Retracted Article: Convenient synthesis of pyrimidine 2′-deoxyribonucleoside monophosphates with important epigenetic marks at the 5-position†
Received
28th April 2020
, Accepted 23rd June 2020
First published on 23rd June 2020
Abstract
Methyl groups of thymine and 5-methylcytosine (5mC) bases in DNA undergo endogenous oxidation damage. Additionally, 5mC residues can be enzymatically deaminated or oxidized through either genetic alterations or the newly identified epigenetic reprogramming pathway. Several methods have been developed to measure the formation of modified DNA nucleobases including 32P-postlabeling. However, the postlabeling method is often limited by the absence of authentic chemical standards. The synthesis of monophosphate standards of nucleotide oxidation products is complicated by the presence of additional functional groups on the modified bases that require complex protection and deprotection strategies. Due to the emerging interest in the pyrimidine oxidation products, the corresponding protected 3′-phosphoramidites needed for solid-phase oligonucleotide synthesis have been reported, and several are commercially available. We report here an efficient synthesis of 3′-monophosphates from 3′-phosphoramidites and the subsequent enzymatic conversion of 3′-monophosphates to the corresponding 5′-monophosphates using commercially available enzymes.
Introduction
The importance of DNA oxidation and hydrolysis in the development of human diseases is increasingly recognized.1–3 Recently a biochemical pathway has been identified in which 5-methylcytosine (5mC), an important component of the epigenetic landscape in higher organisms, can be enzymatically oxidized to the corresponding 5-hydroxymethyl (5hmC), formyl (5foC) and carboxyl (5caC) analogs, mediated by ten eleven translocation (TET) enzymes (Scheme 1).4–9 This epigenetic reprogramming pathway is essential to normal development and differentiation, yet, defective in most human cancer cells.4–11 The spontaneous hydrolysis of 5mC to T at methylated CpG islands has long been considered the mechanism underlying the single most frequent type of single-base mutation found in human cancer cells.12–14 This pathway might also include enzymatic deamination to the corresponding uracil analogs followed by glycosylase removal and DNA repair (Scheme 1).8,9 In addition to the active demethylation pathway, an alternative proposal for restoration of cytosine (C) involves the direct C–C bond cleavage at the 5-position to remove the substitution.15 The potential coexistence of the enzymatic 5mC modification pathway and established endogenous DNA damage and repair pathways creates significant analytical challenges.
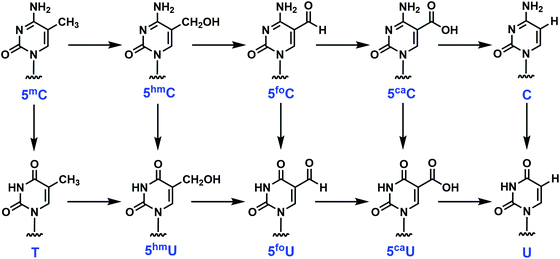 |
| | Scheme 1 Endogenous chemical or enzymatic conversions among pyrimidine analogs. 5-Methylcytosine (5mC), which is formed in DNA during epigenetic programming, can be oxidized to several 5-substituted cytosine derivatives. These cytosine derivatives can also undergo deamination to form uracil derivatives. The enzyme-mediated oxidation and deamination of 5mC is part of an epigenetic reprogramming pathway in mammalian cells. 5mC, 5-methylcytosine; 5hmC, 5-hydroxymethylcytosine; 5foC, 5-formylcytosine; 5caC, 5-carboxylcytosine; 5hmU, 5-hydroxymethyluracil; 5foU, 5-formyluracil; 5-caU, 5-carboxyluracil. | |
Multiple methods have been developed to quantify the abovementioned epigenetic modifications including chromatographic separation by HPLC, GC and TLC and identification and quantification by mass spectrometry, UV detection or 32P-labeling. The 32P-labeling approach has proven to be very sensitive for measuring a multitude of DNA damage products;16–19 however, this approach is often limited by the absence of authentic standards. While MS methods have emerged as extremely powerful and sensitive approaches to the measurement and unequivocal identification of DNA damage products, 32P-labeling strategies provide an approach for following sequential conversions of specific nucleotides through multiple chemical or enzymatic pathways in complicated matrices including intact cells and cell extracts and at specific sequence positions within the oligonucleotides.
In the case of the modified pyrimidines examined here, chemical synthesis of the 2′-deoxynucleosides can be challenging, and conversion to the corresponding monophosphates is complicated by the additional functional groups present on the modified bases that must be selectively protected prior to chemical phosphorylation.20–23 Due to the growing interest in the modified pyrimidines, several groups have reported the synthesis of phosphoramidite derivatives needed for the solid-phase assembly of oligonucleotides containing these modifications (Table 1), and some of these modified phosphoramidites are now commercially available. The investment made in developing methods for the protection and deprotection of the 3′-phosphoramidites could be leveraged to provide a convenient source for both the corresponding 3′- and 5′-monophosphates. Here we report a novel and versatile synthesis of 3′-dNMPs using nucleoside phosphoramidites as building blocks. With the availability of fully characterized dNMPs, not only the composition of synthetic oligonucleotides can be determined, but also the 32P-labeling strategy can be applied to this group of biologically important pyrimidine analogs.
Table 1 Characterization of 3′-dNMPs
| 3′-dNMPs |
Elemental composition [M − H]− |
Exact mass, m/z [M − H]− |
Measured mass, m/z [M − H]− |
λ
max (nm) |
Extinction coefficient (103 M−1 cm−1) |
Retention timea (min) |
|
The retention times were obtained on an Aquasil C18 column using an ammonium acetate (20 mM, pH 5.5) – methanol solvent system.
|
| 3′-cadUMP |
C10H12N2O10P− |
351.0230 |
351.0236 |
271 |
12.3 ± 0.5 |
3.25 |
| 3′-cadCMP |
C10H13N3O9P− |
350.0389 |
350.0390 |
280 |
10.6 ± 0.5 |
3.52 |
| 3′-dCMP |
C9H13N3O7P− |
306.0491 |
306.0490 |
269 |
9.7 ± 0.7 |
5.78 |
| 3′-hmdCMP |
C10H15N3O8P− |
336.0597 |
336.0586 |
274 |
12.5 ± 0.7 |
6.22 |
| 3′-dUMP |
C9H12N2O8P− |
307.0331 |
307.0345 |
261 |
13.7 ± 0.8 |
6.71 |
| 3′-hmdUMP |
C10H14N2O9P− |
337.0437 |
337.0444 |
263 |
11.9 ± 0.5 |
6.97 |
| 3′-mdCMP |
C10H15N3O7P− |
320.0648 |
320.0643 |
277 |
10.2 ± 0.3 |
10.17 |
| 3′-fodUMP |
C10H12N2O9P− |
335.0280 |
335.0289 |
275 |
17.2 ± 0.3 |
10.88 |
| 3′-dTMP |
C10H14N2O8P− |
321.0488 |
321.0493 |
267 |
10.3 ± 0.6 |
11.47 |
| 3′-fodCMP |
C10H13N3O8P− |
334.0440 |
334.0424 |
283 |
14.1 ± 0.3 |
13.27 |
Results and discussion
Synthesis of 3′-dNMPs
Chemical coupling of phosphoramidite with 2-cyanoethanol.
The facile activation of phosphoramidite monomer in the presence of a weakly acidic azole catalyst and the subsequent trapping of the resulting tetrazolyl phosphoramidite intermediate with different nucleophiles set the stage for a straightforward synthesis of phosphite triester. Indeed, when mixed with 1H-tetrazole in CH3CN, the diisopropylamino group of phosphoramidite was readily protonated and rapidly displaced by the hydroxyl group of 2-cyanoethanol despite the reduced nucleophilicity due to the presence of the electron-withdrawing cyano group. This reaction proceeds extremely fast: complete consumption of the phosphoramidite starting material was observed after 15 to 30 min at room temperature (monitored by TLC). Ease of purification is another important feature of this coupling reaction: after regular aqueous workup, the organic solvent was removed under reduced pressure to afford the desired phosphite triester as a white to pale-yellow foam. These crude products were subjected to the next step without further purification.
Oxidation and detritylation.
In modern solid-phase oligonucleotide synthesis, oxidation of phosphite triester is usually accomplished by aqueous iodine in combination with pyridine. However, for solution-phase reactions, separation of byproduct from phosphate triester could be problematic. We thus seek a “traceless” yet mild oxidizing agent. H2O2 in CH3CN has been used to oxidize thymidine phosphoramidites to the corresponding phosphoramidates in quantitative yields. This method was optimized for our 3′-dNMPs synthesis. Deoxynucleoside-3′-phosphite triester was treated with 3% H2O2 in CH3CN (v/v = 1/5) for 15 min at room temperature before the solvent was evaporated in vacuo, which led to the formation of 3′-phosphate triester without further purification. The oxidation step allowed good functional group tolerance (Table 1) with full conversion within minutes. The subsequent detritylation was carried out in 80% acetic acid for about 30 min, again at room temperature. Removal of the solvent provided deoxynucleoside-3′-phosphate triester with a free 5′-hydroxyl group. The decent solubility of these compounds in organic solvent allows them to be chromatographically purified on silica gel using CH2Cl2 and methanol.
The formation of deoxynucleoside-3′-phosphate triester with a free 5′-hydroxyl group was further confirmed by NMR analysis. 1H NMR showed complete disappearance of signals corresponding to trityl group protons, and 31P signals shifted from around 148 ppm (doublet) in phosphoramidites to −3.5 ppm (singlet) in deoxynucleoside-3′-phosphate triesters.
Deprotection strategies.
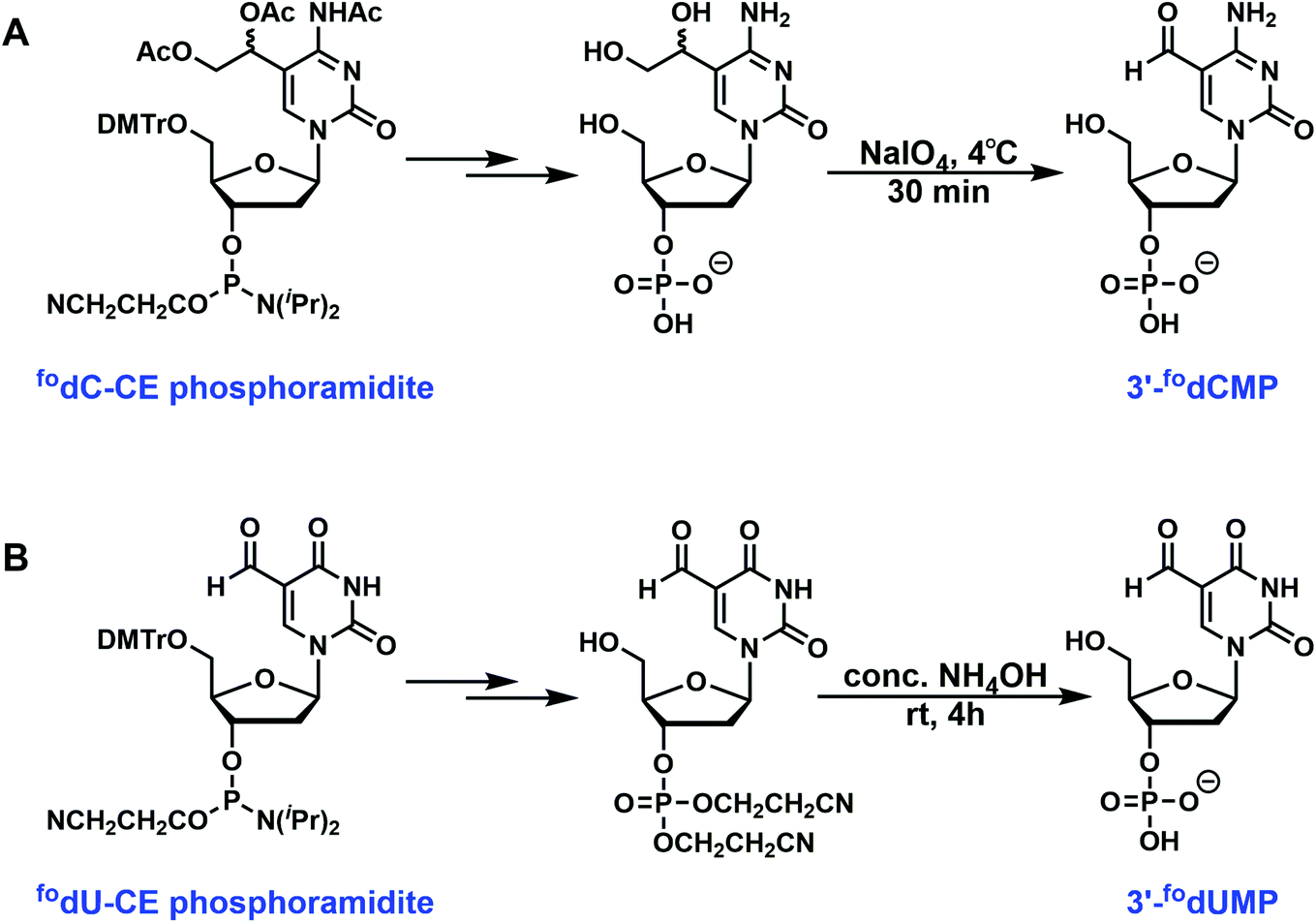
Removal of cyanoethyl groups from phosphate and removal of protecting group from bases can be achieved in concentrated aqueous NH4OH at elevated temperature (60 °C). The solvent was then evaporated under reduced pressure, and the crude product was ready for purification. However, for several phosphoramidites, the presence of special protecting groups or sensitive functionality requires alternative or additional deprotection strategies. For 5-carboxy-dC- and dU-CE phosphoramidites, the carboxylate exists as either an ethyl or a methyl ester. Deprotections were carried out in 0.4 M methanolic NaOH (methanol/water = 4/1) for 17 h at room temperature before being neutralized with acetic acid. Commercially available 5-formyl-dC-CE phosphoramidite contains acetylated diol instead of a formyl group. NH4OH treatment removes the acetyl group to generate 1,2-diol, which requires further oxidative cleavage by NaIO4 to unmask the formyl functionality. This has been performed with 50 mM (final concentration) NaIO4 solution at 4 °C for 30 min. The reaction was then quenched with the addition of excess ethylene glycol. The 5-formyl-dU-CE phosphoramidite synthesized in our lab bears unprotected formyl group, which was compatible with concentrated NH4OH for 4 h at room temperature.
Purification.
Purification for most of the 3′-dNMPs was quite straightforward. The crude product was dissolved in water, washed with ethyl acetate to remove the side product from deprotection. The aqueous layer was then concentrated to provide the desired 3′-dNMP. For 3′-cadCMP, 3′-cadUMP and 3′-fodCMP, HPLC purification was performed to separate 3′-monophosphate from the inorganic salt generated during deprotection step (see “Materials and methods” section for HPLC conditions).
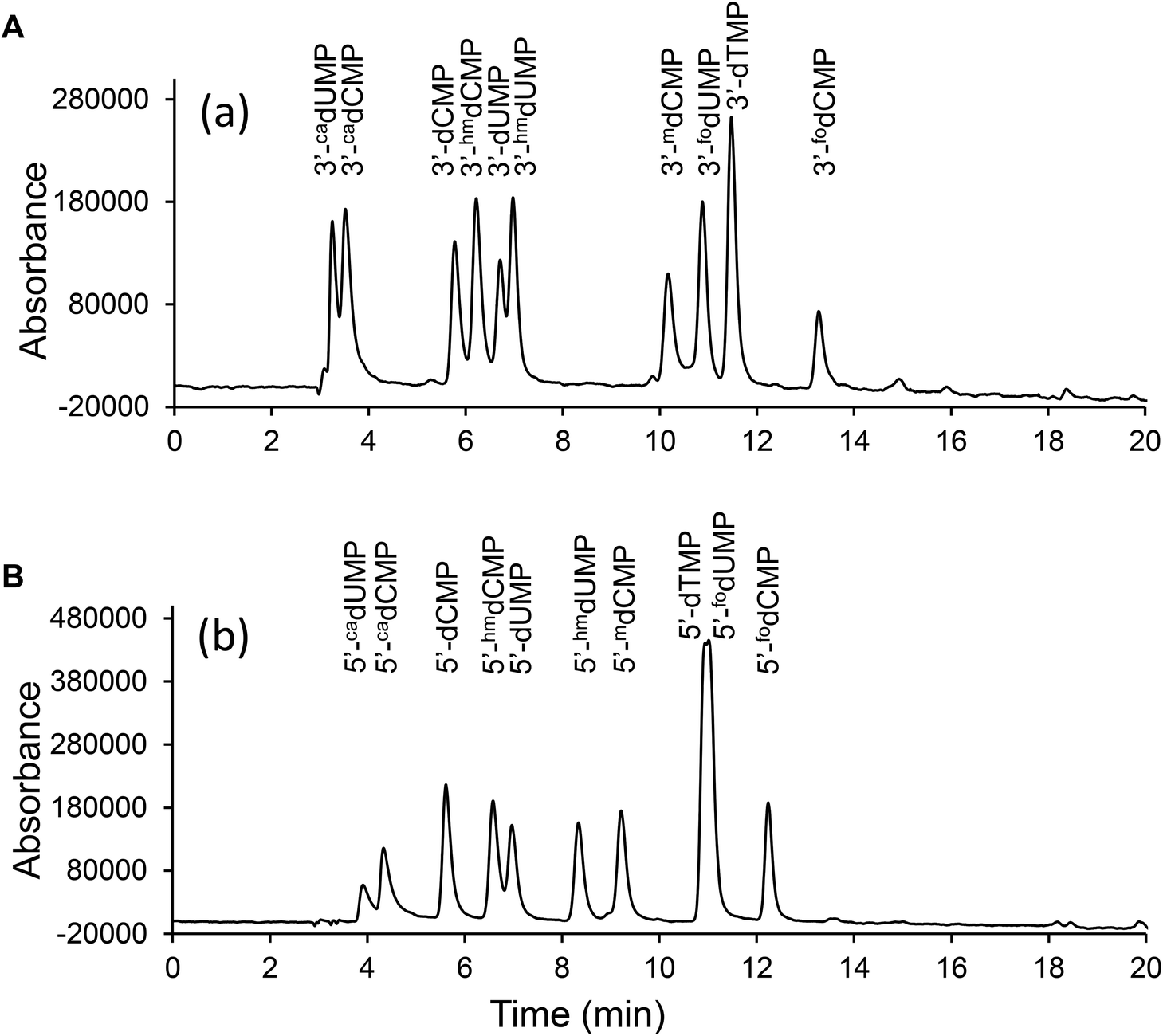
The aforementioned synthetic protocols of 3′-dNMPs have several beneficial features. First of all, phosphoramidites are either commercially available or easily accessible, which enables a straightforward construction of 3′-dNMPs. Secondly, already-existing protecting groups in phosphoramidites prevent undesired side reactions. Reactions were easily monitored by TLC, and the 3′-dNMPs were obtained in 70–90% yields with minimal purification (most synthesis only requires one chromatographic isolation). The method is, therefore, suitable for large scale synthesis for a broad class of compounds. Corresponding MS (Table 1), UV (Table 1), HPLC (Table 1, Fig. 3 and S1†) and NMR (ESI†) data are provided for each of the ten 3′-monophosphates prepared here.
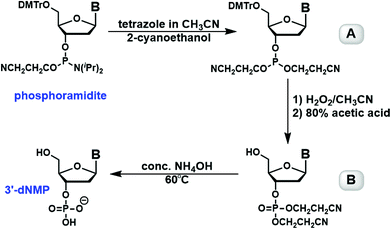 |
| | Fig. 1 Conversion of a phosphoramidite to a 3′-dNMP. The phosphoramidite is coupled with 2-cyanoethanol in the presence of tetrazole in CH3CN to generate the phosphite triester A, which is then oxidized with H2O2 and acetic acid to create the phosphate triester B. Alkaline deprotection of B leads to the formation of the corresponding 3′-dNMP. | |
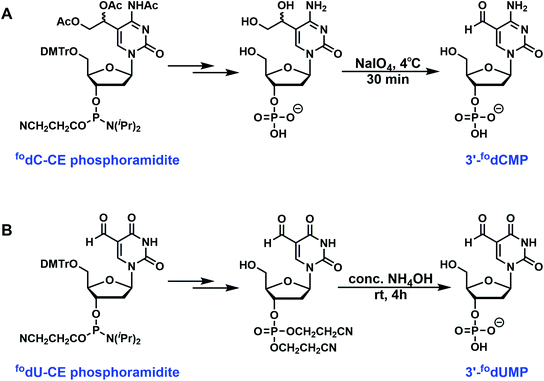 |
| | Fig. 2 Special deprotection strategies. (A) Commercially available 5-formyl-dC-CE phosphoramidite contains acetylated diol instead of a formyl group. NH4OH treatment removes the acetyl group to generate 1,2-diol, which requires further oxidative cleavage by NaIO4 to unmask the formyl functionality. (B) The 5-formyl-dU-CE phosphoramidite synthesized in our lab bears unprotected formyl group, which was compatible with concentrated NH4OH for 4 h at room temperature. | |
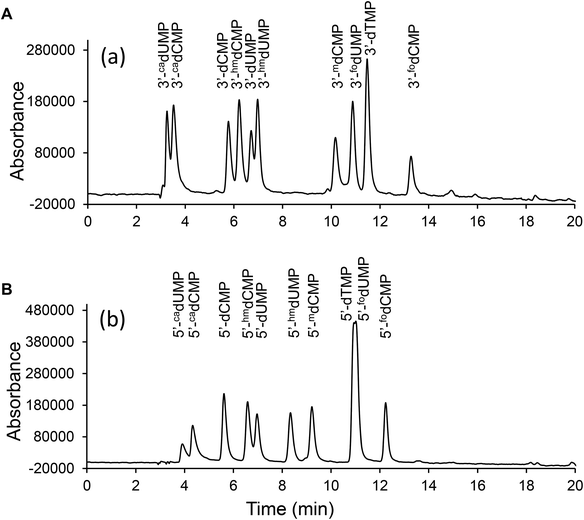 |
| | Fig. 3 HPLC separation of the 3′-dNMPs and 5′-dNMPs. The HPLC chromatograms of (A) 3′-dNMPs and (B) 5′-dNMPs were obtained on an Aquasil C18 column using ammonium acetate (20 mM, pH 5.5) – methanol solvent system. The above chromatograms were obtained with UV absorbance at 275 nm. | |
Enzymatic conversion of 3′-dNMP to 5′-dNMP
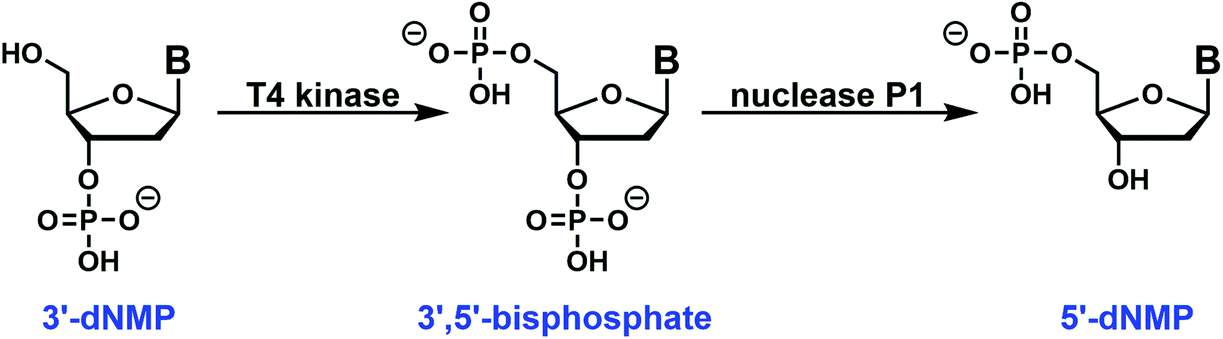
The easy access to the 3′-dNMPs made the facile synthesis of 5′-dNMPs possible. This enzymatic conversion was inspired by phosphoglycerate mutase, a glycolytic enzyme.24 It catalyzes the conversion of 3-phosphoglycerate to 2-phosphoglycerate through a 2,3-bisphosphoglycerate intermediate.24,25 In our two-enzyme coupled synthesis, the 3′-dNMP was first incubated with ATP and a T4 kinase lacking the 3′-phosphatase activity to generate the corresponding 3′,5′-bisphosphate (Fig. 4).26,27 Subsequently, the 3′,5′-bisphosphate was dephosphorylated by nuclease P1 to afford the 5′-dNMP.28 Both T4 kinase and nuclease P1 demonstrated excellent functional group tolerance as 5′-dNMPs carrying various substituents were generated in decent to good yields by this robust sequence (Table 2). Similar to the 3′-dNMPs, the 5′-dNMPs can be readily resolved by HPLC (Fig. 3).
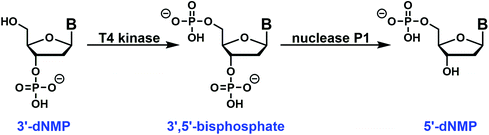 |
| | Fig. 4 Enzymatic conversion of a 3′-dNMP to a 5′-dNMP: a chemically synthesized 3′-dNMP is initially converted to the 3′,5′-bisphosphate with T4 kinase. Following HPLC purification, the dephosphorylation of the bisphosphate by Nuclease P1 results in the formation of 5′-dNMP. Substitution with 32P-enriched ATP in the T4 kinase step can be used to generate the corresponding labeled 5′-dNMP. | |
Table 2 Characterization of 5′-dNMPs
| 5′-dNMPs |
Yielda (%) |
Retention timeb (min) |
|
Yield for two steps starting from the 3′-dNMP.
The retention times were obtained on an Aquasil C18 column using an ammonium acetate (20 mM, pH 5.5) – methanol solvent system.
|
| 5′-cadUMP |
80.2 |
3.91 |
| 5′-cadCMP |
79.2 |
4.33 |
| 5′-dCMP |
53.2 |
5.61 |
| 5′-hmdCMP |
85.7 |
6.58 |
| 5′-dUMP |
54.0 |
6.97 |
| 5′-hmdUMP |
81.5 |
8.34 |
| 5′-mdCMP |
56.2 |
9.22 |
| 5′-dTMP |
83.3 |
10.94 |
| 5′-fodUMP |
59.3 |
11.01 |
| 5′-fodCMP |
31.5 |
12.24 |
Determination of synthetic oligonucleotide composition using dNMP standards
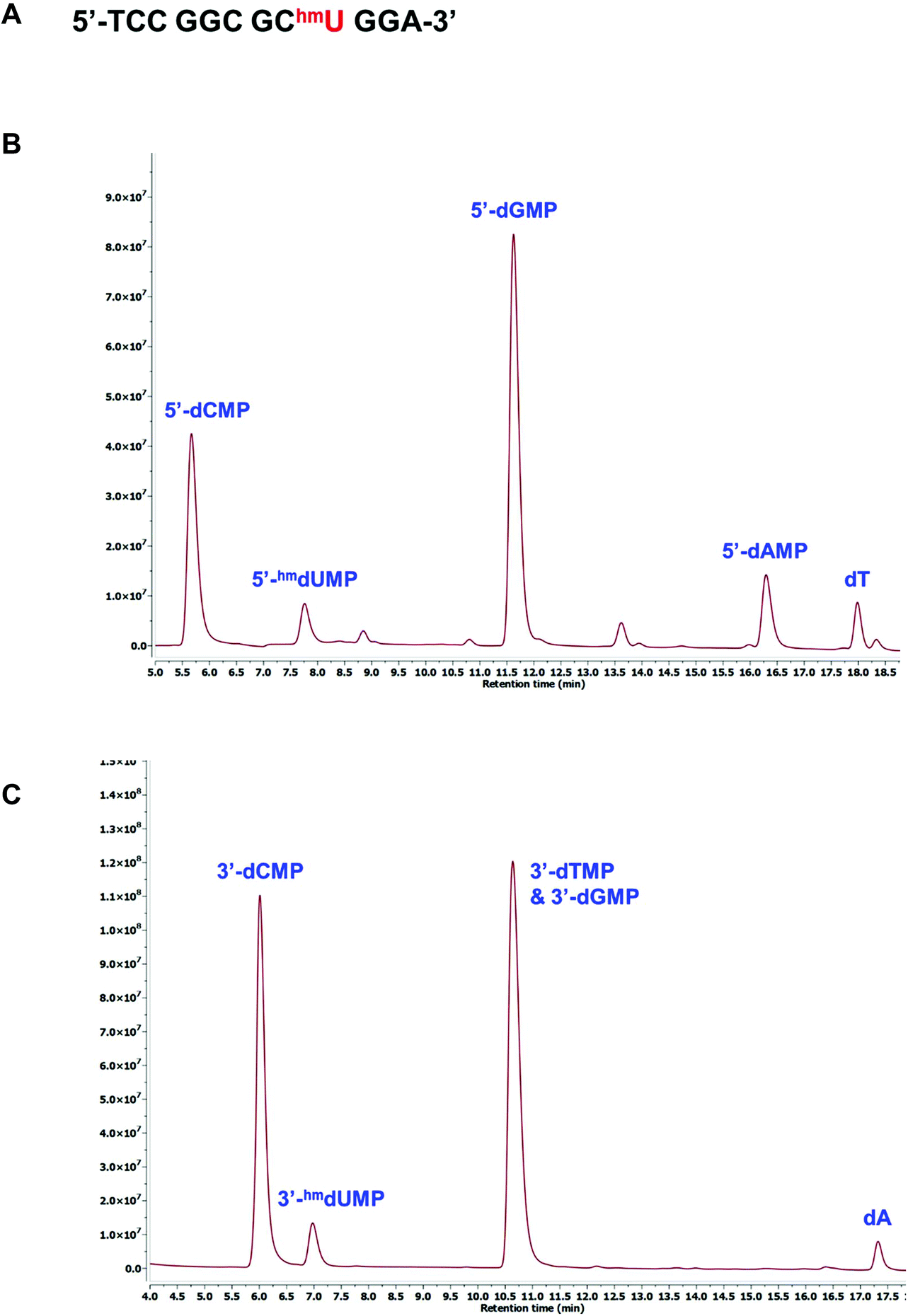
The presence of modified pyrimidines in synthetic oligonucleotides can be confirmed using either the 3′- or 5′-dNMP standards. A 12mer oligonucleotide containing an internal 5hmU residue was generated by standard solid-phase DNA synthesis (Fig. 5A). This oligonucleotide was incubated with snake venom phosphodiesterase I, an exonuclease capable of successively hydrolyzing 5′-dNMPs from the 3′-terminus of a DNA sequence.29,30 The digested sample was then injected onto HPLC for further analysis. Comparison with the 5′-dNMP standards indicated that in addition to G, T, C and A, 5hmU was also present in the sequence (Fig. 5B). The composition of the oligonucleotide can also be determined by treating the same 12mer with bovine spleen phosphodiesterase II, an exonuclease that can remove 3′-dNMPs sequentially from the 5′-terminus.31 HPLC analysis suggested the formation of 3′-hmdUMP along with other nucleoside monophosphates (Fig. 5C).
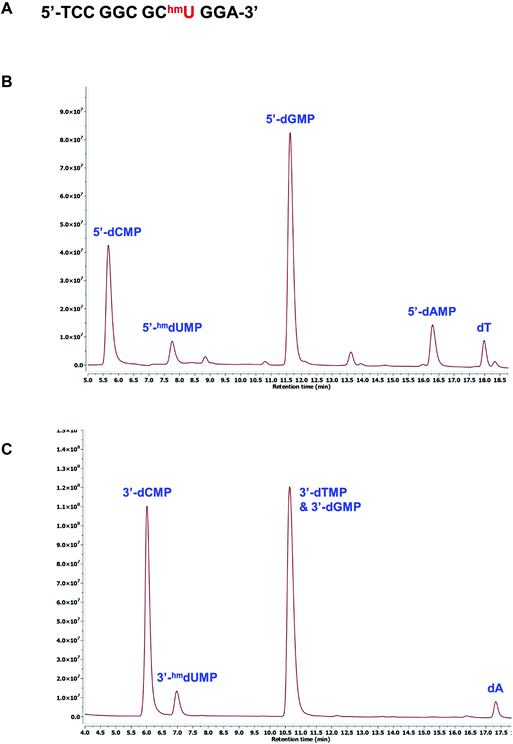 |
| | Fig. 5 Confirmation of 5hmU in an oligonucleotide sequence. (A) Sequence of the single-stranded oligonucleotide. HPLC chromatograms of the dNMPs resulting from the digestion of 12mer with (B) snake venom phosphodiesterase I, or (C) bovine spleen phosphodiesterase II. | |
Probing conversions of nucleobase using dNMP standards
The availability of the dNMP standards should facilitate experiments probing both chemical and enzymatic conversions of target residues in oligonucleotides. We demonstrate this by examining the chemical reactivity of 5hmC in a duplex oligonucleotide. Bisulfite sequencing has become extremely powerful in distinguishing cytosine from 5mC residues and thus tracking methylation patterns in genomic DNA.32–34 However, 5mC and 5hmC cannot be distinguished by this method.35 It has been reported that 5hmC can be oxidized to 5foC by KRuO4.36–38 Subsequently, bisulfite treatment can convert 5foC to uracil (U).39 This two-step sequence could serve as an alternative approach in detecting 5hmC level (Fig. 6A). In order to evaluate this conversion sequence directly, a 12mer oligonucleotide containing a 5hmC residue at the 5′-position was 32P-labeled, annealed to the unlabeled 24mer complementary strand and then ligated to an unlabeled 12mer oligonucleotide to form a duplex (Fig. 6B). A fraction of the duplex was digested with DNase I and snake venom phosphodiesterase I. The resulting 5′-dNMPs were then resolved by HPLC with an in-line radio-detector. The 32P label was found exclusively in the 5′-hmdCMP (Fig. 6C). A second fraction of the duplex was treated with KRuO4 and digested as described above. The formation of 5foC was readily detected (Fig. 6D). The complete conversion of 5hmC to U was observed when the duplex was incubated with KRuO4 followed by bisulfite treatment and enzymatic digestion. In this case, the radioactivity was detected in 5′-dUMP exclusively (Fig. 6E). While analytical methods have been used to confirm the conversion of 5hmC to U,36 ours is the first study to directly follow the transformations in on oligonucleotide with 32P-labeling. The selective 32P-labeling of a target base, followed by enzymatic digestion and HPLC analysis, provides a method to monitor either enzymatic or chemical conversions, even with crude cellular extracts.
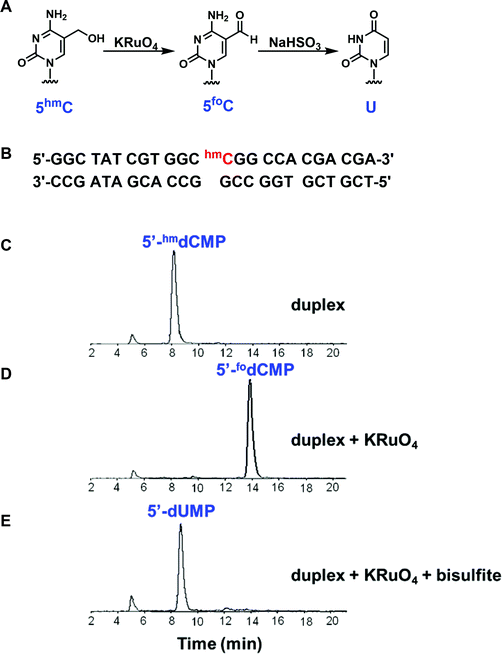 |
| | Fig. 6 Conversion of 5hmC in an oligonucleotide sequence. (A) Chemical modifications of the 5hmC-containing oligonucleotide. (B) Sequence of the duplex. HPLC chromatograms of the 5′-dNMPs resulting from the digestion of (C) duplex, (D) duplex treated with KRuO4, and (E) duplex treated with KRuO4 and bisulfite. The signals were detected by a radio-detector. | |
Conclusions
Phosphoramidites prepared for the synthesis of oligonucleotides containing biologically important modified bases can be conveniently converted to the corresponding 3′-dNMPs. 5′-dNMPs are then obtained through enzyme-mediated phosphorylation and dephosphorylation. Authentic 3′- and 5′-dNMP standards, such as those described here, are essential for the validation of highly sensitive analytical methods using 32P-labeling. We have demonstrated this approach by following the conversion of a selectively-labeled 5hmC residue in an oligonucleotide to 5foC, and then on to U, using Rh oxidation and bisulfite conversion methods currently employed to sequence 5mC and 5hmC residues in DNA. This approach can be extended to study future base modification strategies needed to follow additional base conversions in DNA, including those involved with epigenetic reprogramming.
Materials and methods
Materials
Reagent grade solvents including dichloromethane, methanol, and ethyl acetate were purchased from Fisher Scientific (Pittsburgh, PA) and used without further purification. Anhydrous grade solvents including tetrahydrofuran, pyridine, trimethylamine, and acetonitrile were purchased from Sigma-Aldrich (St Louis, MO) and used as received unless otherwise noted. Di-tert-butylsilyl bis(trifluoromethanesulfonate), dimethylformamide, 5-iodo-2′-deoxyuridine, 4,4′-dimethoxytrityl chloride, tris(dibenzylideneacetone)dipalladium(0), triphenylphosphine, toluene, imidazole, tributyltin hydride, HF-pyridine, diisopropylethylamine, 2-cyanoethanol, 2-cyanoethyl N,N,N′,N′-tetraisopropylphosphordiamidite, and carbon monoxide were purchased from Sigma-Aldrich (St Louis, MO). Commercially available phosphoramidites and sublimed 1H-tetrazole in anhydrous acetonitrile were purchased from Glen Research (Sterling, VA). Thin layer chromatography (TLC) was performed on precoated silica gel 60 F254, 5 cm × 20 cm, 250 μM thick plates purchased from EMD (Gibbstown, NJ). Snake venom phosphodiesterase I and bovine spleen phosphodiesterase II were purchased from Worthington Biochemical. T4 polynucleotide kinase and T4 DNA ligase were purchased from New England Biolabs. Nuclease P1 was purchased from US Biologicals.
1H, 13C, and 31P NMR spectra were obtained using a Bruker Ultrashield 300 MHz spectrometer (Manning Park, MA). HPLC analysis was performed with a ThermoFinnigan (Waltham, MA) Surveyor HPLC system with a photodiode array and a single quad mass spectrometer detector. For the analysis of 32P-labeled monophosphates, a similar HPLC system was interfaced with a Berthold Technologies FlowStar LB513 radioactivity detector (Wildbad, Germany). Oligonucleotides were synthesized with a Millipore Expedite nucleic acids synthesis system (Billerica, MA). High-resolution mass spectra were obtained with either a Waters Micromass Q-tof Ultima (Milford, MA) or a Thermo Scientific Q-Exactive Hybrid Quadrupole Orbitrap (Waltham, MA).
General procedures for deoxynucleoside-3′-monophosphate synthesis
Coupling.
To a solution of phosphoramidite in anhydrous CH3CN were added 0.45 M tetrazole in CH3CN (1.5 equiv.) and 2-cyanoethanol (1.5 equiv.). The reaction was stirred at room temperature and monitored by TLC. Upon completion (usually 15 to 30 min), the reaction mixture was diluted with ethyl acetate with 1% TEA. The organic phase was washed with 5% NaHCO3, water, brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to afford phosphite triester A (Fig. 1).
Oxidation and detritylation.
Phosphite triester A was dissolved in a mixture of CH3CN and 3% H2O2 (v/v = 5/1), stirred at room temperature for 15 min before the solvent was evaporated under reduced pressure. The residue was redissolved in 80% acetic acid, kept at room temperature for another 30 min before the solvent was evaporated. The crude product was purified by column chromatography to afford phosphate triester B.
Deprotection.
Phosphate triester B was treated with conc. NH4OH at 60 °C overnight. The solvent was evaporated under reduced pressure. The residue was redissolved in water, and extracted with ethyl acetate 3 times. The aqueous layer was concentrated in vacuo to afford the desired deoxynucleoside-3′-monophosphate.
Special deprotection strategies
5-Carboxyl-2′-deoxycytidine (5cadC) and 5-carboxyl-2′-deoxyuridine (5cadU).
For 5cadC or 5cadU, intermediate B was treated with 0.4 M of NaOH in methanol/water (v/v = 4/1) at room temperature for 17 h before it was neutralized with acetic acid. The solvent was then evaporated under reduced pressure. The residue was redissolved in water and extracted with ethyl acetate 3 times. The aqueous layer was concentrated in vacuo to afford 3′-cadCMP or 3′-cadUMP.
5-Formyl-2′-deoxycytidine (5fodC).
After NH4OH deprotection, the 1,2-diol requires further oxidation with NaIO4 to unveil the aldehyde functional group (Fig. 2A). It was dissolved in cold distilled water (4 °C) and treated with an equal volume of cold (4 °C) 100 mM of NaIO4 solution for 30 min at 4 °C before it was quenched with 8 equiv. of ethylene glycol with respect to NaIO4.
5-Formyl-2′-deoxyuridine (5fodU).
The NH4OH deprotection was carried out at room temperature for 4 h before the solvent was evaporated under reduced pressure (Fig. 2B). This mild deprotection allows the convenient isolation of sensitive 3′-fodUMP.
Enzymatic conversion of a 3′-dNMP to the corresponding 5′-dNMP
Each 3′-dNMP was enzymatically converted to the corresponding 5′-dNMP using a series of two enzymatic reactions. In the first step, the 3′-dNMP was converted to the 3′,5′-bisphosphate. In this reaction, 400 nmol of the 3′-dNMP was combined with 20 μL 1 M Tris-HCl, pH 7.0, 20 μL 0.1 M MgCl2, 20 μL 50 mM DTT, 40 μL 20 mM ATP, and 20 μL T4 kinase (10 U μL−1) lacking 3′-phosphatase activity in a total volume of 200 μL as described previously.26,27 The reaction mixture was incubated at 37 °C, and progress was followed by HPLC analysis of a 5 μL aliquot of the reaction. In some cases, the 3′-dNMP co-eluted with ATP. However, the ATP can be reduced by incubation with 1 μL of 1 U μL−1 apyrase for 10 min prior to HPLC analysis.
The 3′,5′-bisphosphate generated in the above reaction was purified by HPLC as described below and converted to the 5′-dNMP with nuclease P1 as described previously.28 HPLC fractions containing the bisphosphate were combined and dried. The bisphosphate was incubated in water (50 μL), buffer (25 μL 0.25 M NaOAc, pH 5.0, 15 μL 2 mM ZnCl2) and nuclease P1 (10 μL 5 μg μL−1) at 37 °C. The reaction was monitored by HPLC, and the final product 5′-dNMP isolated by HPLC and characterized by UV and mass spectrometry as described below.
High-resolution mass spectrometry (HRMS) analysis.
The 3′-dNMPs were characterized by high-resolution mass spectrometry on a Thermo Scientific Q-Exactive Hybrid Quadrupole Orbitrap mass spectrometer operating in the negative ion mode. The 3′-dNMPs were dissolved in acetonitrile/water (v/v = 1/1) to a final concentration of 20 μM and directly infused into the ESI source at a rate of 5 μL min−1. The source temperature was at 320 °C and electrospray high voltage was −3.5 kV. In each case, the measured mass was within 5 ppm of the calculated (theoretical) mass. The results are given in Table 1.
HPLC analysis and purification.
The 3′-dNMPs, 5′-dNMPs and 3′,5′-bisphosphates were analyzed by LC-ESI-MS on Surveyor MSQ and were chromatographed by Surveyor LC system (Thermo Finnigan, Waltham, MA) with a flow of 1 mL min−1 over an Aquasil C18 column (Thermo Scientific, Waltham, MA). Eluting buffers were buffer A (20 mM NH4OAc, pH 5.5) and buffer B (methanol). The gradient was 0 → 10 min, 100% → 90% buffer A, 0 → 10% buffer B; 10 → 15 min, 90% → 80% buffer A, 10% → 20% buffer B; 15 → 21 min, 80% → 52% buffer A, 20 → 48% buffer B; 21 → 22 min, 52% → 40% buffer A, 48 → 60% buffer B; 22 → 24 min, 40% buffer A, 60% buffer B; 24 → 25 min, 40% → 100% buffer A, 60% → 0% buffer B; 25 → 35 min, 100% buffer A and 0% buffer B. The elution was scanned from 210 nm to 600 nm (Surveyor PDA Detector, Thermo Finnigan, Waltham, MA). The chromatographic eluent was directly injected into the ion source with a 1/5 splitting. Ions were scanned by use of a negative polarity mode over a full-scan range of m/z 100–500.40 The separation of the 3′-dNMPs and 5′-dNMPs is shown in Fig. 3, and the retention times are given in Tables 1 and 2. The nucleotides were detected with a diode array detector that provided the UV spectrum of each analyte.
Determination of extinction coefficient
Determination of 3′-dNMP concentration by 1H NMR.
In a typical measurement, 3′-dNMP was dissolved in 700 μL of 99.96% D2O. To this solution was added 1 or 2 μL of 99.9% DMSO as an internal standard. The sample was then transferred to a Wilmad 528 NMR tube capped with a rubber septum and sealed with parafilm. Proton NMR spectra were recorded on a Bruker Ultrashield 300 MHz spectrometer using a published protocol with modifications. In brief, spectra were acquired with 64 scans, 90° pulse, 35 s relaxation delay, SW = 6000 Hz. Integration was carried out using Bruker XWin NMR software. For 3′-dNMPs, base proton(s) and H1′ signals were used for quantitative comparison; thus, several measurements contribute to the average. The DMSO methyl integral was set to be 1.000, the averaged relative intensity of 3′-dNMP peaks gave the concentration of 3′-dNMP.
Determination of extinction coefficient.
UV absorbance was recorded on a Cary 100 Bio UV/Vis spectrophotometer blanked against 100 mM phosphate buffer (pH 7). NMR samples were diluted with 100 mM phosphate buffer (pH 7) so that Amax readings were between 0.5 and 1.0 to ensure high sensitivity. UV measurements were performed on the same day as NMR measurements to minimize the possibility of evaporation or sample degradation. The extinction coefficient was calculated using eqn (1):| | 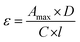 | (1) |
where ε is the extinction coefficient at optimal wavelength, Amax is the absorbance at optimal wavelength, D is the dilution factor, C is the concentration of 3′-dNMP, and l is the path length of the sample, which is 1 cm.
Oligonucleotide synthesis and purification.
Oligonucleotides were prepared by solid-phase synthesis method as described previously.41–43 Following synthesis and deprotection, they were purified by HPLC using Hamilton PRP-1 column and a gradient of 0 to 25% CH3CN in 10 mM of potassium phosphate buffer (pH 6.8). Oligonucleotides were then detritylated with 80% aqueous acetic acid at room temperature for 30 min. Following detritylation, they were purified by HPLC using a Vydac C18 column and a gradient of 0 to 70% CH3CN in water. Oligonucleotide purity was examined by MALDI-TOF.44
Exonuclease digestion.
A 12mer oligonucleotide (2 nmol) containing an internal 5hmU residue (5′-TCC GGC GChmU GGA-3′) was treated with snake venom phosphodiesterase I in a buffer containing 25 mM Tris-HCl (pH 8), 5 mM MgCl2, and 50 mM NaCl. The sample was incubated at 37 °C for 1 h before being injected onto HPLC and analyzed as described above. Similarly, the 12mer was also incubated with bovine spleen phosphodiesterase II in a buffer solution containing 50 mM Tris-HCl (pH 7.5) and 5 mM MgCl2. The sample was kept at 37 °C for 30 min before being injected onto HPLC and analyzed as described above.
Ligation and duplex formation.
A 12mer oligonucleotide containing 5hmC at the 5′-end (5′-hmCGG CCA CGA CGA-3′) was 32P-labeled with T4 polynucleotide kinase and [γ-32P]-ATP. The 32P-labeled oligonucleotide was isolated with a spin column and then annealed with a 24mer complementary strand (Fig. 5B). The 32P-labeled strand of the duplex was then ligated with another 12mer oligonucleotide (5′-GGC TAT CGT GGC-3′) using T4 DNA ligase to generate the 24mer duplex.
Conversion of 5hmC to 5foC in oligonucleotide.
The 24mer oligonucleotide duplex (83 pmol) described above, containing a unique labeled 5hmC, was denatured by heating with 40 mM NaOH at 37 °C for 30 min and immediately cooled to 0 °C in ice. The denatured oligo was incubated with KRuO4 (0.53 mM) in an alkaline solution for 2 h at 0 °C as described before.36–38 The oligonucleotide was then purified with a Sephadex G50 spin column, enzymatically digested to the corresponding 5′-dNMPs using DNase I and snake venom phosphodiesterase I. The liberated 5′-dNMPs were mixed with unlabeled 5′-dNMP standards and separated by HPLC with both UV and radioactive detectors.
Conversion of 5hmC to U in oligonucleotide.
After being treated with KRuO4, the oligonucleotide was incubated in 1.5 M sodium bisulfite and heated at 95 °C for 5 min, followed by incubation at 60 °C for 3 h in the dark as described before.45,46 The oligo was then purified by the spin column, digested and analyzed by HPLC as described above.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported in part by start-up funding from VCU (to Y. C.) and from NSF (CHE-1846785 to Y. C.).
References
- E. Mullaart, P. H. Lohman, F. Berends and J. Vijg, Mutat. Res., 1990, 237, 189–210 CAS.
- B. N. Ames, L. S. Gold and W. C. Willett, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 5258–5265 CrossRef CAS PubMed.
- P. Lonkar and P. C. Dedon, Int. J. Cancer, 2011, 128, 1999–2009 CrossRef CAS PubMed.
- S. Kriaucionis and N. Heintz, Science, 2009, 324, 929–930 CrossRef CAS PubMed.
- M. Tahiliani, K. P. Koh, Y. Shen, W. A. Pastor, H. Bandukwala, Y. Brudno, S. Agarwal, L. M. Iyer, D. R. Liu, L. Aravind and A. Rao, Science, 2009, 324, 930–935 CrossRef CAS PubMed.
- S. Ito, A. C. D'Alessio, O. V. Taranova, K. Hong, L. C. Sowers and Y. Zhang, Nature, 2010, 466, 1129–1133 CrossRef CAS PubMed.
- M. Munzel, D. Globisch and T. Carell, Angew. Chem., Int. Ed., 2011, 50, 6460–6468 CrossRef PubMed.
- Y. Fu and C. He, Curr. Opin. Chem. Biol., 2012, 16, 516–524 CrossRef CAS PubMed.
- L. Shen and Y. Zhang, Curr. Opin. Cell Biol., 2013, 25, 289–296 CrossRef CAS PubMed.
- C. Liu, L. Liu, X. Chen, J. Shen, J. Shan, Y. Xu, Z. Yang, L. Wu, F. Xia, P. Bie, Y. Cui, X. W. Bian and C. Qian, PLoS One, 2013, 8, e62828 CrossRef CAS PubMed.
- H. Yang, Y. Liu, F. Bai, J. Y. Zhang, S. H. Ma, J. Liu, Z. D. Xu, H. G. Zhu, Z. Q. Ling, D. Ye, K. L. Guan and Y. Xiong, Oncogene, 2013, 32, 663–669 CrossRef CAS PubMed.
- P. A. Jones, W. M. Rideout 3rd, J. C. Shen, C. H. Spruck and Y. C. Tsai, Bioessays, 1992, 14, 33–36 CrossRef CAS PubMed.
- S. Tornaletti and G. P. Pfeifer, Oncogene, 1995, 10, 1493–1499 CAS.
- V. Rusmintratip and L. C. Sowers, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 14183–14187 CrossRef CAS PubMed.
- A. Schon, E. Kaminska, F. Schelter, E. Ponkkonen, E. Korytiakova, S. Schiffers and T. Carell, Angew. Chem., 2020, 59, 5591–5594 CrossRef PubMed.
- N. J. Gorelick, Mutat. Res., 1993, 288, 5–18 CrossRef CAS PubMed.
- M. V. Reddy and K. Randerath, Carcinogenesis, 1986, 7, 1543–1551 CrossRef CAS PubMed.
- M. Zeisig, T. Hofer, J. Cadet and L. Moller, Carcinogenesis, 1999, 20, 1241–1245 CrossRef CAS PubMed.
- D. H. Phillips and V. M. Arlt, Nat. Protoc., 2007, 2, 2772–2781 CrossRef CAS PubMed.
- D. K. Rogstad, J. Heo, N. Vaidehi, W. A. Goddard 3rd, A. Burdzy and L. C. Sowers, Biochemistry, 2004, 43, 5688–5697 CrossRef CAS PubMed.
- K. Sato, W. Hirose and A. Matsuda, Curr. Protoc. Nucleic Acid Chem., 2008, 1.21.1–1.21.19 Search PubMed , chapter 1, unit 1 21.
- Q. Dai and C. He, Org. Lett., 2011, 13, 3446–3449 CrossRef CAS PubMed.
- A. S. Schroder, J. Steinbacher, B. Steigenberger, F. A. Gnerlich, S. Schiesser, T. Pfaffeneder and T. Carell, Angew. Chem., Int. Ed., 2014, 53, 315–318 CrossRef PubMed.
- L. A. Fothergill-Gilmore and H. C. Watson, Adv. Enzymol. Relat. Areas Mol. Biol., 1989, 62, 227–313 CAS.
- H. I. Fraser, M. Kvaratskhelia and M. F. White, FEBS Lett., 1999, 455, 344–348 CrossRef CAS PubMed.
- J. R. Lillehaug and K. Kleppe, Biochemistry, 1975, 14, 1221–1225 CrossRef CAS PubMed.
- E. Hilario, Mol. Biotechnol., 2004, 28, 77–80 CrossRef CAS PubMed.
- D. H. Phillips, A. Hewer and V. M. Arlt, Methods Mol. Biol., 2005, 291, 3–12 CAS.
-
M. Laskowski, in The Enzymes, ed. P. D. Boyer, Academic Press, 1971, vol. 4, pp. 313–328 Search PubMed.
- N. W. Ho and P. T. Gilham, Biochim. Biophys. Acta, 1973, 308, 53–58 CrossRef CAS.
-
A. Bernardi and G. Bernardi, in The Enzymes, ed. P. D. Boyer, Academic Press, 1971, vol. 4, pp. 329–336 Search PubMed.
- M. Frommer, L. E. McDonald, D. S. Millar, C. M. Collis, F. Watt, G. W. Grigg, P. L. Molloy and C. L. Paul, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 1827–1831 CrossRef CAS PubMed.
- A. Chatterjee, E. J. Rodger, I. M. Morison, M. R. Eccles and P. A. Stockwell, Methods Mol. Biol., 2017, 1537, 249–277 CrossRef CAS PubMed.
- F. Leti, L. Llaci, I. Malenica and J. K. DiStefano, Methods Mol. Biol., 2018, 1706, 233–254 CrossRef CAS PubMed.
- Y. Huang, W. A. Pastor, Y. Shen, M. Tahiliani, D. R. Liu and A. Rao, PLoS One, 2010, 5, e8888 CrossRef PubMed.
- M. J. Booth, M. R. Branco, G. Ficz, D. Oxley, F. Krueger, W. Reik and S. Balasubramanian, Science, 2012, 336, 934–937 CrossRef CAS PubMed.
- W. Mao, J. Hu, T. Hong, X. Xing, S. Wang, X. Chen and X. Zhou, Org. Biomol. Chem., 2013, 11, 3568–3572 RSC.
- P. Schuler and A. K. Miller, Angew. Chem., Int. Ed., 2012, 51, 10704–10707 CrossRef PubMed.
- L. Jin, W. Wang, D. Hu and J. Lu, Phys. Chem. Chem. Phys., 2014, 16, 3573–3585 RSC.
- O. Shimelis, X. Zhou, G. Li and R. W. Giese, J. Chromatogr. A, 2004, 1053, 143–149 CrossRef CAS PubMed.
-
M. J. Gait, Oligonucleotide synthesis : a practical approach, IRL Press, Oxford Oxfordshire, Washington, DC, 1984 Search PubMed.
- J. Fujimoto, L. Tran and L. C. Sowers, Chem. Res. Toxicol., 1997, 10, 1254–1258 Search PubMed.
- V. Guerniou, D. Gasparutto, S. Sauvaigo, A. Favier and J. Cadet, Nucleosides, Nucleotides Nucleic Acids, 2003, 22, 1073–1075 CrossRef CAS PubMed.
- Z. Cui, J. A. Theruvathu, A. Farrel, A. Burdzy and L. C. Sowers, Anal. Biochem., 2008, 379, 196–207 CrossRef CAS PubMed.
- R. P. Darst, C. E. Pardo, L. Ai, K. D. Brown and M. P. Kladde, Curr. Protoc. Mol. Biol., 2010, 7.9.1–7.9.17 CrossRef PubMed , chapter 7, unit 7 9 1–17.
- Y. Li and T. O. Tollefsbol, Methods Mol. Biol., 2011, 791, 11–21 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob00884b |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *bc
*bc