015, 51, 10624–10627 RSC.
- M. Morais, J. P. M. Nunes, K. Karu, N. Forte, I. Benni, M. E. B. Smith, S. Caddick, V. Chudasama and J. R. Baker, Org. Biomol. Chem., 2017, 15, 2947–2952 RSC.
- P. Bryant, M. Pabst, G. Badescu, M. Bird, W. McDowell, E. Jamieson, J. Swierkosz, K. Jurlewicz, R. Tommasi, K. Henseleit, X. Sheng, N. Camper, A. Manin, K. Kozakowska, K. Peciak, E. Laurine, R. Grygorash, A. Kyle, D. Morris, V. Parekh, A. Abhilash, J. W. Choi, J. Edwards, M. Frigerio, M. P. Baker and A. Godwin, Mol. Pharm., 2015, 12, 1872–1879 CrossRef CAS PubMed.
- O. Koniev, I. Dovgan, B. Renoux, A. Ehkirch, J. Eberova, S. Cianférani, S. Kolodych, S. Papot and A. Wagner, MedChemComm, 2018, 9, 827–830 RSC.
- A. Maruani, M. E. B. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 6645 CrossRef CAS PubMed.
- A. Maruani, P. A. Szijj, C. Bahou, J. C. F. Nogueira, S. Caddick, J. R. Baker and V. Chudasama, Bioconjugate Chem., 2020, 31, 520–529 CrossRef CAS PubMed.
- S. J. Walsh, S. Omarjee, W. R. J. D. Galloway, T. T.-L. Kwan, H. F. Sore, J. S. Parker, M. Hyvönen, J. S. Carroll and D. R. Spring, Chem. Sci., 2019, 10, 694–700 RSC.
- N. S. Robertson, S. J. Walsh, E. Fowler, M. Yoshida, S. M. Rowe, Y. Wu, H. F. Sore, J. S. Parker and D. R. Spring, Chem. Commun., 2019, 55, 9499–9502 RSC.
- V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Angew. Chem., Int. Ed., 2009, 48, 9879–9883 CrossRef CAS PubMed.
- Y. Wu, A. Kaur, E. Fowler, M. M. Wiedmann, R. Young, W. R. J. D. Galloway, L. Olsen, H. F. Sore, A. Chattopadhyay, T. T. L. Kwan, W. Xu, S. J. Walsh, P. De Andrade, M. Janecek, S. Arumugam, L. S. Itzhaki, Y. H. Lau and D. R. Spring, ACS Chem. Biol., 2019, 14, 526–533 CrossRef CAS PubMed.
- M. T. W. Lee, A. Maruani, J. R. Baker, S. Caddick and V. Chudasama, Chem. Sci., 2016, 7, 799–802 RSC.
- H. Pye, M. A. Butt, H. W. Reinert, A. Maruani, J. P. M. Nunes, J. S. Marklew, M. Qurashi, L. Funnell, A. May, I. Stamati, R. Hamoudi, J. R. Baker, M. E. B. Smith, S. Caddick, M. P. Deonarain, G. Yahioglu, V. Chudasama and L. B. Lovat, Photochem. Photobiol. Sci., 2016, 15, 1227–1238 RSC.
- J. D. Bargh, A. Isidro-Llobet, J. S. Parker and D. R. Spring, Chem. Soc. Rev., 2019, 48, 4361–4374 RSC.
- J. D. Bargh, S. J. Walsh, A. Isidro-Llobet, S. Omarjee, J. S. Carroll and D. R. Spring, Chem. Sci., 2020, 11, 2375–2380 RSC.
- C. Bahou, E. A. Love, S. Leonard, R. J. Spears, A. Maruani, K. Armour, J. R. Baker and V. Chudasama, Bioconjugate Chem., 2019, 30, 1048–1054 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob00907e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |