
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diarylethene moiety as an enthalpy-entropy switch: photoisomerizable stapled peptides for modulating p53/MDM2 interaction†
Alexander V.
Strizhak‡
ab,
Oleg
Babii‡
 c,
Sergii
Afonin‡
c,
Iuliia
Bakanovich
ab,
Teodors
Pantelejevs
d,
Wenshu
Xu
a,
Elaine
Fowler
a,
Rohan
Eapen
e,
Krishna
Sharma
a,
Maxim O.
Platonov
b,
Vasyl V.
Hurmach
bf,
Laura
Itzhaki
e,
Marko
Hyvönen
d,
Anne S.
Ulrich
*cg,
David R.
Spring
*a and
Igor V.
Komarov
*fh
c,
Sergii
Afonin‡
c,
Iuliia
Bakanovich
ab,
Teodors
Pantelejevs
d,
Wenshu
Xu
a,
Elaine
Fowler
a,
Rohan
Eapen
e,
Krishna
Sharma
a,
Maxim O.
Platonov
b,
Vasyl V.
Hurmach
bf,
Laura
Itzhaki
e,
Marko
Hyvönen
d,
Anne S.
Ulrich
*cg,
David R.
Spring
*a and
Igor V.
Komarov
*fh
aUniversity Chemical Laboratory, University of Cambridge, Lensfield Road, CB2 1EW Cambridge, UK. E-mail: Spring@ch.cam.ac.uk
bEnamine Ltd, Vul. Chervonotkatska 78, 02094 Kyiv, Ukraine
cInstitute of Biological Interfaces (IBG-2), Karlsruhe Institute of Technology (KIT), POB 3640, 76021 Karlsruhe, Germany. E-mail: Anne.Ulrich@kit.edu
dDepartment of Biochemistry, University of Cambridge, 80 Tennis Court Road, CB2 1GA Cambridge, UK
eDepartment of Pharmacology, University of Cambridge, Tennis Court Road, CB2 1PD Cambridge, UK
fTaras Shevchenko National University of Kyiv, Vul. Volodymyrska 60, 01601 Kyiv, Ukraine
gInstitute of Organic Chemistry (IOC), KIT, Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany
hLumobiotics GmbH, Auer Str. 2, 76227, Karlsruhe, Germany. E-mail: Igor.Komarov@lumobiotics.com
First published on 11th May 2020
Abstract
Analogs of the known inhibitor (peptide pDI) of the p53/MDM2 protein–protein interaction are reported, which are stapled by linkers bearing a photoisomerizable diarylethene moiety. The corresponding photoisomers possess significantly different affinities to the p53-interacting domain of the human MDM2. Apparent dissociation constants are in the picomolar-to-low nanomolar range for those isomers with diarylethene in the “open” configuration, but up to eight times larger for the corresponding “closed” isomers. Spectroscopic, structural, and computational studies showed that the stapling linkers of the peptides contribute to their binding. Calorimetry revealed that the binding of the “closed” isomers is mostly enthalpy-driven, whereas the “open” photoforms bind to the protein stronger due to their increased binding entropy. The results suggest that conformational dynamics of the protein-peptide complexes may explain the differences in the thermodynamic profiles of the binding.
Introduction
Reversibly photoisomerizable (i.e. “photoswitchable”) bioactive compounds and molecular systems are the subjects of intense research in the recent decade.1 Half a century after photoregulation of the biological activity of an azobenzene derivative was first reported,2 translational researches seem to be close to real applications of the photoswitchable compounds in pharmacology.3 The term “photopharmacology” has even been coined4 for the medicinal use of such compounds, although there are no approved photoswitchable drugs available yet.5 A driving force for research toward their medicinal applications is the general belief that the safety of drugs can be improved by exerting spatiotemporal control over their activity using light. Indeed, the safety advantage at the efficacy doses for some representative photoswitchable drug candidates has already been convincingly demonstrated in proof-of-principle studies in vivo.6Most of the reported photoswitchable biologically active compounds are small molecules that exploit photoisomerization of azobenzene building blocks.1,7 Due to the significant structural change of the cis- or trans-azobenzene unit upon isomerization, the photoisomers of azobenzene derivatives differ significantly in geometry, which can, therefore, substantially affect their biological activity.8 Extensive research has been directed to overcoming two major drawbacks of these azobenzene-based biologically active compounds, namely (i) the high rate of in vivo reduction of the azo group, and (ii) the intrinsic thermal instability of the cis-photo-forms.7c At the same time, it is known that photoisomerizable compounds of another type, the diarylethene (DAE) derivatives (Fig. 1), are reasonably inert in vivo and thermally stable in their both photoforms.9 The photochromism and thermal stability of diarylethenes have been known for almost thirty years.10 However, until the last decade, no reports on the photocontrolled biologically active DAE-based compounds were published. It has been suggested11 that the evidently small structural changes upon photoisomerization12 may have discouraged an active search for biologically active photoswitchable DAE derivatives.
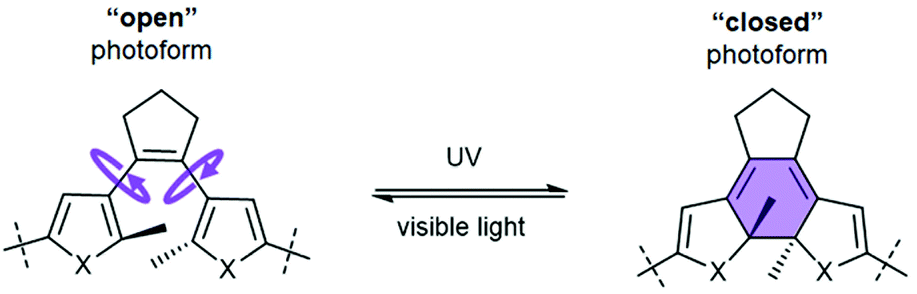 | ||
| Fig. 1 Photoisomerizable DAE moiety (X = S, NH, O), illustrating the nomenclature of the photoisomers used in this work, and characteristic changes in the molecular flexibility associated with photoisomerization. | ||
In 2012, α-helical DNA-binding peptides were described, cross-linked by a DAE-derived bridge between two non-neighbouring side chains. In these constructs, both helicity and DNA binding affinity varied reversibly upon photoisomerization.13 This study demonstrated for the first time that structural contexts – where DAE photoisomerization should lead to considerable changes in binding affinity to a target – can be found in polypeptides. Further studies demonstrated appreciable effects of DAE photoisomerization in other types of peptide structure, such as a cyclic β-hairpin14 and a helix-hinge-helix motif.15
Two recent works, on a small-molecule DAE-containing inhibitor of phosphoribosyl isomerase A from Mycobacterium tuberculosis,16 and on a similarly designed protein kinase C inhibitor,17 pointed to an essential DAE property that is important for the efficiency of photocontrol, namely, the differential rigidity/flexibility of the two DAE photoforms (Fig. 1). Those derivatives bearing a flexible (“open”) DAE unit can adapt their conformation to the binding target and tend to show higher inhibitory activity than the rigid (“closed”) photoisomers. We reasoned that this characteristic property of the DAE moiety could be usefully applied to design photoswitchable inhibitors of protein–protein interactions (PPIs).11
PPIs are the key players in signal transduction and regulatory cascades in every living cell; therefore, disease-associated PPIs are regarded as important targets in drug discovery.18 It has been observed that conformationally flexible mediators of PPIs often show higher inhibitory capacity than their rigid counterparts.19 Addition of constrains to the ligands, on the contrary, can lead to less favourable changes in binding entropy.20 These findings suggest that light-induced changes in the conformational flexibility/rigidity of DAE-derived PPI modulators could provide an effective means for controlling their inhibitory efficiency.
Here, we provide evidence for the validity of this hypothesis, based on our research of DAE-derived photocontrolled peptides that are capable of inhibiting the interaction between p53 and the E3 ubiquitin ligase murine double minute 2 homolog (MDM2). This interaction is a part of a complex interplay between the tumour suppressor protein p53 and its two natural inhibitors (oncogenic proteins MDM2 and MDMX), and is regarded as an important target for anticancer therapeutics.21 p53/MDM2/X PPI is one of the best-studied and most targeted intracellular PPIs related to cancer progression. Several compounds that disrupt the p53/MDM2/X interaction are under clinical trials, including peptides.22 The dual peptidic MDM2/MDMX antagonist ALRN-6924 was the first stapled helical peptide to enter clinical trials23 (now in Phase I/II24). Nevertheless, the toxicity associated with p53 activation has been reported as a critical issue on the way toward clinical application of the MDM2/MDMX antagonists.21b This fact encouraged us to develop photoswitchable p53/MDM2 antagonists, possibly having an improved safety profile.
In the present study, we report on a series of compounds which demonstrate efficient photocontrol of binding to the p53-interacting domain of the human MDM2, with Ki in the picomolar-to-submicromolar range. To the best of our knowledge, these are the first examples of DAE-containing peptidic PPI modulators. These new photocontrollable compounds complement the repertoire of the azobenzene-stapled PPI inhibitors which have been described in the literature, e.g. against the Bak(Bid)/Bcl-x(L) interaction (involved in the regulation of the programmed cell death),25 β-arrestin/β2-adaptin PPI (involved in the regulation of clathrin-mediated endocytosis),19c,26 and the WDR5–MLL1 interaction27 (involved in transcriptional regulation and leukemogenesis).
Results and discussion
Compound design
We have used peptidic templates for the design of our MDM2 antagonists first of all because peptides were shown to be highly efficient modulators of PPIs.28 As the medium-sized peptide molecules (10–12 amino acid residues) are much larger than the DAE moiety, incorporation of this photoswitch should not disturb the peptide pharmacophores too much, at least compared to small-molecule templates. This feature provides numerous options for varying the position of the DAE fragment, and, correspondingly, allows for optimizing the control of the biological activity by light. It has also been shown that cyclic peptides can serve as a convenient platform for molecular grafting, such as to mimic functional motifs of biologically active proteins,29 and they are usually more metabolically stable compared to their linear analogues.22bWe considered two general design strategies for our molecules, schematically illustrated in Fig. 2. In one strategy, the residues at the PPI interface that are critical for the interaction are grafted onto a cyclic β-hairpin scaffold, equipped with a DAE moiety at one of the turn positions. The other strategy uses an α-helix scaffold, stabilized by cross-linking (DAE-stapling) of two appropriate amino acid side chains. We have recently demonstrated the utility of β-hairpin scaffolds in the development of photoswitchable membranolytic peptides.6a,14 In the present work, we thus followed the second strategy, exploring the α-helical DAE-stapled peptide geometry. Our choice has been stimulated by the fact that many known efficient PPI modulators are stapled helical peptides.22a The amino acid sequences of these peptides are usually derived from the protein interaction surfaces, which often contain specific α-helical regions.30 The modular design of stapled peptides allows for the straightforward incorporation of a photoisomerizable moiety, as was shown earlier for the azobenzene-stapled PPI inhibitors.25a,31
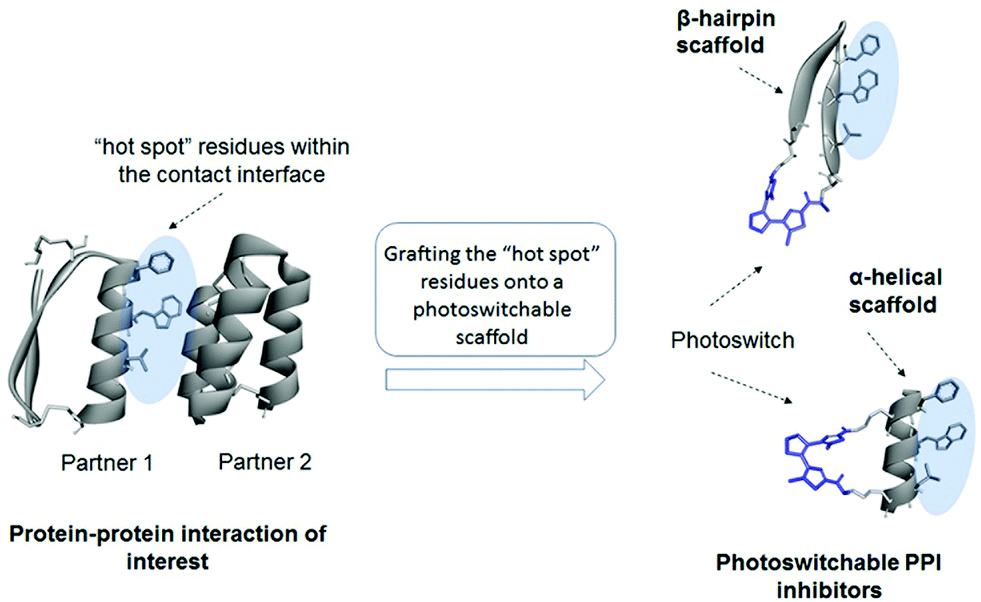 | ||
| Fig. 2 General design strategies for the photoswitchable PPI modulators considered in this work. | ||
The known stapled helical peptides reported as efficient p53/MDM2/X interaction inhibitors32 have usually been derived from linear sequences discovered by phage display. One such sequence, the pDI (Ac-LTFEHYWAQLTS-NH2), was reported in 2007.33 This peptide and its derivatives were proven to be very potent MDM2 antagonists. Hence, we decided to use the pDI as an initial peptide template for our design.
It is known from X-ray34 and computational35 studies that p53 binds to MDM2 with an essential contribution from three residues within the so-called “p53 hot spot”: Phe, Trp, and Leu located on the α-helical frame of the p53 N-terminal domain. Therefore, these residues and their spatial positions should be retained in the analogues. In ref. 32d, the authors used a pDI-derived sequence 1a (Fig. 3a, Ac-LTF-(Orn(N3))-EYWAQL-(Orn(N3))-S-NH2; Orn(N3): L-azidoornithine) to prepare the stapled p53/MDM2 interaction inhibitors using the azide–alkyne cycloaddition (“click”) reaction. The Orn(N3) residues occupy the (i, i + 7) positions in 1a and do not interfere with the “p53 hot spot” Phe3, Trp7 and Leu10 when the molecule of 1a adopts an ideal α-helix (Fig. 3b). This design turned out to be successful32d for the non-photoswitchable peptides; therefore, we used precursor 1a and its homolog 1b to prepare our DAE-stapled photoswitchable peptides.
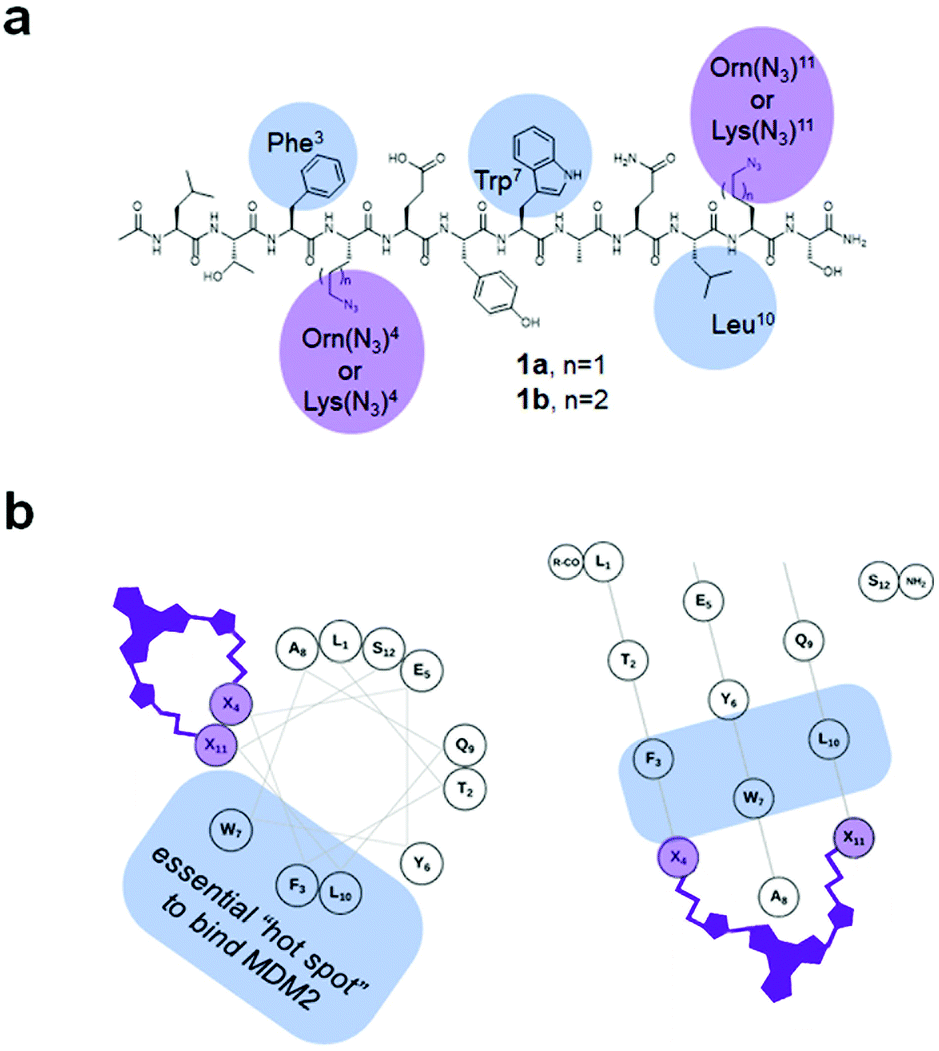 | ||
| Fig. 3 (a) Structural formulae of the precursors 1a,b used to prepare the stapled peptides. The amino acid residues discussed in the text are highlighted. (b) Helical wheel (left) and helical mesh36 (right) representations of a 1. The (Orn(N3) residues are denoted as “X”, showing the relative spatial position of the installed DAE-derived staple. | ||
To conjugate the diarylethene unit as a staple to the bis-L-azidoornithine-substituted linear peptide precursors 1a,b, we synthesized two bis-alkynes 2,3 (Fig. 4).
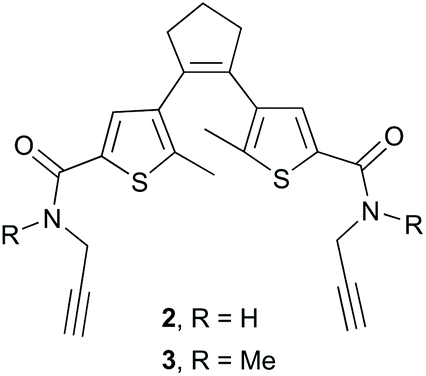 | ||
| Fig. 4 Bis-alkyne-substituted DAE-derived precursors for peptide stapling. | ||
Combining the precursors 1a,b and 2,3 by a Cu-catalyzed “click” reaction yielded target peptides 4–7 with slightly different sizes and flexibilities of the stapling linkers (Fig. 5).
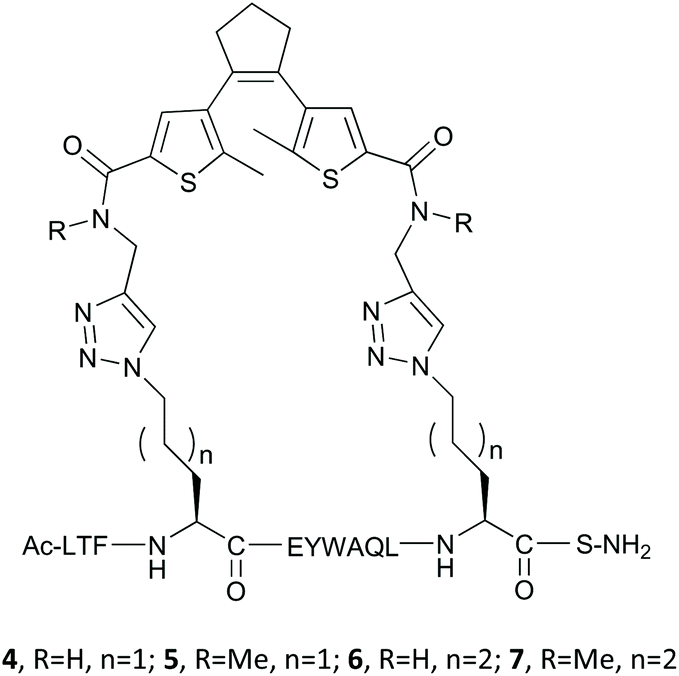 | ||
| Fig. 5 Stapled DAE-containing peptides studied in this work. | ||
Peptide synthesis
The linear precursors 1a,b were synthesized manually using standard Fmoc solid-phase chemistry on a low-loaded (0.33 mmol g−1) Rink amide resin (see ESI† for details). After cleavage, 1a,b were purified by a preparative HPLC prior to the “click” reactions. For stapled peptide synthesis we used two-component “double-click” protocol elaborated before (see ESI† for details).37 Corresponding linear peptides 1a,b were coupled with the building blocks 2,3 in a water/tert-butanol 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture (at 0.1 mM concentration, 0.01 mmol of 1a,b was used) at room temperature, in the presence of copper sulfate, sodium ascorbate, and tris(3-hydroxypropyltriazolyl-methyl) amine during 1–3 days to obtain the stapled peptides 4–7 with 54–70% yield after HPLC purification.
:1 mixture (at 0.1 mM concentration, 0.01 mmol of 1a,b was used) at room temperature, in the presence of copper sulfate, sodium ascorbate, and tris(3-hydroxypropyltriazolyl-methyl) amine during 1–3 days to obtain the stapled peptides 4–7 with 54–70% yield after HPLC purification.
Photochromic behaviour
The absorption spectra of the synthesized peptides are characterized by the features that are typical for DAE derivatives.9,18 Representative examples are illustrated in Fig. 6. Notably, the long-wavelength maxima of the “open” photoforms of peptides 4 and 6, which contain non-methylated amide bonds flanking the DAE fragments were shifted bathochromically by ∼40 nm compared to the corresponding N-Me derivatives 5 and 7 (Fig. 6).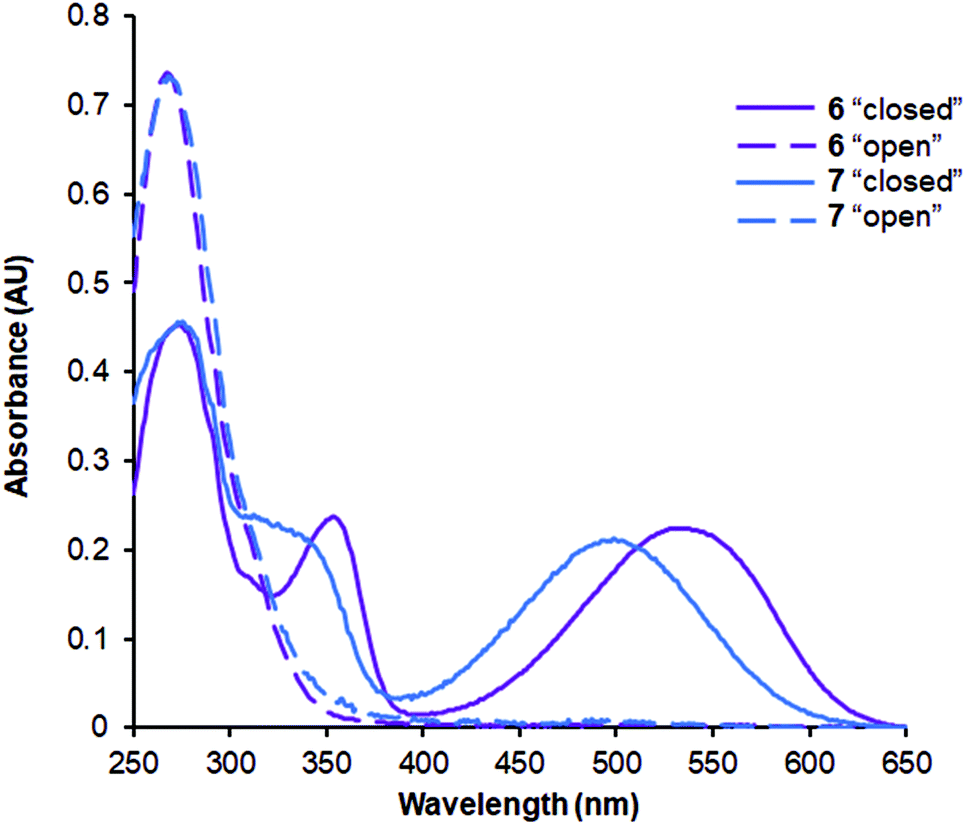 | ||
| Fig. 6 Representative absorption spectra of the DAE-stapled peptides. The spectra were recorded at the concentration of 20 μM in 10 mM phosphate buffer (pH 7.4) in 10 mm quartz cuvette at ambient temperature. | ||
To evaluate the photochromic properties of 4–7, they were dissolved in degassed 50% water/acetonitrile or in 8 M urea at a concentration of 0.5 mM, and irradiated with UV light (254 nm, ∼20 mW cm−2). The progress of the photoisomerization reaction was monitored by analytical HPLC. In the water/acetonitrile mixture, compounds 4–7 showed moderate conversion to the “closed” form, with a 20–30% conversion achieved after 30–40 min of continuous irradiation. However, in the urea solution the photoswitching was more efficient, reaching the photostationary states with 80–90% of the “closed” photoform after 10–20 min of irradiation. In this chaotropic environment the peptides are presumably less structured, which provides the DAE moiety with a higher population of the antiparallel rotamer needed for the photoconversion. We have previously observed this effect also for the DAE-modified cyclic β-hairpin peptides.38 No by-products were found when irradiation was carried out under an argon atmosphere. In contrast, fast degradation of the peptides was observed in the presence of oxygen, suggesting the involvement of reactive oxygen species in this process.
To test the reverse, i.e. the “closed” to “open” transformation, the same solutions were subjected to irradiation generated by different LED sources. All compounds were converted to the “open” photoforms quantitatively by irradiation with green light (λmax = 528 nm, Fig. 7a), also without any detectable formation of by-products according to analytical HPLC. In our experimental set-up, complete photoconversion by this light source (1.8 mW cm−2 at 4 cm distance to the peptide solution) was reached for all compounds in ∼16 min. Besides, we found that appreciable photoconversion into the “open” forms, especially for the non-methylated analogues 4 and 6, can be induced by orange (λmax = 600 nm) or even red light (λmax = 625 nm), under comparable light exposures (Fig. 7b and c). This is an important feature for the possible future use of such compounds in vivo, because light with a longer wavelength penetrates tissues of live organisms more deeply and without causing damage.39
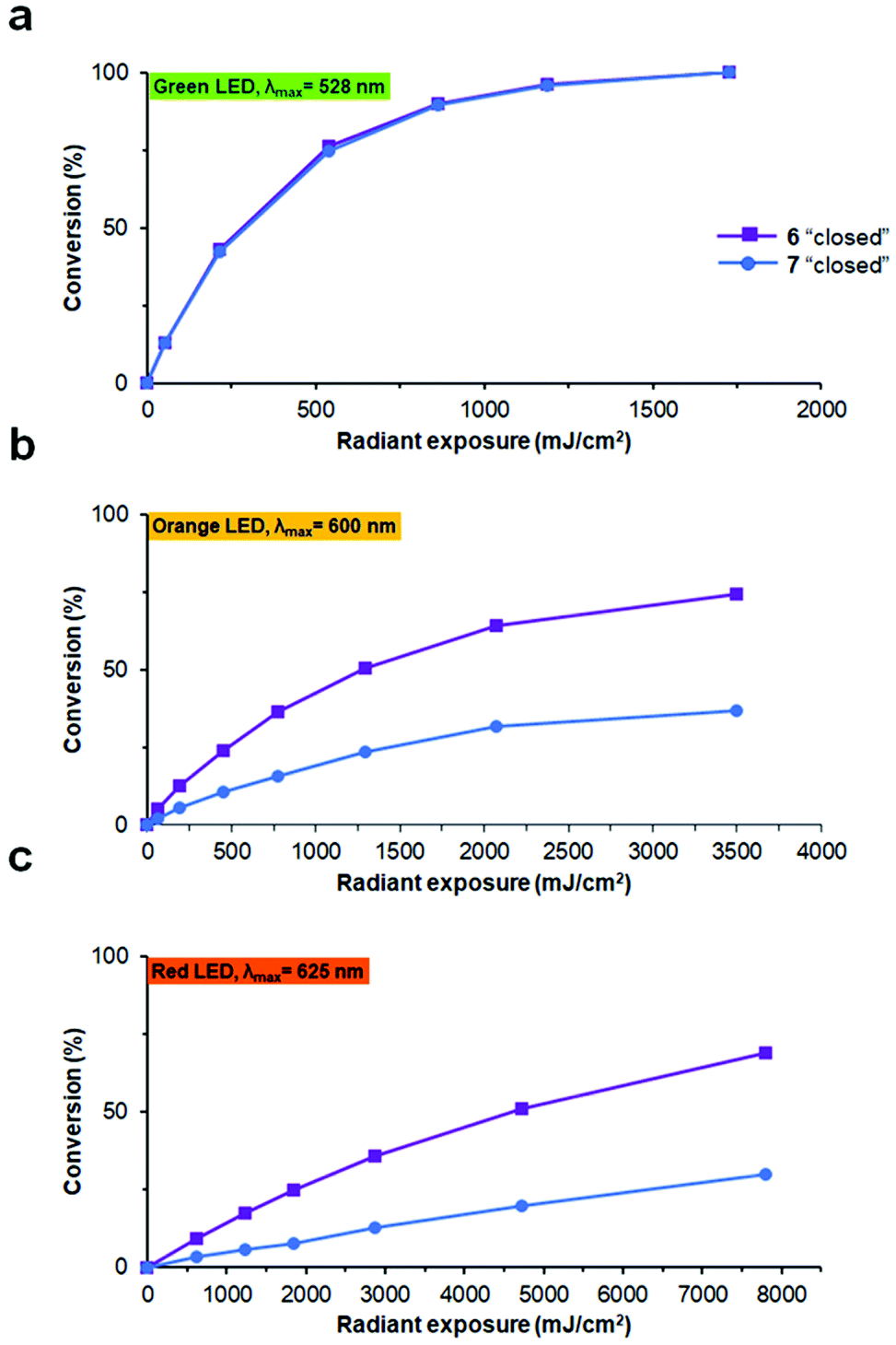 | ||
| Fig. 7 “Closed” to “open” photoconversion of peptides 6 and 7 by light of different wavelengths: (a) 528 nm; (b) 600 nm; (c) 625 nm, plotted as a function of light radiant exposure. The experiments were performed at a concentration of 20 μM in 10 mM phosphate buffer, pH 7.4, in 10 mm light-path cuvettes at ambient temperature. The conversion was monitored at the absorption maximum for each peptide. | ||
For the following experiments, all of the peptides 4–7 were obtained in their pure “open” and “closed” photoforms by preparative HPLC.
The efficiency of binding to MDM2
First, the binding affinity to MDM2 was assessed for peptides 4–7 and compared with that for the pDI. In order to compare the results with the literature data,40 we used the p53-interacting domain of human MDM2 (MDM26–125) for the assays. The value of the dissociation constant (Kd) for pDI was determined using an assay based on direct tryptophan fluorescence quenching. Apparent dissociation constants (Ki) for 4–7 were measured in the presence of pDI as a tracer, using a similar competition assay.41 The use of both Trp fluorescence-based assays was possible, because the only binding partner possessing a tryptophan fluorescence was pDI. The p53 binding domain MDM26–125 contains no Trp residues, and the Trp fluorescence of 4–7 turned out to be negligible under the measurement conditions due to strong intramolecular quenching by the DAE chromophore. The assay was sensitive enough, allowing us to perform measurements at low concentrations of the binding partners (1 μM, see further details in the ESI†), which was essential to avoid their aggregation. We found a Kd of 3.0 ± 1.0 nM for pDI; this value is in agreement with the literature data.32b Our measurements also revealed that all DAE-modified peptides have Ki values in the picomolar-to-low nanomolar range (Table 1), comparable with the most powerful known MDM2 antagonists,22a,32b including the known non-photoswitchable stapled peptides.32d,42| Peptide | K i (Kd) (nM) | Closed/open Ki ratiod | ΔH (kJ mol−1) | −TΔS (kJ mol−1) |
|---|---|---|---|---|
| a Determined by a competitive Trp fluorescence quenching assay. b Determined by a direct Trp fluorescence quenching assay. c Determined by a fluorescence polarization competition assay. d The data of the Trp fluorescence quenching assay were used. | ||||
| 4 “open” | 4.0 ± 0.6a | 8.3 | −24.6 ± 1.6 | −23.3 ± 2 |
| 4 “closed” | 33.0 ± 3.9a | −36.0 ± 1.7 | −6.7 ± 2.1 | |
| 5 “open” | 0.8 ± 0.1a | 2.5 | −33.0 ± 1.2 | −18.9 ± 1.5 |
| 1.6 ± 0.2c | ||||
| 5 “closed” | 2.0 ± 0.3a | −48.5 ± 1.9 | −1.4 ± 2.3 | |
| 6 “open” | 4.9 ± 0.6a | 5.5 | −18.6 ± 2.5 | −28.7 ± 2.7 |
| 5.7 ± 0.3c | ||||
| 6 “closed” | 26.8 ± 4.6a | −28.6 ± 2.0 | −14.6 ± 2.6 | |
| 7 “open” | 1.9 ± 0.2a | 3.8 | −26.9 ± 1.2 | −22.8 ± 1.5 |
| 0.4 ± 0.1c | ||||
| 7 “closed” | 7.2 ± 1.6a | −34.0 ± 2.4 | −12.5 ± 3.0 | |
| pDI | (3.0 ± 1.0b) | −53.3 ± 3.7 | 4.7 ± 4.7 | |
We were also pleased to find that the photoisomers of each DAE-modified peptide were systematically different: the binding efficiency was stronger (the Ki lower) for all the “open” photoforms compared to the corresponding “closed” photoisomers. In order to confirm the data for our most efficient inhibitors, we additionally conducted Ki measurements using a fluorescence polarization (FP) competition assay. A known fluorescent inhibitor, 5′-TAMRA-RFMDYWEGL-NH2, was used as a tracer; the results are also listed in Table 1. These results were in agreement with the data obtained by the Trp fluorescence quenching assay for the same compounds, and indeed confirmed the very high efficiency of the binding for the peptides 5–7 in the “open” photoforms.
Structural characterization
Next, we explored the conformational preferences of 4–7 in solution by circular dichroism (CD) spectroscopy. To evaluate the impact of photoisomerization on the secondary structure in these experiments, we first acquired CD spectra of the light-sensitive “closed” photoforms kept in darkness. The samples were then irradiated with visible light (λmax = 550 nm, irradiance ∼20 mW cm−2, 10 min), achieving in situ complete conversion to the “open” isomers, and CD spectra were measured again under no light protection. We also acquired the CD spectrum of the template peptide pDI for the comparison. The results are illustrated in Fig. 8 (see ESI† for experimental details).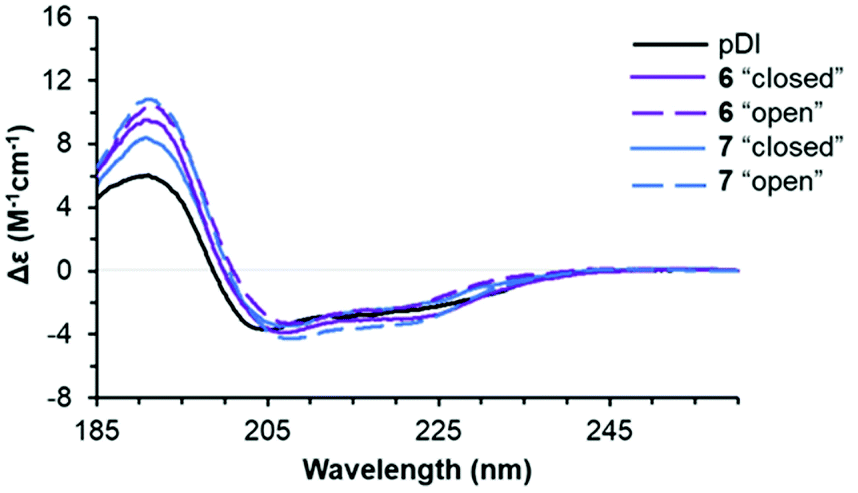 | ||
| Fig. 8 Representative far-UV CD spectra of pDI and the two photoforms of peptides 6 and 7 (50 μM peptide solutions in phosphate buffer, supplemented with 0.005% Tween 20 at 25 °C). Spectra of the “closed” photoforms were recorded in 1 mm light-path cuvettes, the spectra of the corresponding “open” photoforms were measured after the in situ photoisomerization by irradiation with 550 nm light. | ||
We compared the peptide conformations under conditions close to the binding experiment (10 mM phosphate buffer, pH 7.4, 0.005% Tween 20). We did not detect any isosbestic point between pDI and 4–7 in the aqueous buffer. Hence, in solution, equilibrium must exist between more than two conformations of the peptides. Indeed, spectral deconvolution (Table 2) revealed some non-negligible (20–40%) contributions from non-helical structures. Overall, however, the helicity increased upon photoisomerization from the “closed” to the “open” photoform (from 33–43% to 41–46%), and a decrease in the non-structured fraction (from 26–29% to 21–26%) was observed at the same time. These results suggested that 4–7 are more helical than pDI, and that the “open” photoforms are more structured than the “closed” isomers.
| Peptide | α r | α D | β r | β D | T | U | NRMSDb |
|---|---|---|---|---|---|---|---|
| a α r – regular α-helix; αD – distorterd α-helix; βr – regular β-strand; βD – distorted β-strand; T – turns; U – unstructured. b Normalized root mean square deviation. | |||||||
| 4 “open” | 27 | 16 | 8 | 7 | 17 | 24 | 0.021 |
| 4 “closed” | 19 | 14 | 12 | 10 | 19 | 26 | 0.014 |
| 5 “open” | 28 | 18 | 9 | 7 | 16 | 21 | 0.008 |
| 5 “closed” | 27 | 16 | 11 | 8 | 16 | 25 | 0.005 |
| 6 “open” | 27 | 14 | 11 | 9 | 13 | 26 | 0.005 |
| 6 “closed” | 25 | 14 | 8 | 7 | 16 | 29 | 0.008 |
| 7 “open” | 30 | 16 | 8 | 7 | 13 | 26 | 0.005 |
| 7 “closed” | 27 | 16 | 8 | 7 | 15 | 27 | 0.010 |
| 1 a | 33 | 21 | 7 | 6 | 10 | 23 | 0.008 |
| pDI | 23 | 17 | 12 | 8 | 16 | 24 | 0.005 |
Noticeably, the conformational differences between the “closed” and “open” isomers did not correlate with the differences in Ki. Our data suggested, therefore, that the percentage of α-helix in aqueous solutions is a poor predictor of MDM2 binding for our stapled peptides. This conclusion is further corroborated by the results from a recent paper by Lane, Sawyer et al.,44 who studied the impact of helix-perturbing residues on the affinity of their peptides to MDM2 and also found no direct correlation.
A possible explanation of the facts described above may be based on the assumption that both photoforms of the peptides bind to the target protein partly involving their linkers. The involvement of linkers of stapled peptides in the binding to target proteins was reported earlier in the literature.32b,c,45 The binding of our DAE linkers, irrespective of the photostate, could contribute to the observed high efficiency of binding, and at the same time attenuate the role of the “hot spot” residues and hence the helicity. In order to test this hypothesis, we carried out additional experimental and computational studies of the MDM26–125 complexes with one of our best binding compounds, peptide 5.
First, we obtained crystals of MDM26–125 in complex with the “open” form of 5, which is suitable for co-crystallisation. Attempts to crystallise the “closed” form were not successful, possibly due to residual “open” form peptide forming upon exposure to ambient light and resulting in a non-homogenous composition. In the solved structure (PDB ID: 6Y4Q), the crystallographic unit cell has P1 symmetry and contains two protein and two ligand molecules each, resulting in two 1:1 complexes per asymmetric unit. The peptide binds as expected, forming an α-helix, with the Phe3, Trp7 and Leu10 “hot-spot” residues residing in the known lipophilic pockets on the protein (Fig. 9a). The linker points outside the binding pocket on its N-proximal half, whereas the remaining half forms several interactions with the protein, confirming our hypothesis that the linker directly contributes to the interaction. Edge-to-face π-stacking interactions are observed between the triazole and thiophene moieties of the linker and Phe55 of the protein. The triazole ring also forms hydrogen bonds to a nearby water molecule, which, in turn, forms an H-bond with Gln59. Unexpectedly, however, the DAE moiety was observed participating in the crystal packing and forming contacts with the complex from a neighbouring unit cell (Fig. 9b).
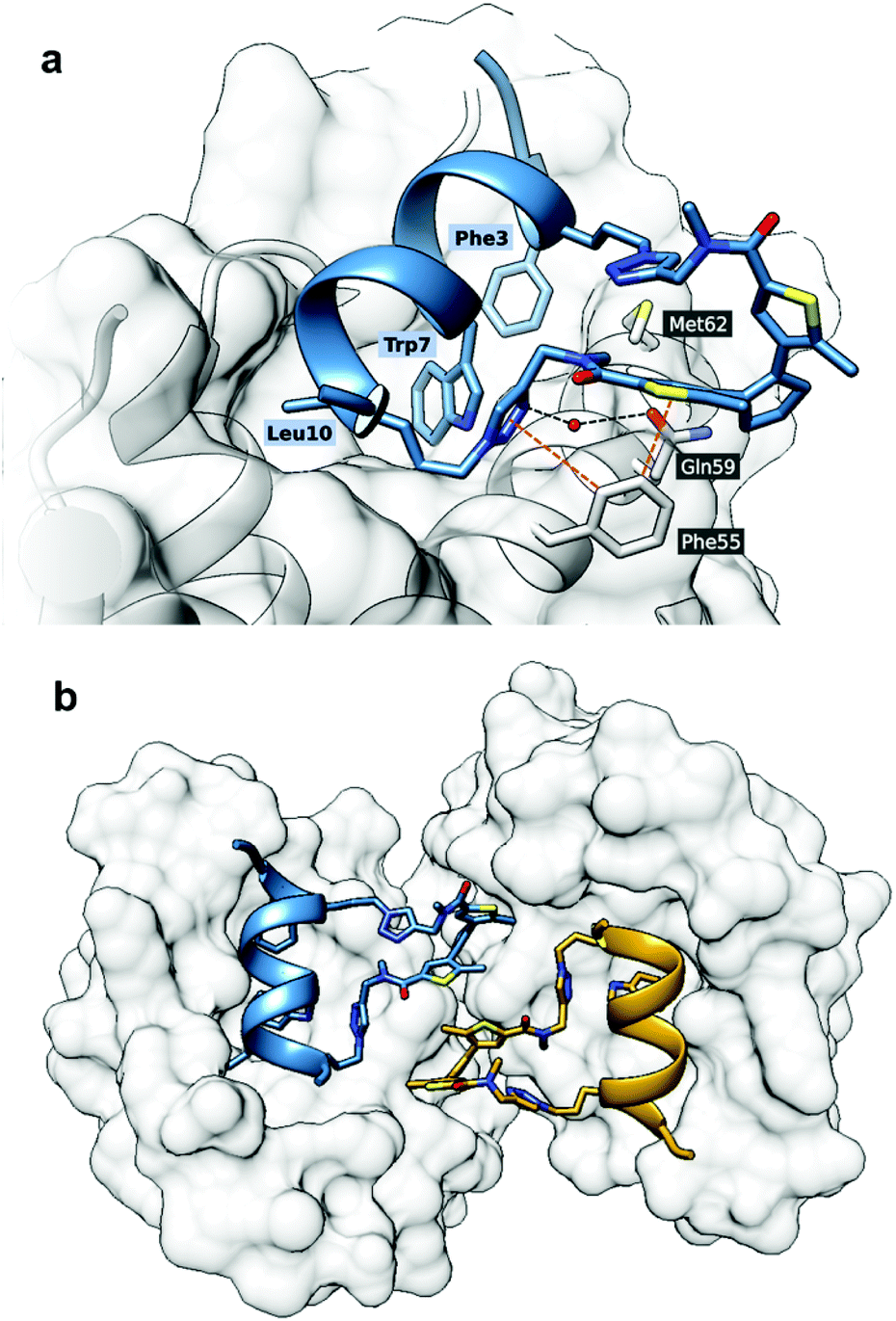 | ||
| Fig. 9 Crystal structure of MDM2 with 5“open” (PDB ID: 6Y4Q). (a) The binding mode of 5“open” in complex with MDM26–125 as deduced from X-ray crystallographic model. The peptide and linker are shown in blue; MDM26–125 surface is depicted in grey. Orange dashed lines represent π-stacking interactions, black dashed lines are hydrogen bonds; (b) the linker of the peptide 5 forms contacts with the nearby complex in the asymmetric unit. | ||
It is challenging to assert whether a similar binding mode would be present in solution, as we did not observe other crystal forms. We can only speculate that the presence of such dimers in the dilute solutions used by us in the Ki measurements is rather improbable.
Next, a computational study was performed in two steps. The first step was the molecular docking of both photoisomers of 5 to the MDM26–125 molecule. Conformationally flexible ligands and a rigid protein were used at this stage. The pharmacophore model of the protein binding site was derived from our X-ray data above. We used an algorithm for systematic docking (SDOCK+) implemented in the QXP docking software, which was capable of reproducing the ligand conformation with minimum root mean square deviation (RMSD) in comparison to the crystallographic data.46 Complexes with the “open” and “closed” photoforms of ligand 5 were calculated separately. The second step consisted of a molecular dynamics (MD) simulation of the complexes obtained by docking, using the GROMACS 5.1.3 software tool and a Charmm36 force field (see ESI† for computational details).47 Each complex was separately simulated five times, and the results of all simulations were consistent.
Our MD simulations demonstrated a good stability of the 5/MDM26–125 complexes within the simulation time (100 ns), independently as to whether the ligand was in the “open” or in the “closed” photoform. The essential Trp7, Phe3 and Leu10 residues of the ligands interacted with the corresponding protein sites in all cases, but surprisingly, this binding mode slightly changed during the simulation time. In particular, interaction of Leu10 with its initial binding site was not preserved during the entire simulation period. Importantly, the simulations identified numerous contacts between the stapling linker of peptide 5“open” and the protein (Fig. 10a), confirming our hypothesis about an involvement of the linker in the binding. One end of the linker in the complexes did not interact with MDM26–125, it pointed away from the binding pocket. The nearest observed distances between the atoms of the linker and protein were 6.7 Å and 7.0 Å, for the open and closed photophorms, respectively. The other end of the linker, in the case of the 5“open”, interacted with the protein at Lys51 and Phe55 by unstable hydrogen bonds, aromatic stacking of the DAE with Phe55, and by π-cation interaction with Lys51. Inspection of the complex with 5 in the “closed” photoform, however, showed that this structure differed significantly from the complex with 5“open” (Fig. 10b). The linker in this case interacted with the protein as well, but the number of contacts was reduced. Furthermore, the entire DAE moiety pointed away from the protein surface and did not form any contacts with the protein. The binding mode of the linkers changed slightly during the 100 ns simulation, as they left their initial binding pocket. Still, in both cases the interactions with the protein mentioned above were stable throughout the whole simulation period.
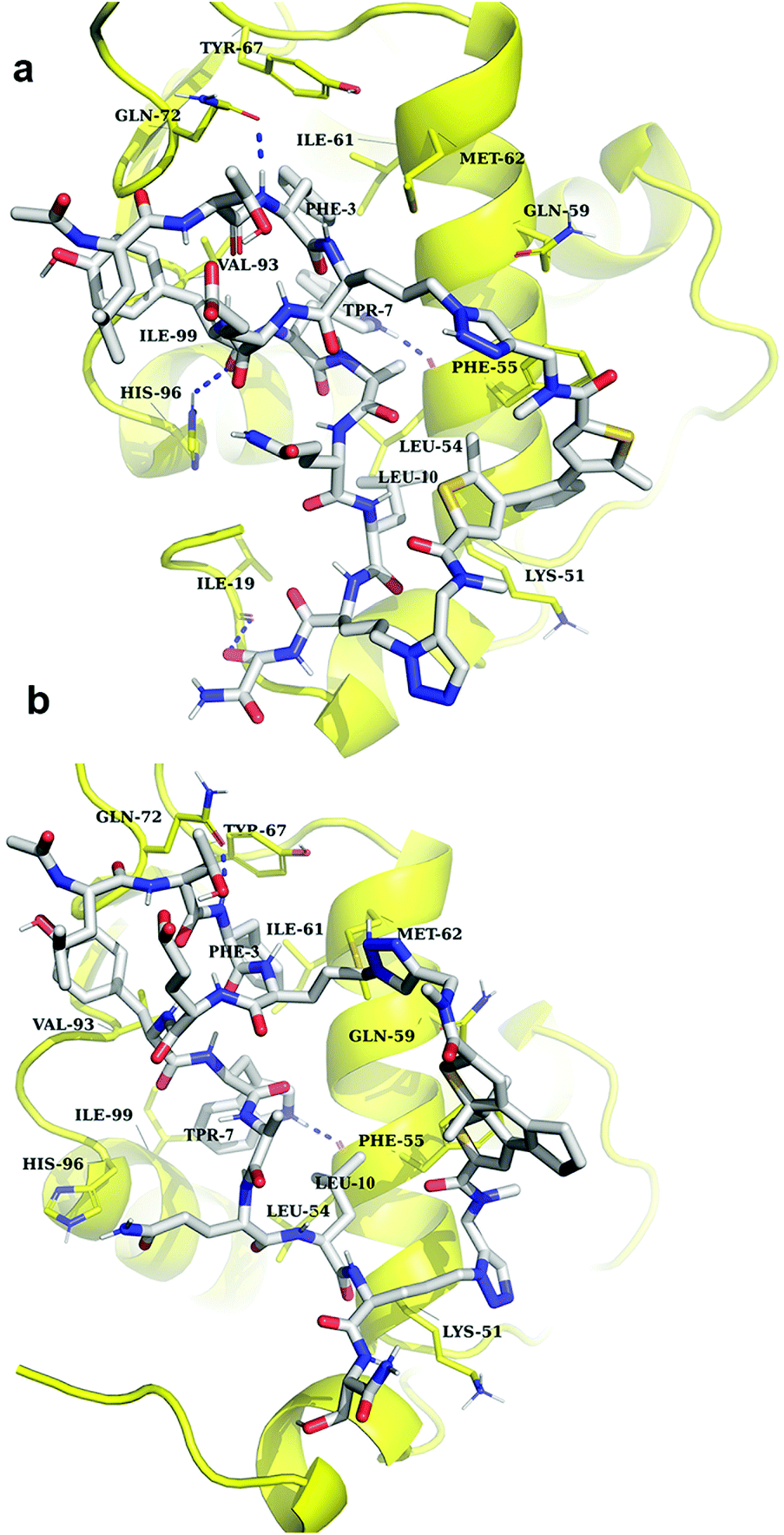 | ||
| Fig. 10 Snapshots (at 50th ns) of MD simulations of the 5/MDM26–125 complexes: (a) complex with the “open” photoform of 5; (b) complex with the “closed” photoform of 5. The ligand is shown in grey/blue/red colours; the protein is drawn in yellow. Amino acid residues of the protein involved in binding are denoted with the three-letter code. | ||
Estimation of enthalpy and entropy of binding
It is widely recognized that the thermodynamic signature of binding is crucial in the recognition event of an active ligand, and that both enthalpic and entropic control may define the overall selectivity of binding.48 A growing body of evidence suggests that PPIs are characterized by a rather complex interplay of enthalpy and entropy of the binding event. Peptide ligands that interfere with PPIs can interact with the target protein either by enthalpy- or by entropy-driven mechanisms.49 Which mechanism will be dominant depends largely on the conformational dynamics of the peptide. Even subtle changes in the rigidity/flexibility balance of their molecular structure may significantly affect both thermodynamic parameters of the free binding energy and, consequently, efficiency and selectivity of the binding process.50 We have thus suggested11,51 that the two photoisomers of DAE-containing peptides may differ so prominently in their ability to modulate PPIs, because they differ fundamentally in their flexibility/rigidity (Fig. 1). The more flexible “open” photoform should generally facilitate the formation of a particular conformation that is required for binding. On the other hand, the rigid “closed” DAE form would rather be expected to destabilize such specific conformation in the desired structural context. We have already observed such an effect of the DAE moiety when incorporated into the backbones of cyclic β-hairpin peptides and in linear α-helix-DAE-α-helix conjugates. In the case of the DAE-stapled peptides 4–7, the CD spectra described above suggested a similar influence of the DAE photoswitch on the stability of the α-helix, in agreement with the results of Fujimoto et al.13 In particular, the “open” photoforms of our peptides in water were more structured in the peptidic part than the “closed” photoforms (Table 2). Consequently, the “open” photoforms may not pay an excessive entropic penalty upon binding, as they are pre-organized for binding and should therefore have better affinities. The hypothesis about this effect of preorganization is still a subject of hot debate in the literature,19b,20 however, it has been experimentally proven in the case of some stapled p53/MDM2 inhibitors.49In order to gain insight into the thermodynamic parameters of the 4–7/MDM26–125 binding, we performed isothermal titration calorimetry (ITC) measurements. The ITC data could shed light on the relative contributions of enthalpy and entropy to the overall free energy of binding.52 Unfortunately, the sensitivity of the method did not allow us to reliably estimate the Kd values at low concentration (<20 μM) of the peptides. For a reliable determination of Kd, the so-called c-value (c = [MDM26–125]n/Kd, where [MDM26–125] is the concentration of the protein and n is the stoichiometry of the reaction) should be between 1 and 1000.53 Even at low concentrations, where aggregation of the binding partners was not observed, the c-values for our peptides are expected to be much higher than 1000. The enthalpies of binding, ΔH, however, could be measured by ITC with sufficient confidence even in such cases. Combining these ΔH values with the dissociation constants determined by other assays, one can get the corresponding entropies of binding, ΔS, using the well-known relation ΔG = ΔH − TΔS = −RT × ln(Kd−1), as was demonstrated on many examples.53,54 Therefore, we decided to fit our ITC data and the apparent values of Kd from the Trp fluorescence quenching assay to the single-site binding model to obtain the corresponding binding entropies.55 The thermodynamic parameters of the 4–7/MDM26–125 binding thus obtained are listed in Table 1. These data, illustrated for clarity in Fig. 11, revealed a striking difference for the two photoisomeric states: while the binding of the “closed” forms seems to be enthalpy-driven, the “open” isomers bind with a substantial contribution of entropy. The difference is particularly pronounced for peptides 4 and 6, i.e. those compounds for which we had measured the most significant differences in Ki values between the two photoforms. It is known that different ligands can have significantly different thermodynamic signatures of binding to MDM2,49 as determined by their conformational landscape.56 In our peptides, the DAE photoswitch seems to provide a means to control these features in one and the same molecule, simply by the use of light.
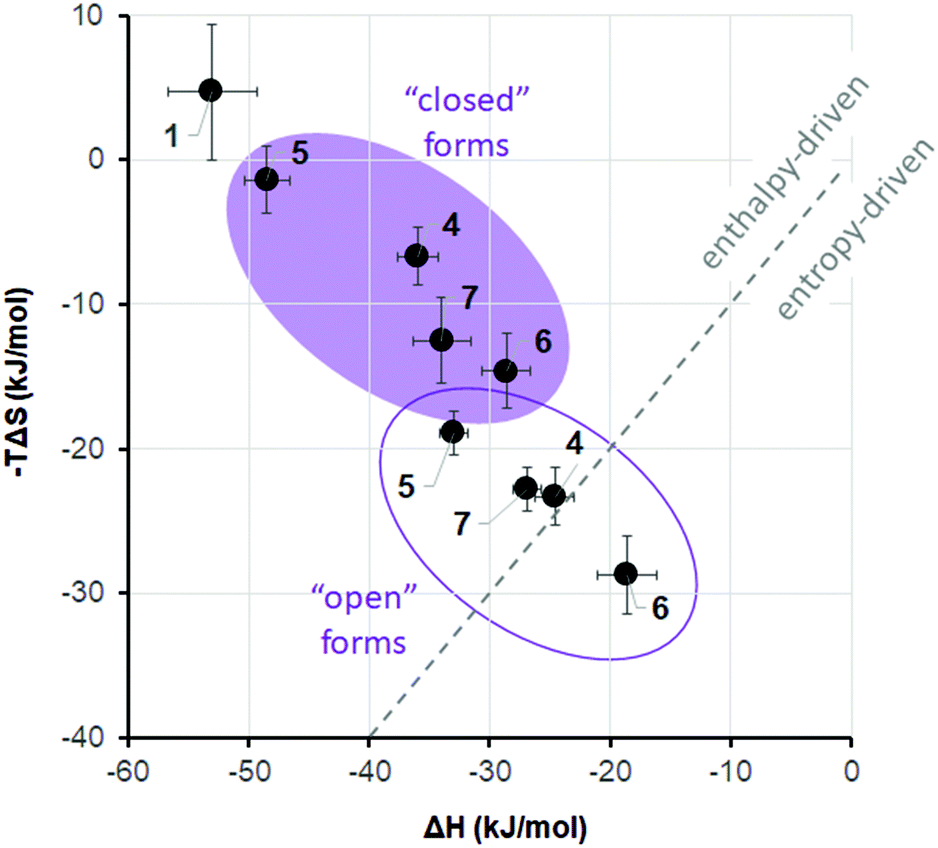 | ||
| Fig. 11 Thermodynamics of binding of the peptides 6–9 to MDM26–125. | ||
Contribution of conformational dynamics to binding
Although the thermodynamic parameters that we estimated for the 4–7/MDM26–125 binding seem to indicate that a preorganization of the “open” forms of our molecules can be the reason of their more favourable binding entropy and binding energy compared to the corresponding “closed” forms, the overall picture is not as simple. As was pointed out by several authors,20,49 there are many examples of seemingly counter-intuitive deviations from this paradigm. In our case, this became obvious when we observed no apparent correlation between the binding affinities on the one hand and the structural differences between the photoisomers on the other. We suspected that this may be due to the overall conformational dynamics of the complexes, which contribute to the binding entropy and play a significant role in the photoswitching efficiency of the compounds 4–7.The conformational dynamics can make the structure–activity correlations not as straightforward. For example, binding of some peptide-derived ligands to their protein targets was shown not to benefit from conformation-stabilizing stapling57 or rigidification;58 on the contrary, conformationally flexible, less structured ligands may actually have a higher affinity.59 To compare the conformational dynamics of the binding partners in our case, we analysed the MD trajectories of MDM26–125 complexes with 5 in the “open” and “closed” photoforms. Conformational movements of the ligands and the protein are reflected by their all-atom root mean square fluctuations (RMSFs), calculated over the entire simulation period. As can be seen from Fig. 12a, the RMSFs for 5“open” were higher than for 5“closed” in the simulated 5/MDM26–125 complexes, which means that 5“open” has much more conformational freedom. Notably, the 5“open” RMSFs exceed those of 5“closed” not only in the area of the linker, but also in the α-helical peptidic region. In turn, this enhanced mobility seems to contribute to the more favourable binding entropy of 5“open”. We further suggest that this may well be a result of the enhanced molecular flexibility of the DAE moiety in the “open” configuration, as it lacks the rigidifying bond between the two thiophene rings (see Fig. 1).
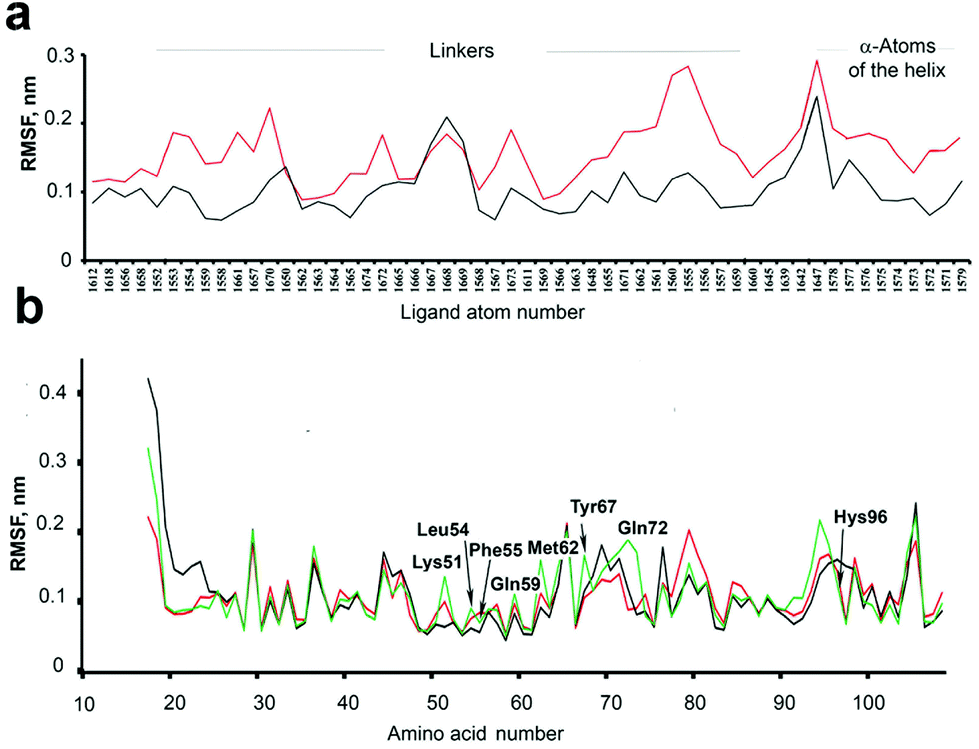 | ||
| Fig. 12 (a) RMSF of the atoms of 5 when complexed with MDM26–125. Red trace – the complex with the “open” form of the ligand, black trace – with the “closed” form of 5. Shown are all atoms for the linker, and representative atoms (α-atoms of the amino acid residues) in the peptidic part. (b) RMSF of the free MDM26–125 (green) and in complex with 5“open” (red) and 5“closed” (black). Amino acid residues comprising the binding pocket are shown with their three-letter code. | ||
The freedom of conformational motion of a bound ligand can modulate the overall conformational dynamics of the protein–ligand complex, which is known to govern the overall Gibbs energy of the interaction.60 Some insights into the protein dynamics can also be gained from the MD simulation results. The RMSF differences for some of the protein atoms in the complexes 5“open”/MDM26–125 and 5“closed”/MDM26–125 are quite pronounced and depend on the protein region. Some protein moieties are more mobile (RMSF are larger) in the complex with 5“open”, but there are also some regions that are more dynamic in the 5“closed”/MDM26–125 complex (compare the red and black traces in Fig. 12b). It is also interesting to compare the RMSF of the ligand-free protein (which we have simulated separately, green trace in Fig. 12b) and of the protein in the complexes with 5. The overall changes in the protein conformational landscape upon complexation of the ligands are too complex to allow drawing any specific conclusions, but it is evident that these changes are significantly different for the complexes with 5“open” and 5“closed”, corroborating the observed Ki difference for the photoisomers. These findings warrant further studies into the contribution of conformational dynamics to protein–ligand interactions, and DAE-modified ligands may indeed represent valuable model systems in this field.
Conclusions
A “double-click” macrocyclization reaction of the linear precursors 1a,b possessing L-azidoornithine/L-azidolysine residues in (i, i + 7)-positions with the bis-alkyne DAE-derived linkers 2 and 3 can be used to obtain efficiently photoswitchable peptides, capable of photoregulating PPI inhibition. We demonstrated here that an up to 8-fold difference in the affinity of the two photoforms of an MDM2 binding peptide can be achieved by rational design, with the affinity of one of the photoisomers being in the picomolar range. Generally, the more active, “open” photoisomers tend to bind to the protein better due to a more favourable binding entropy, while the binding of the “closed” photoisomers is predominantly enthalpy-driven. The efficiency of photoregulation seems to be affected by a partial involvement of the linker moiety in the binding. Notably, the more efficient MDM26–125 binders are those peptide photoisomers that are generated by visible light, and both photoisomers are perfectly thermally stable. These properties may boost the further development of DAE-based PPI modulators as possible drug candidates for photopharmacology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge EU funding by the EU H2020-MSCA-RISE-2015 through the PELICO project (grant 690973). This work was also supported by the DFG-GRK 2039 (S.A., A.S.U.) and by the BMBF-VIP + (O.B., A.S.U.). TP's doctoral studentship was supported by the Medical Research Council (UK). We thank Prof. Dr Burkhard Luy and Dr Claudia Muhle-Goll (KIT) for access to the ITC instrument. We also thank Diamond Light Source for access to macromolecular crystallography beam line i03 (proposal 18548) and for the data that contributed to these results. We are also grateful for access to and support by the X-ray Crystallographic and Biophysical Research Facility at the Department of Biochemistry, University of Cambridge.Notes and references
- (a) W. Szymański, J. M. Beierle, H. A. V. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114 CrossRef PubMed; (b) K. Hüll, J. Morstein and D. Trauner, Chem. Rev., 2018, 118, 10710 CrossRef PubMed; (c) Z. L. Pianowski, Chem. – Eur. J., 2019, 25, 5128 CrossRef CAS PubMed.
- (a) H. Kaufman, S. M. Vratsanos and B. F. Erlanger, Science, 1968, 162, 1487 CrossRef CAS PubMed; (b) J. Bieth, S. M. Vratsanos, N. Wassermann and B. F. Erlanger, Proc. Natl. Acad. Sci. U. S. A., 1969, 64, 1103 CrossRef CAS PubMed.
- M. M. Lerch, M. J. Hansen, G. M. van Dam, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2016, 55, 10978 CrossRef CAS PubMed.
- (a) W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178 CrossRef CAS PubMed; (b) J. Broichhagen, J. A. Frank and D. Trauner, Acc. Chem. Res., 2015, 48, 1947 CrossRef CAS PubMed.
- See, however, D. Schmidt, T. Rodat, L. Heintze, J. Weber, R. Horbert, U. Girreser, T. Raeker, L. Bußmann, M. Kriegs, B. Hartke and C. Peifer, ChemMedChem, 2018, 13, 2415 CrossRef CAS PubMed . The drug described in this paper, although photoswitchable, has no medical application of its photoswitching properties yet. .
- (a) O. Babii, S. Afonin, L. V. Garmanchuk, V. V. Nikulina, T. V. Nikolaienko, O. V. Storozhuk, D. V. Shelest, O. I. Dasyukevich, L. I. Ostapchenko, V. Iurchenko, S. Zozulya, A. S. Ulrich and I. V. Komarov, Angew. Chem., Int. Ed., 2016, 55, 5493 CrossRef CAS PubMed; (b) S. Afonin, O. Babii, A. Reuter, V. Middel, M. Takamiya, U. Strähle, I. V. Komarov and A. S. Ulrich, Beilstein J. Org. Chem., 2020, 16, 39 CrossRef CAS PubMed; (c) O. Babii, S. Afonin, T. Schober, L. V. Garmanchuk, L. I. Ostapchenko, V. Yurchenko, S. Zozulya, O. Tarasov, I. Pishel, A. S. Ulrich and I. V. Komarov, Future Drug Discov., 2020, 2, FDD28 CrossRef.
- (a) M. Schoenberger, A. Damijonaitis, Z. Zhang, D. Nagel and D. Trauner, ACS Chem. Neurosci., 2014, 5, 514 CrossRef CAS PubMed; (b) M. Borowiak, W. Nahaboo, M. Reynders, K. Nekolla, P. Jalinot, J. Hasserodt, M. Rehberg, M. Delattre, S. Zahler, A. Vollmar, D. Trauner and O. Thorn-Seshold, Cell, 2015, 162, 403 CrossRef CAS PubMed; (c) R. J. Mart and R. K. Allemann, Chem. Commun., 2016, 52, 12262 RSC; (d) L. Albert and O. Vázquez, Chem. Commun., 2019, 55, 10192 RSC.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422 RSC.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174 CrossRef CAS PubMed.
- M. Irie, Jpn. J. Appl. Phys., 1989, 28, 215 CrossRef CAS.
- I. V. Komarov, S. Afonin, O. Babii, T. Schober and A. S. Ulrich, Chem. – Eur. J., 2018, 24, 11245 CrossRef CAS PubMed.
- (a) S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nature, 2007, 446, 778 CrossRef CAS PubMed; (b) M. Irie, Bull. Chem. Soc. Jpn., 2008, 81, 917 CrossRef CAS; (c) M. Irie, Proc. Jpn. Acad., Ser. B, 2010, 86, 472 CrossRef CAS PubMed.
- K. Fujimoto, M. Kajino, I. Sakaguchi and M. Inouye, Chem. – Eur. J., 2012, 18, 9834 CrossRef CAS PubMed.
- O. Babii, S. Afonin, M. Berditsch, S. Reiβer, P. K. Mykhailiuk, V. S. Kubyshkin, T. Steinbrecher, A. S. Ulrich and I. V. Komarov, Angew. Chem., Int. Ed., 2014, 53, 3392 CrossRef CAS PubMed.
- K. Fujimoto, T. Maruyama, Y. Okada, T. Itou and M. Inouye, Tetrahedron, 2013, 69, 6170 CrossRef CAS.
- B. Reisinger, N. Kuzmanovic, P. Löffler, R. Merkl, B. König and R. Sterner, Angew. Chem., Int. Ed., 2014, 53, 595 CrossRef CAS PubMed.
- D. Wilson, J. W. Li and N. R. Branda, ChemMedChem, 2017, 12, 284 CrossRef CAS PubMed.
- (a) J. A. Wells and C. L. McClendon, Nature, 2007, 450, 1001 CrossRef CAS PubMed; (b) A. Mullard, Nat. Rev. Drug Discovery, 2012, 11, 173 CrossRef CAS PubMed.
- (a) J. P. Davidson, O. Lubman, T. Rose, G. Waksman and S. F. Martin, J. Am. Chem. Soc., 2002, 124, 205 CrossRef CAS PubMed; (b) S. F. Martin and J. H. Clements, Annu. Rev. Biochem., 2013, 82, 267 CrossRef CAS PubMed; (c) A. Martín-Quirós, L. Nevola, K. Eckelt, S. Madurga, P. Gorostiza and E. Giralt, Chem. Biol., 2015, 22, 31 CrossRef PubMed.
- D. G. Udugamasooriya and M. R. Spaller, Biopolymers, 2008, 89, 653 CrossRef CAS PubMed.
- (a) P. Chène, Nat. Rev. Cancer, 2003, 3, 102 CrossRef PubMed; (b) L. T. Vassilev, Trends Mol. Med., 2007, 13, 23 CrossRef CAS PubMed; (c) A. Burgess, K. M. Chia, S. Haupt, D. Thomas, Y. Haupt and E. Lim, Front. Oncol., 2016, 6, 1 Search PubMed; (d) S. Wang, Y. Zhao, A. Aguilar, D. Bernard and C. Y. Yang, Cold Spring Harbor Perspect. Med., 2017, a026245 CrossRef PubMed.
- (a) N. Estrada-Ortiz, C. G. Neochoritis and A. Dömling, ChemMedChem, 2016, 11, 757772 CrossRef PubMed; (b) T. K. Sawyer, A. W. Partridge, H. Y. K. Kaan, Y.-C. Juang, S. Lim, C. Johannes, T. Y. Yuen, C. Verma, S. Kannan, P. Aronica, Y. S. Tan, B. Sherborne, S. Ha, J. Hochman, S. Chen, L. Surdi, A. Peier, B. Sauvagnat, P. J. Dandliker, C. J. Brown, S. Ng, F. Ferrer and D. P. Lane, Bioorg. Med. Chem., 2018, 26, 2807 CrossRef CAS PubMed.
- V. Tisato, R. Voltan, A. Gonelli, P. Secchiero and G. Zauli, J. Hematol. Oncol., 2017, 10, 133 CrossRef PubMed.
- (a) https://clinicaltrials.gov/ct2/show/NCT02264613 ; (b) https://www.cancer.gov/about-cancer/treatment/clinical-trials/intervention/mdm2mdmx-inhibitor-alrn-69 .
- (a) S. Kneissl, E. J. Loveridge, C. Williams, M. P. Crump and R. K. Allemann, ChemBioChem, 2008, 9, 3046 CrossRef CAS PubMed; (b) P. Wysoczanski, R. J. Mart, E. J. Loveridge, C. Williams, S. B.-M. Whittaker, M. P. Crump and R. K. Allemann, J. Am. Chem. Soc., 2012, 134, 7644 CrossRef CAS PubMed; (c) R. J. Mart, R. J. Errington, C. L. Watkins, S. C. Chappell, M. Wiltshire, A. T. Jones, P. J. Smith and R. K. Allemann, Mol. BioSyst., 2013, 9, 2597 RSC.
- L. Nevola, A. Martín-Quirós, K. Eckelt, N. Camarero, S. Tosi, A. Llobet, E. Giralt and P. Gorostiza, Angew. Chem., Int. Ed., 2013, 52, 7704 CrossRef CAS PubMed.
- L. Albert, J. Xu, R. Wan, V. Srinivasan, Y. Dou and O. Vázquez, Chem. Sci., 2017, 8, 4612 RSC.
- (a) L. Nevola and E. Giralt, Chem. Commun., 2015, 51, 3302 RSC; (b) T. A. F. Cardote and A. Ciulli, ChemMedChem, 2016, 11, 787 CrossRef CAS PubMed; (c) A. D. Cunningham, N. Qvit and D. Mochly-Rosen, Curr. Opin. Struct. Biol., 2017, 44, 59 CrossRef CAS PubMed.
- (a) T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 13020 CrossRef CAS PubMed; (b) C. K. Wang and D. J. Craik, Nat. Chem. Biol., 2018, 14, 417 CrossRef CAS PubMed.
- M. R. Arkin and J. A. Wells, Nat. Rev. Drug Discovery, 2004, 3, 301 CrossRef CAS PubMed.
- (a) V. Borisenko and G. A. Woolley, J. Photochem. Photobiol., A, 2005, 173, 21 CrossRef CAS; (b) L. Guerrero, O. S. Smart, C. J. Weston, D. C. Burns, G. A. Woolley and R. K. Allemann, Angew. Chem., Int. Ed., 2005, 44, 7778 CrossRef CAS PubMed.
- (a) C. J. Brown, S. T. Quah, J. Jong, A. M. Goh, P. C. Chiam, K. H. Khoo, M. L. Choong, M. A. Lee, L. Yurlova, K. Zolghadr, T. L. Joseph, C. S. Verma and D. P. Lane, ACS Chem. Biol., 2013, 8, 506 CrossRef CAS PubMed; (b) Y. S. Chang, B. Graves, V. Guerlavais, C. Tovar, K. Packman, K.-H. To, K. A. Olson, K. Kesavan, P. Gangurde, A. Mukherjee, T. Baker, K. Darlak, C. Elkin, Z. Filipovic, F. Z. Qureshi, H. Cai, P. Berry, E. Feyfant, X. E. Shi, J. Horstick, D. A. Annis, A. M. Manning, N. Fotouhi, H. Nash, L. T. Vassilev and T. K. Sawyer, Proc. Natl. Acad. Sci. U. S. A., 2013, E3445 CrossRef CAS PubMed; (c) S. M. Q. Chee, J. Wongsantichon, Q. S. Tng, R. Robinson, T. L. Joseph, C. Verma, D. P. Lane, C. J. Brown and F. J. Ghadessy, PLoS One, 2014, e104914 CrossRef PubMed; (d) Y. H. Lau, Y. Wu, M. Rossmann, B. X. Tan, P. de Andrade, Y. S. Tan, C. Verma, G. J. McKenzie, A. R. Venkitaraman, M. Hyvönen and D. R. Spring, Angew. Chem., Int. Ed., 2015, 54, 15410 CrossRef CAS PubMed; (e) K. Sharma, A. V. Strizhak, E. Fowler, W. Xu, B. Chappell, H. F. Sore, W. R. J. D. Galloway, M. N. Grayson, Y. H. Lau, L. S. Itzhaki and D. R. Spring, ACS Omega, 2020, 5, 1157 CrossRef CAS PubMed.
- B. Hu, D. M. Gilkes and J. Chen, Cancer Res., 2007, 67, 8810 CrossRef CAS PubMed.
- P. H. Kussie, S. Gorina, V. Marechal, B. Elenbaas, J. Moreau, A. J. Levine and N. P. Pavletich, Science, 1996, 274, 948 CrossRef CAS PubMed.
- I. Massova and P. A. Kollman, J. Am. Chem. Soc., 1999, 121, 8133 CrossRef CAS.
- S. Reißer, S. Prock, H. Heinzmann and A. S. Ulrich, Biochem. Mol. Biol. Educ., 2018, 46, 403 CrossRef PubMed.
- Y. H. Lau, Y. Wu, P. de Andrade, W. R. Galloway and D. R. Spring, Nat. Protoc., 2015, 10, 585 CrossRef CAS PubMed.
- C. Schweigert, O. Babii, S. Afonin, T. Schober, J. Leier, N. C. Michenfelder, I. V. Komarov, A. S. Ulrich and N. A. Unterreiner, ChemPhotoChem, 2019, 3, 403 CrossRef CAS.
- (a) R. Weissleder, Nat. Biotechnol., 2001, 19, 316 CrossRef CAS PubMed; (b) S. Samanta, A. A. Beharry, O. Sadovski, T. M. McCormick, A. Babalhavaeji, V. Tropepe and G. A. Woolley, J. Am. Chem. Soc., 2013, 135, 9777 CrossRef CAS PubMed.
- Y. H. Lau, P. de Andrade, S.-T. Quah, M. Rossmann, L. Laraia, N. Sköld, T. J. Sum, P. J. E. Rowling, T. L. Joseph, C. Verma, M. Hyvönen, L. S. Itzhaki, A. R. Venkitaraman, C. J. Brown, D. P. Lane and D. R. Spring, Chem. Sci., 2014, 5, 1804 RSC.
- A. Yammine, J. Gao and A. H. Kwan, Sci. Rep., 2019, 9, e3253 CrossRef CAS PubMed.
- C. J. Brown, S. T. Quah, J. Jong, A. M. Goh, P. C. Chiam, K. H. Khoo, M. L. Choong, M. A. Lee, L. Yurlova, K. Zolghadr, T. L. Joseph, C. S. Verma and D. P. Lane, ACS Chem. Biol., 2013, 8, 506 CrossRef CAS PubMed.
- N. Sreerama and R. W. Woody, Anal. Biochem., 2000, 287, 252 CrossRef CAS PubMed.
- A. W. Partridge, H. Y. K. Kaan, Y.-C. Juang, A. Sadruddin, S. Lim, C. J. Brown, S. Ng, D. Thean, F. Ferrer, C. Johannes, T. Y. Yuen, S. Kannan, P. Aronica, Y. S. Tan, M. R. Pradhan, C. S. Verma, J. Hochman, S. Chen, H. Wan, S. Ha, B. Sherborne, D. P. Lane and T. K. Sawyer, Molecules, 2019, 24, 2292 CrossRef CAS PubMed.
- S. Baek, P. S. Kutchukian, G. L. Verdine, R. Huber, T. A. Holak, K. W. Lee and G. M. Popowicz, J. Am. Chem. Soc., 2012, 134, 103 CrossRef CAS PubMed.
- G. L. Warren, C. W. Andrews, A. M. Capelli, B. Clarke, J. LaLonde, M. H. Lambert, M. Lindvall, N. Nevins, S. F. Semus, S. Senger, G. Tedesco, I. D. Wall, J. M. Woolven, C. E. Peishoff and M. S. Head, J. Med. Chem., 2006, 49, 5912 CrossRef CAS PubMed.
- J. Huang and A. D. MacKerell Jr., J. Comput. Chem., 2013, 34, 2135 CrossRef CAS PubMed.
- M. Varese, S. Guardiola, J. García and E. Giralt, ChemBioChem, 2019, 20, 2981 CrossRef CAS PubMed.
- C. J. Brown, S. G. Dastidar, S. T. Quah, A. Lim, B. Chia and C. S. Verma, PLoS One, 2011, 6, e24122 CrossRef CAS PubMed.
- (a) B. C. Doak, J. Zheng, D. Dobritzsch and J. Kihlberg, J. Med. Chem., 2016, 59, 2312 CrossRef CAS PubMed; (b) S. Jeganathan, M. Wendt, S. Kiehstaller, D. Brancaccio, A. Kuepper, N. Pospiech, A. Carotenuto, E. Novellino, S. Hennig and T. N. Grossmann, Angew. Chem., Int. Ed., 2019, 58, 17351 CrossRef CAS PubMed.
- O. Babii, S. Afonin, T. Schober, I. V. Komarov and A. S. Ulrich, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 2505 CrossRef CAS PubMed.
- (a) R. Grünberg, M. Nilges and J. Leckner, Structure, 2006, 14, 683 CrossRef PubMed; (b) K. Henzler-Wildman and D. Kern, Nature, 2007, 450, 964 CrossRef CAS PubMed; (c) K. K. Frederick, M. S. Marlow, K. G. Valentine and A. J. Wand, Nature, 2007, 448, 325 CrossRef CAS PubMed.
- M. M. Pierce, C. S. Raman and B. T. Nall, Methods, 1999, 19, 213 CrossRef CAS PubMed.
- R. Perozzo, G. Folkers and L. Scapozza, J. Recept. Signal Transduction, 2004, 24, 1 CrossRef CAS PubMed.
- Strictly speaking, this approach could be incorrect if the tracer peptide pDI would influence the kon and/or koff for the binding of the peptides, for example, by allosteric MDM2 modulation. In this case, the Kd for the peptides would be different from their Ki measured in the presence of pDI. The literature (see, for example,M. Zdzalik, K. Pustelny, S. Kedracka-Krok, K. Huben, A. Pecak, B. Wladyka, S. Jankowski, A. Dubin, J. Potempa and G. Dubin, Cell Cycle, 2010, 9, 4584) and our own data where the corresponding Kd and Ki values are in mutual agreement revealed that such an influence of the tracer is not substantial.
- S. Yadahalli, J. Li, D. P. Lane, S. Gosavi and C. S. Verma, Sci. Rep., 2017, 7, 1 CrossRef CAS PubMed.
- T. Okamoto, K. Zobel, A. Fedorova, C. Quan, H. Yang, W. J. Fairbrother, D. C. S. Huang, B. J. Smith, K. Deshayes and P. E. Czabotar, ACS Chem. Biol., 2013, 8, 297 CrossRef CAS PubMed.
- E. A. Kumar, Q. Chen, S. Kizhake, C. Kolar, M. Kang, C.-en A. Chang, G. E. O. Borgstahl and A. Natarajan, Sci. Rep., 2013, 3, 1639 CrossRef PubMed.
- E. Danelius, M. Pettersson, M. Bred, J. Min, M. B. Waddell, R. K. Guy, M. Grøtli and M. Erdelyi, Org. Biomol. Chem., 2016, 14, 10386 RSC.
- (a) S.-R. Tzeng and C. G. Kalodimos, Nature, 2012, 488, 236 CrossRef CAS PubMed; (b) M.-H. Seo, J. Park, E. Kim, S. Hohng and H.-S. Kim, Nat. Commun., 2014, 5, 1 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob00831a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |