
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMerging singlet-oxygen induced furan oxidations with organocatalysis: synthesis of enantiopure cyclopentanones and hydrindanes†
Dimitris
Kalaitzakis
 ,
Manolis
Sofiadis
,
Vasileios
Tsopanakis
,
Tamsyn
Montagnon
and
Georgios
Vassilikogiannakis
*
,
Manolis
Sofiadis
,
Vasileios
Tsopanakis
,
Tamsyn
Montagnon
and
Georgios
Vassilikogiannakis
*
Department of Chemistry, University of Crete, Vasilika Vouton, 71003 Iraklion, Crete, Greece. E-mail: vasil@uoc.gr
First published on 24th March 2020
Abstract
A new methodology is described herein which converts simple and readily accesible furan substrates into complex enantio-enriched carbocyclic skeletons through the implementation of a simple one-pot procedure. Singlet oxygen furan photoxygenation affords an enedione which then participates in an organocatalysed double-Michael reaction with an enal to furnish a cyclopentanone structure with up to four new contiguous stereogenic centres. The enantioselectivity and diastereoselectivity of this process are both excellent. If desired, further aldol-annulation steps can be appended to the cascade reaction sequence to afford key enantiopure hydrindane motifs.
Introduction
Sustainability in chemistry1 is a complex paradigm that is in reality very difficult to achieve because it encompasses so many seemingly disparate facets. From using benign2 and abundant reagents and feedstocks3 to both atom-4 and step-economy5 (concepts united in “ideal” chemistry6), a host of different reaction characteristics affect its overall measurement.7 In our work, we have made substantial progress towards achieving the goal by using the traceless oxidant –singlet oxygen, 1O2–, to orchestrate cascade sequences which turn simple, readily accessible and relatively “flat” molecules into complex three-dimensional target scaffolds over the course of one synthetic operation.8 During this research, we have managed to solve a number of problems and meet additional challenges, such as using water as a solvent,9 applying the new methodologies within fragile biological molecules8e or scaling-up our photochemical processes.10 However, one fundamental pillar remained that, unless incorporated into the strategy, would limit its utility because of the homochiral world we live in: we needed to turn these racemic synthetic technologies into enantioselective ones. By combining the enantioselective bond forming power of organocatalysis,11 which often also scores high in sustainability metrics, with such cascades,12 we have now achieved a big step forward towards meeting this fundamental challenge.Singlet oxygen reactions have been paired with organocatalysis in the past in single step, simple, functional group interconversions;13–15 more specifically, stereoselective α-hydroxylation of ketones,13 aldehydes14 and 1,3-dicarbonyl compounds.15 The only multistep procedures are still a form of enantioselective hydroxylation, but in this case they proceed via a Kornblum–DeLaMare rearrangement (diene → 4-hydroxy-2-enone).16 We are targeting something different, a more general framework building reaction cascade wherein multiple bonds are diastereo- and enantioselectively constructed in a single synthetic operation.
Recently, we achieved the syntheses of a number of fused N-containing [5,5]-bicycles12via organocatalysed reactions of 4-pyrrolin-2-ones,17 but we now wanted to target some all-carbon skeletons with greater ubiquity and value, namely, enantiopure cyclopentanones, as well as important fused bicyclic congeners, and we hoped to do this by commandeering the enedione (of type 1i, Scheme 1) produced upon photooxygenation of furan substrates. Enediones should be extremely versatile due to their multisite and multi-type reactivity; features that lend themselves to their participation in domino reactions and/or complexity building sequences.8a,17 However, they have not been used as frequently as one might expect,18 given these desirable characteristics because of two intrinsically linked properties of theirs: firstly, there are few general syntheses of these intermediates,8 and, secondly, they are relatively unstable, which makes their storage and handling more complicated than it would otherwise be. In addition, tunability – the ability to control the site and type of reaction – has not yet been broadly achieved.19 In contrast, 4-oxo-alkyl-2-enoates (keto-ene-ester/acid) and amido-ene-keto/ester/acid have been widely studied due to their enhanced stability and more predictable reactivity patterns.20,21 We knew that we could make a wide variety of enediones in situ through mild furan photoxygenation protocols8 (1 → 1i, Scheme 1), thus, overcoming the problems described earlier. Crucially, furans, with a full range of substitution patterns, are readily accessible22 (including from biomass3). So we hoped that we could now build into our singlet-oxygen initiated cascade reaction sequences subsequent organo-catalysed double-Michael reactions (1i + 2 → 3, Scheme 1) that would furnish the desired cyclopentanones; regioselectively, diastereoselectively and enantioselectively. The efficient and stereoselective synthesis of cyclopentanes and cyclopentanones continues to be a very important challenge.23 If this initial target, creating up to four new contiguous stereocenters controllably, was successfully attained, we then hoped to extend the sequence to encompass an aldol-annulation (3 → 4, reminiscent of the Hajos–Parrish–Eder–Sauer–Wiechert reaction24), thus, also giving us stereoselective access to key hydrindane motifs24,25 (4, Scheme 1). Hydrindanes are core units of a large number of structurally diverse natural product targets.
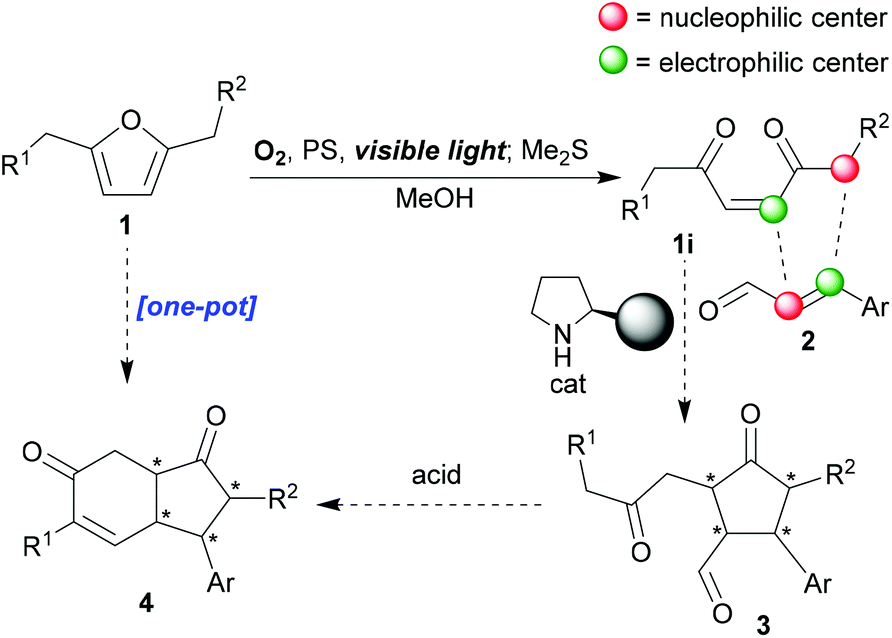 | ||
| Scheme 1 Proposed synthetic strategy for enantioselective access to cyclopentanones and hydrindane scaffolds. | ||
Results and discussion
We chose to start our investigation using the commercially available Jørgensen–Hayashi catalyst26 which had already proved so successful in many iminium catalysis scenarios (including hydrindane synthesis25). cis-Enedione 1ai was synthesised from furan 1a using the standard photoxygenation protocol that we had developed previously;8d–m we then tested its reaction with enal 2a under a range of conditions (Scheme 2). With regard to solvents, protic solvents proved to be the most efficient. Moreover, we were gratified to find that the “green” EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O mixture proved to be the best system tested (EtOH being a green solvent with attractive properties27). The enantiomeric and diastereomeric excesses obtained in these initial experiments were impressive. Furthermore, it was revealed that both geometrical isomers 1ai and 1aii gave the same product. When pure enedione 1ai was subjected to the organocatalyst (20 mol%) in EtOH:H2O (9:1), after 30 minutes complete isomerisation (cis → trans) had occurred. Exactly the same isomerisation was observed under the reaction conditions within 30 minutes. Finally, the optimized conditions were applied to a one-pot protocol starting directly from furan 1a which impressively afforded 3a in 65% overall isolated yield (with excellent dr and ee, Scheme 2C).
:H2O mixture proved to be the best system tested (EtOH being a green solvent with attractive properties27). The enantiomeric and diastereomeric excesses obtained in these initial experiments were impressive. Furthermore, it was revealed that both geometrical isomers 1ai and 1aii gave the same product. When pure enedione 1ai was subjected to the organocatalyst (20 mol%) in EtOH:H2O (9:1), after 30 minutes complete isomerisation (cis → trans) had occurred. Exactly the same isomerisation was observed under the reaction conditions within 30 minutes. Finally, the optimized conditions were applied to a one-pot protocol starting directly from furan 1a which impressively afforded 3a in 65% overall isolated yield (with excellent dr and ee, Scheme 2C).
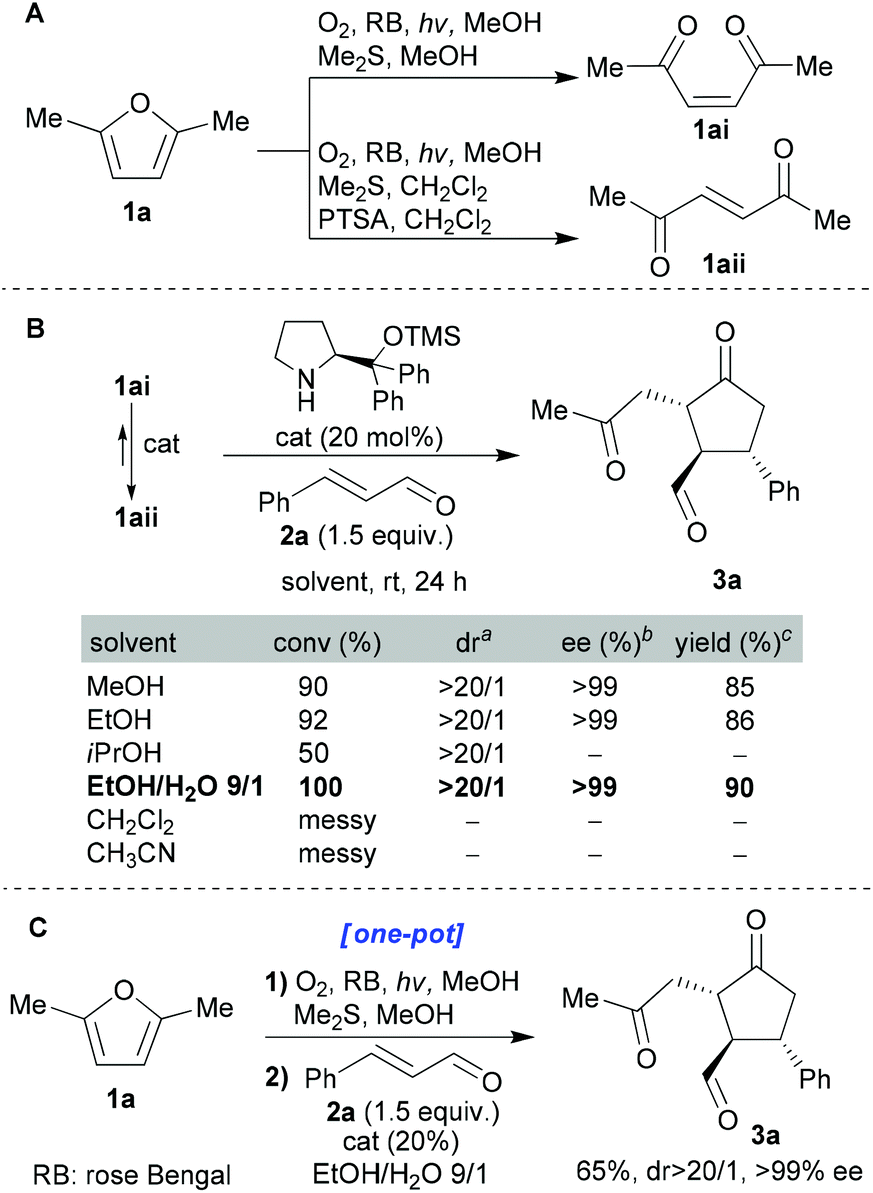 | ||
| Scheme 2 A) Singlet oxygen triggered transformation of 1a into the enediones 1ai and 1aii. (B) Optimization of the organocatalytic conditions. aDetermined by 1H-NMR of the crude reaction mixture. bDetermined by chiral HPLC analysis after reduction of the aldehyde group of 3a (isolated) using NaBH4. cYields of isolated 3a. (C) One-pot transformation of furan 1a to enantio-enriched 3a. All reactions were performed on a 0.1 mmol scale. | ||
These conditions were then applied to various enals (Scheme 3). All the ee values were measured using chiral HPLC analysis after reduction of the aldehyde group using NaBH4 (see the ESI† for full details). It can be seen that the dr and ee values were consistently extremely high and that isolated yields were excellent (59–65% for the one-pot processes and 79–95% for the stepwise variant) given the degree to which the complexity was enhanced through this one-pot process. Finally, the use of aliphatic enal 2-hexenal led to a messy reaction mixture containing a small amount of the desired cyclopentanone.
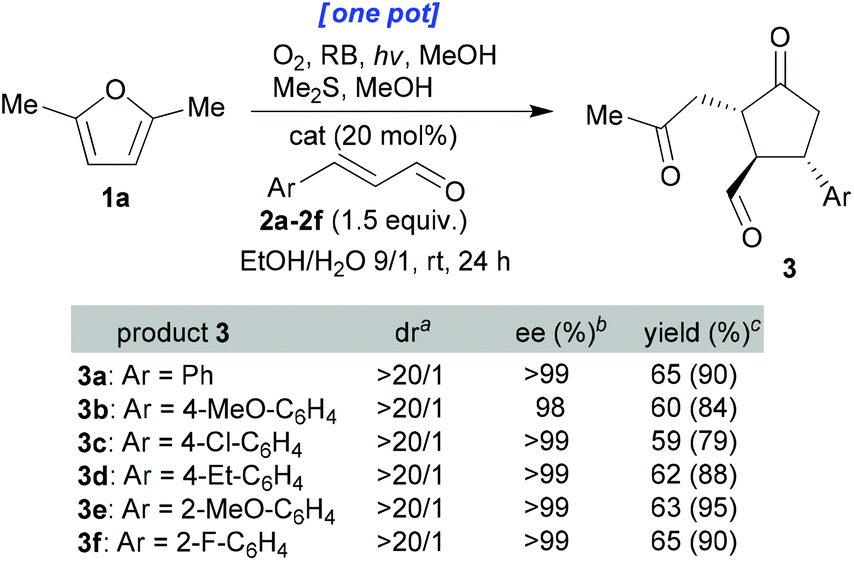 | ||
| Scheme 3 One-pot synthesis of optically pure cyclopentanones of type 3 from furan 1a. All reactions were performed on a 0.1 mmol scale. aDetermined by 1H-NMR of the crude reaction mixture. bDetermined by chiral HPLC analysis after reduction of the aldehyde group of the isolated 3 using NaBH4. cYields of isolated products 3. In parentheses are the isolated yields of products 3 when the reaction started from pure 1ai. | ||
We next turned our attention to extending the reaction sequence by adding an aldol-annulation step. To this end, isolated cyclopentanones 3a, 3c, and 3e were heated (70 °C) in DCE with PTSA (0.7 equiv.) for 8 hours (Scheme 4A). These conditions gave the desired fused products 4a, 4c and 4e with excellent yields, dr and ee (80–84, >20/1 and >99%, respectively). cis-Hydrindanes 4a, 4c and 4e were formed despite the starting aldehydes having a trans relationship at the corresponding centres (easily explained by the epimerisability of the centre next to the ketone when heated in the presence of an acid). This reaction was then successfully transferred into the one-pot protocol (1a → 4a, 4c, 4e; Scheme 4B). When the final aldol/dehydration step is included in our one-pot sequences, without isolation and purification of the intermediates, we observed that isolated yields of the final products were a bit better when methylene blue was used instead of rose Bengal. For this reason, we decided to use methylene blue in these one-pot protocols.
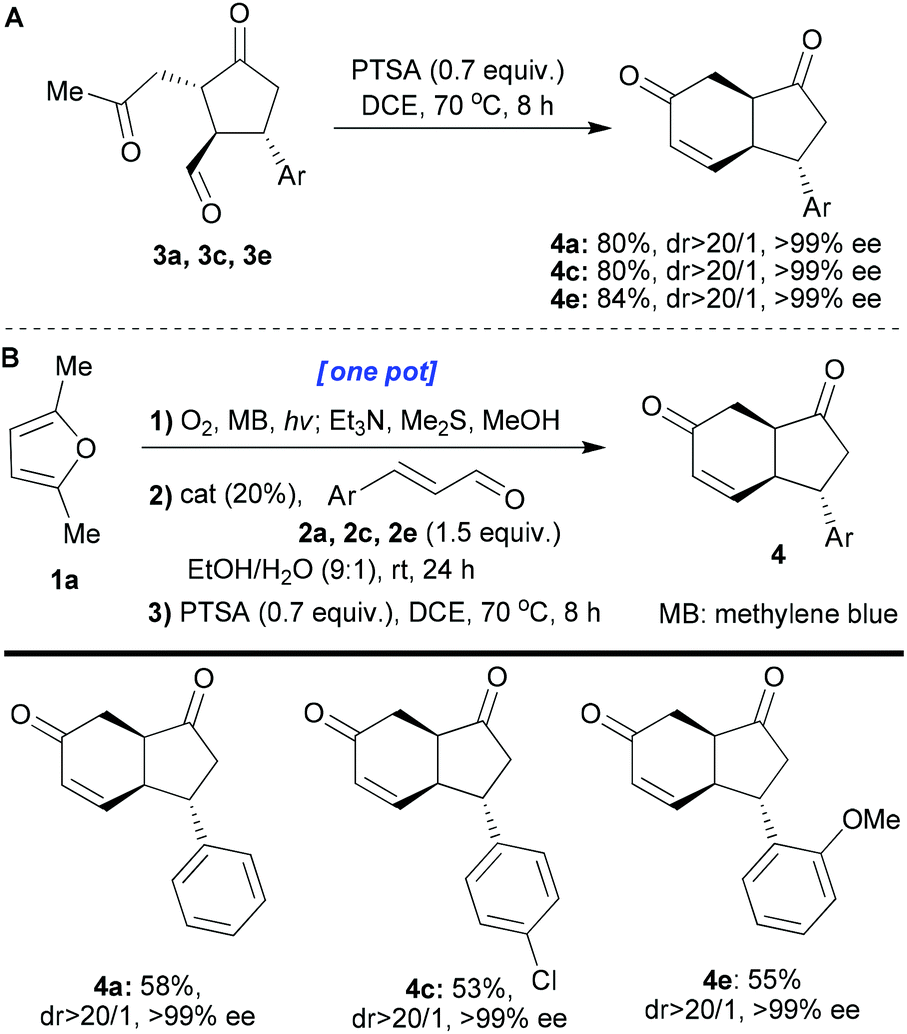 | ||
| Scheme 4 A) Synthesis of hydrindanes 4 starting from cyclopentanones 3. (B) One-pot synthesis of hydrindanes 4 starting from furan 1a. All the reactions were performed on a 0.1 mmol scale. The yields refer to isolated compounds. The dr values were measured by 1H-NMR of the crude reaction mixture. The ee values were determined by chiral HPLC analysis. | ||
In the next part of our study, unsymmetrical furans were investigated. In this case, while stereoselectivity remained paramount, good regioselectivity in the organo-catalysed step also needed to be attained. Enedione 1bi was prepared and shown to exhibit identical behaviour to enedione 1ai in terms of its cis → trans isomerisation (in EtOH:H2O 9:1, complete in 30 min) catalysed by the organocatalyst (20 mol%, Scheme 5). The previously optimised protocol (shown in Schemes 2 and 3) was, therefore, applied to furan 1b with notable results (entry 1 of the table shown in Scheme 5). The major product was 5a (5a/5a′ = 3/1, yield 52%, for isolated 5a) wherein the position α to a ketone that was more substituted (1bii, the position shown in red, Scheme 5) had reacted preferentially over the less substituted one (1bii, shown in orange, Scheme 5). Moreover, we managed to improve the regioselectivity by further promoting the formation of the product that comes from the thermodynamic enolate (increasing the temperature and decreasing the equivalence of enal 2a employed, entries 2–4 in the table within Scheme 5). In this way a ratio of 9/1 could be obtained for 5a/5a′ and the product 5a could be formed as a 1.2/1 mixture of inseparable diastereoisomers (isolated yield of 70% from 1b). In a further mechanistic clarification, the minor isomer 5a′ was isolated, dissolved in EtOH/H2O (9/1, 0.1 M) and an organocatalyst (20 mol%) was added. The resulting solution was heated to 50 °C and stirred for 24 h. No formation of isomer 5a was seen, confirming that the cyclisation is not reversible.
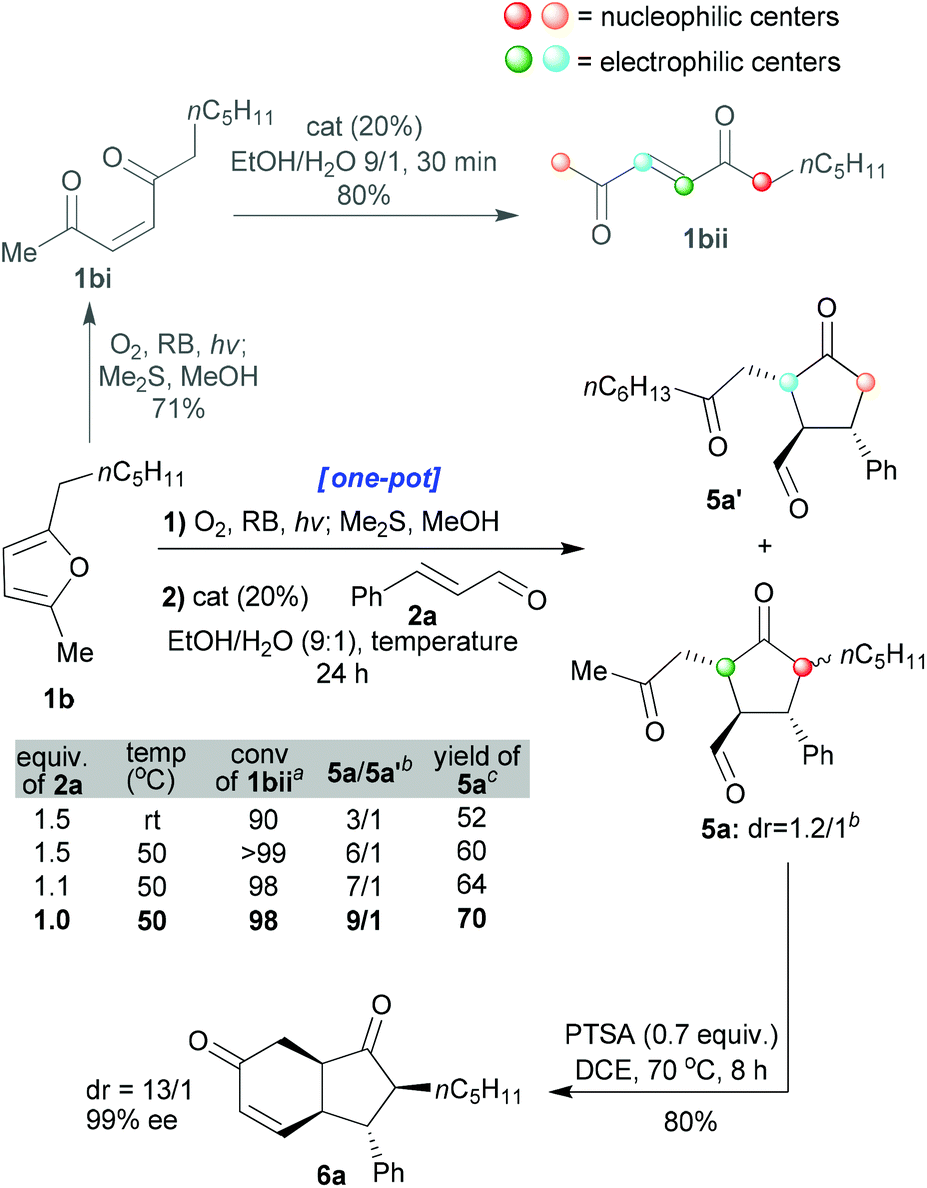 | ||
| Scheme 5 Studies on the stereoselective & regioselective one-pot transformation of 1b into cyclopentanones 5a and 5a′ and subsequent conversion to the carbocycle 6a. αConversion refers to the transformation of intermediate 1bii to 5a and 5a′ and was determined by 1H-NMR of the crude reaction mixture. bDetermined by 1H-NMR of the crude reaction mixture. cIsolated yields of the inseparable diastereomers of product 5a from furan 1b. | ||
Interestingly, the cyclisation reaction of 5a led exclusively to cis-fused 6a with a high distereomeric ratio and excellent enantiomeric excess (dr: 13/1, ee: 99%, and isolated yield: 80%). In this case, two epimerisations had occurred (both sides of the ketone, red and green positions 5a, Scheme 5). The optimised individual steps were then combined in one-pot reaction sequences beginning from a variety of disubstituted furans and employing a variety of enals (1 + 2 → 6, Scheme 6). The results, presented in Scheme 6, show that all the reactions afforded the desired products bearing 4 new stereogenic centres in good yields and with high dr and ee values.
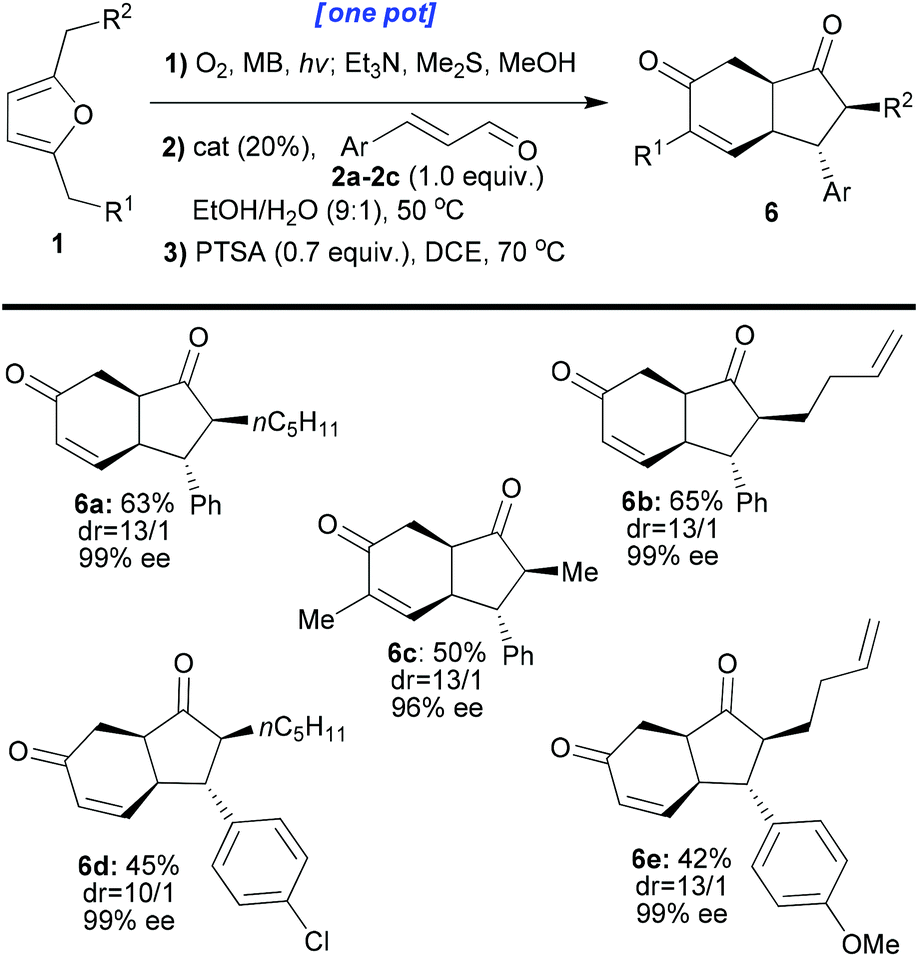 | ||
| Scheme 6 One-pot synthesis of enantio-enriched carbocycles of type 6 from disubstituted furans of type 1. All the reactions were performed on a 0.1 mmol scale. The yields refer to the isolated compounds. The dr values were measured by 1H-NMR of the crude reaction mixture. The ee values were determined by chiral HPLC analysis. | ||
In the final stage of the investigation, and to ensure that the new method could be applied to a wider range of synthetic challenges, we transposed the process into a continuous flow photoreactor28 to facilitate larger scale synthesis of these compounds. The photoreactor we used is our prototype10 based on the principle of irradiating nebulised mixtures. Use of the nebula overcomes issues of poor light penetration (rapid light attenuation, Bouger–Lambert–Beer law) and maximising gas–liquid interfaces for these biphasic reactions which have hindered the development of photochemical continuous flow reactor technologies. A series of test reactions were run in this system starting from either furan 1a or 1b (Scheme 7). In each case, after the photoxygenation step in the photoreactor (conversion = 100% and productivity = 0.35 mmol min−1), the oxidation products were collected in two flasks precharged with methanol and dimethylsulfide to achieve immediate reduction of the intermediate hydroperoxides to the enediones 1ai and 1bi. The solutions were divided and treated under different conditions (adapted from those described in Schemes 3, 4 and 6) to afford a range of products on a larger scale (1ai → 3a, 3c or 4e and 1bi → 6a or 6d) with the expected (high) selectivities.
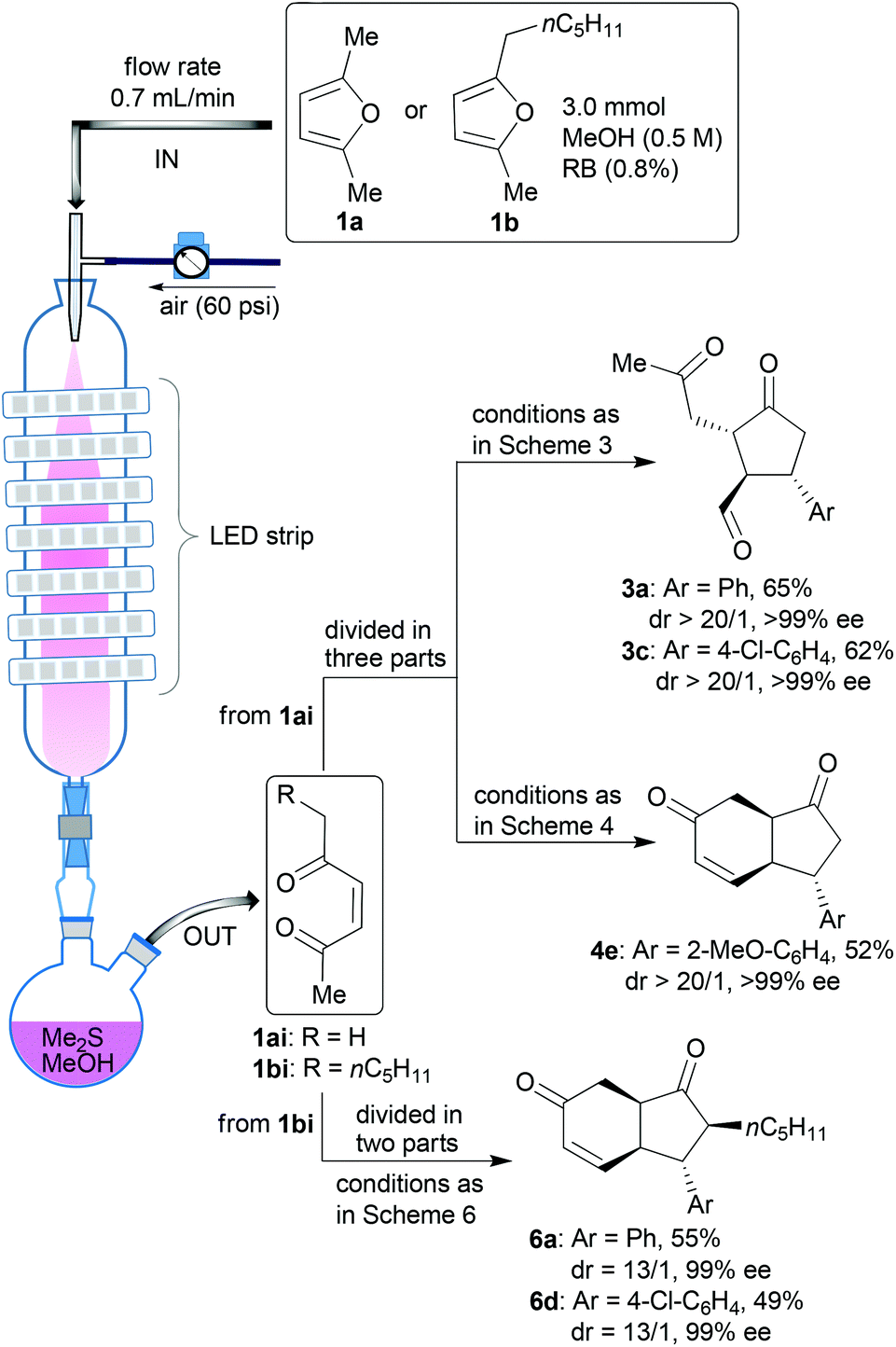 | ||
| Scheme 7 Use of a prototype photoreactor to achieve larger scale reaction sequences. | ||
Conclusions
Herein, we have presented a general, flexible and highly effective method for the regio-, diastereo- and enantioselective synthesis of important carbocycles, namely, enantiopure cyclopentanones and their hydrindane congeners. The protocols developed are all one-pot protocols which involve the use of relatively “flat” furan substrates and controllably transform them into three-dimensionally complex scaffolds with very high levels of stereochemical purity. The method exploits the ready accessibility of furans, variously substituted at will, as well as, their highly sustainable photoxygenation which affords a full range of reactive enedione intermediates ready for further reaction. The newly developed technology has also been successfully scaled-up using a prototype continuous flow photoreactor to further enhance the broad synthetic utility of this chemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to dedicate this article to Prof. John Gallos, our mentor and friend, on the occasion of his retirement.The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007-2013)/ERC grant agreement no. 277588. We thank the Greek General Secretariat of Research and Technology for matching (reward) funds (KA: 4143). We also thank the Alexander S. Onassis Public Benefit Foundation for the Ph.D. fellowship of Manolis Sofiadis (G ZM 063-1/2016-2017).
Notes and references
- (a) For the 12 principles of Green Chemistry, see: P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed (b) C.-J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197 CrossRef CAS PubMed.
- Benign can be defined (see ref that follows) as “a by-product, reagent or solvent that has no known environmental risk associated with it; for example water, low-concentration saline, dilute ethanol, autoclaved cell mass, etc.”. T. Hudlicky, D. A. Frey, L. Koroniak, C. D. Claeboe and L. E. Brammer Jr., Green Chem., 1999, 1, 57 RSC.
- D. Sengupta and R. W. Pike, Chemicals from Biomass; Integrating Bioprocesses into Chemical Production Complexes for Sustainable Development, CRC Press, Boca Raton Florida, 2012 Search PubMed.
- B. A. Trost, Science, 1991, 254, 1471 CrossRef CAS PubMed.
- P. A. Wender and B. L. Miller, Nature, 2009, 460, 197 CrossRef CAS PubMed.
- (a) T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010 RSC; (b) T. Gaich and P. S. Baran, J. Org. Chem., 2010, 75, 4657 CrossRef CAS PubMed.
- For a discussion on metrics, see: A. Lapkin and D. J. C. Constable, Green Chemistry Metrics: Measuring and Monitoring Sustainable Processes, Wiley, UK, 2008 Search PubMed.
- For reviews, see: (a) T. Montagnon, D. Kalaitzakis, M. Triantafyllakis, M. Stratakis and G. Vassilikogiannakis, Chem. Commun., 2014, 50, 15480 RSC; (b) T. Montagnon, D. Noutsias, I. Alexopoulou, M. Tofi and G. Vassilikogiannakis, Org. Biomol. Chem., 2011, 9, 2031 RSC; (c) T. Montagnon, M. Tofi and G. Vassilikogiannakis, Acc. Chem. Res., 2008, 41, 1001 CrossRef CAS PubMed. For leading examples, see: (d) D. Kalaitzakis, M. Triantafyllakis, G. I. Ioannou and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2017, 56, 4020 CrossRef CAS PubMed; (e) E. Antonatou, K. Hoogewijs, D. Kalaitzakis, A. Baudot, G. Vassilikogiannakis and A. Madder, Chem. – Eur. J., 2016, 22, 8457 CrossRef CAS PubMed; (f) D. Kalaitzakis, M. Triantafyllakis, M. Sofiadis, D. Noutsias and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2016, 55, 4605 CrossRef CAS PubMed; (g) D. Kalaitzakis, D. Noutsias and G. Vassilikogiannakis, Org. Lett., 2015, 17, 3596 CrossRef CAS PubMed; (h) D. Kalaitzakis, A. Kouridaki, D. Noutsias, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2015, 54, 6283 CrossRef CAS PubMed; (i) D. Kalaitzakis, T. Montagnon, E. Antonatou, N. Bardají and G. Vassilikogiannakis, Chem. – Eur. J., 2013, 19, 10119 CrossRef CAS PubMed; (j) D. Kalaitzakis, T. Montagnon, I. Alexopoulou and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2012, 51, 8868 CrossRef CAS PubMed; (k) I. Margaros, T. Montagnon and G. Vassilikogiannakis, Org. Lett., 2007, 9, 5585 CrossRef CAS PubMed; (l) T. Georgiou, M. Tofi, T. Montagnon and G. Vassilikogiannakis, Org. Lett., 2006, 8, 1945 CrossRef CAS PubMed; (m) G. Vassilikogiannakis and M. Stratakis, Angew. Chem., Int. Ed., 2003, 42, 5465 CrossRef CAS PubMed.
- (a) D. Kalaitzakis, M. Triantafyllakis, I. Alexopoulou, M. Sofiadis and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2014, 53, 13201 CrossRef CAS PubMed; (b) D. Noutsias, I. Alexopoulou, T. Montagnon and G. Vassilikogiannakis, Green Chem., 2012, 14, 601 RSC; (c) A. Kouridaki, T. Montagnon, M. Tofi and G. Vassilikogiannakis, Org. Lett., 2012, 14, 2374 CrossRef CAS PubMed.
- (a) G. I. Ioannou, T. Montagnon, D. Kalaitzakis, S. A. Pergantis and G. Vassilikogiannakis, ChemPhotoChem, 2018, 2, 860 CrossRef CAS PubMed; (b) G. I. Ioannou, T. Montagnon, D. Kalaitzakis, S. A. Pergantis and G. Vassilikogiannakis, Org. Biomol. Chem., 2017, 15, 10151 RSC; (c) G. I. Ioannou, T. Montagnon, D. Kalaitzakis, S. A. Pergantis and G. Vassilikogiannakis, ChemPhotoChem, 2017, 1, 173 CrossRef.
- (a) S. Bertelsen and K. A. Jørgensen, Chem. Soc. Rev., 2009, 38, 2178 RSC; (b) D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS PubMed; (c) C. F. Barbas, Angew. Chem., Int. Ed., 2008, 47, 42 CrossRef CAS PubMed.
- D. Kalaitzakis, M. Sofiadis, M. Triantafyllakis, K. Daskalakis and G. Vassilikogiannakis, Org. Lett., 2018, 20, 1146 CrossRef CAS PubMed.
- H. Sundén, M. Engqvist, J. Casas, I. Ibrahem and A. Córdova, Angew. Chem., Int. Ed., 2004, 43, 6532 CrossRef PubMed.
- (a) A. Córdova, H. Sundén, M. Engqvist, I. Ibrahem and J. Casas, J. Am. Chem. Soc., 2004, 126, 8914 CrossRef PubMed; (b) I. Ibrahem, G. L. Zhao, H. Sundén and A. Córdova, Tetrahedron Lett., 2006, 47, 4659 CrossRef CAS; (c) D. J. Walaszek, K. Rybicka-Jasińska, S. Smoleń, M. Karczewski and D. Gryko, Adv. Synth. Catal., 2015, 357, 2061 CrossRef CAS.
- (a) X. F. Tang, J. N. Zhao, Y. F. Wu, Z. H. Zheng, S. H. Feng, Z. Y. Yu, G. Z. Liu and Q. W. Meng, Org. Biomol. Chem., 2019, 17, 7938 RSC; (b) Y. Wang, H. Yin, X. Tang, Y. Wu, Q. Meng and Z. Gao, J. Org. Chem., 2016, 81, 7042 CrossRef CAS PubMed; (c) M. Lian, Z. Li, Y. Cai, Q. Meng and Z. Gao, Chem. – Asian J., 2012, 7, 2019 CrossRef CAS PubMed.
- (a) C. Wiegand, E. Herdtweck and T. Bach, Chem. Commun., 2012, 48, 10195 RSC; (b) S. T. Staben, X. Linghu and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 12658 CrossRef CAS PubMed.
- T. Montagnon, D. Kalaitzakis, M. Sofiadis and G. Vassilikogiannakis, Org. Biomol. Chem., 2020, 18, 180 RSC.
- For examples of enedione use in asymmetric organocatalysis, see: (a) D. Trubitsõn, S. Žari, S. Kaabel, M. Kudrjashova, K. Kriis, I. Järving, T. Pehk and T. Kanger, Synthesis, 2018, 50, 314 CrossRef; (b) B. Zhu, S. Qiu, J. Li, M. L. Coote, R. Lee and Z. Jiang, Chem. Sci., 2016, 7, 6060 RSC; (c) M. Ošeka, A. Noole, S. Žari, M. Öeren, I. Järving, M. Lopp and T. Kanger, Eur. J. Org. Chem., 2014, 3599 CrossRef. For examples of the closely related keto–ene–CHO, see: (d) Y. Hayashi, K. Nagai and S. Umemiya, Chem. – Asian J., 2019, 14, 4146 CrossRef CAS PubMed; (e) B. M. Paz, L. Klier, L. Næsborg, V. H. Lauridsen, F. Jensen and K. A. Jørgensen, Chem. – Eur. J., 2016, 22, 16810 CrossRef CAS PubMed; (f) M. He, J. R. Struble and J. W. Bode, J. Am. Chem. Soc., 2006, 128, 8418 CrossRef CAS PubMed.
- In our own chemistry, pendent nucleophiles have frequently been used to control regioselectivity; for examples, see ref. 8a–c.
- For keto–ene–ester/acid examples, see: (a) J.-B. Lin, S.-M. Xu, J.-K. Xie, H.-Y. Li and P.-F. Xu, Chem. Commun., 2015, 51, 3596 RSC; (b) W. Zhou, X. Su, M. Tao, C. Zhu, Q. Zhao and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14853 CrossRef CAS PubMed; (c) S. Žari, M. Kudrjashova, T. Pehk, M. Lopp and T. Kanger, Org. Lett., 2014, 16, 1740 CrossRef PubMed. For amide–enen–keto/ester/acid (n = 1,2) examples, see: (d) S. Muthusamy and V. Kesavan, Eur. J. Org. Chem., 2019, 4046 CrossRef CAS; (e) B.-X. Xiao, W. Du and Y.-C. Chen, Adv. Synth. Catal., 2017, 359, 1018 CrossRef CAS; (f) K. S. Halskov, F. Kniep, V. H. Lauridsen, E. H. Iversen, B. S. Donslund and K. A. Jørgensen, J. Am. Chem. Soc., 2015, 137, 1685 CrossRef CAS PubMed; (g) J. Stiller, P. H. Poulsen, D. C. Cruz, J. Dourado, R. L. Davis and K. A. Jørgensen, Chem. Sci., 2014, 5, 2052 RSC; (h) L. Hong and R. Wang, Adv. Synth. Catal., 2013, 355, 1023 CrossRef CAS; (i) K. Jiang, Z.-J. Jia, X. Yin, L. Wu and Y.-C. Chen, Org. Lett., 2010, 12, 2766 CrossRef CAS PubMed.
- Very recently a paper describing the formal [3 + 2]-cycloaddition between (E)-ethyl 4-oxopent-2-enoate and 2-enals for an elegant synthesis of Corey's lactone has been published, see: N. Umekubo, Y. Suga and Y. Hayashi, Chem. Sci., 2020, 11, 1205 RSC.
- (a) A. Deepthi, B. P. Babu and A. L. Balachandran, Org. Prep. Proced. Int., 2019, 51, 409 CrossRef CAS; (b) A. Blanc, V. Bénéteau, J. M. Weibel and P. Pale, Org. Biomol. Chem., 2016, 14, 9184 RSC; (c) A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084 CrossRef CAS PubMed; (d) X. L. Hou, H. Y. Cheung, T. Y. Hon, P. L. Kwan, T. H. Lo, S. Y. Tong and H. N. C. Wong, Tetrahedron, 1998, 54, 1955 CrossRef CAS.
- For a review of chiral cyclopentenone syntheses, see: (a) S. P. Simeonov, J. P. M. Nunes, K. Guerra, V. B. Kurteva and C. A. M. Afonso, Chem. Rev., 2016, 116, 5744 CrossRef CAS PubMed. For selected examples of organocatalysed cyclopentanone synthesis, see: (b) K. E. Ozboya and T. Rovis, Chem. Sci., 2011, 2, 1835 RSC; (c) A. Ma and D. Ma, Org. Lett., 2010, 12, 3634 CrossRef CAS PubMed. For selected examples of stereoselective organocatalysed cyclopentane synthesis with ≥3 centres controlled, see: (d) M. Laugeois, S. Ponra, V. Ratovelomanana-Vidal, V. Michelet and M. R. Vitale, Chem. Commun., 2016, 52, 5332 RSC; (e) K. S. Halskov, L. Næsborg, F. Tur and K. A. Jørgensen, Org. Lett., 2016, 18, 2220 CrossRef CAS PubMed; (f) B.-C. Hong, N. S. Dange, C.-S. Hsu, J.-H. Liao and G.-H. Lee, Org. Lett., 2011, 13, 1338 CrossRef CAS PubMed; (g) P. V. Pham, K. Ashton and D. W. C. MacMillan, Chem. Sci., 2011, 2, 1470 RSC; (h) G.-L. Zhao, I. Ibrahem, P. Dziedzic, J. Sun, C. Bonneau and A. Córdova, Chem. – Eur. J., 2008, 14, 10007 CrossRef CAS PubMed; (i) B. Tan, Z. Shi, P. J. Chua and G. Zhong, Org. Lett., 2008, 10, 3425 CrossRef CAS PubMed; (j) D. Enders, C. Wang and J. W. Bats, Angew. Chem., Int. Ed., 2008, 47, 7539 CrossRef CAS PubMed; (k) L. Zu, H. Li, H. Xie, J. Wang, W. Jiang, Y. Tang and W. Wang, Angew. Chem., Int. Ed., 2007, 46, 3732 CrossRef CAS PubMed.
- (a) S. G. Davies, R. L. Sheppard, A. D. Smith and J. E. Thomson, Chem. Commun., 2005, 3802 RSC; (b) B. List, L. Hoang and H. J. Martin, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5839 CrossRef CAS PubMed; (c) Z. G. Hajos and D. R. Parrish, J. Org. Chem., 1974, 39, 1615 CrossRef CAS; (d) U. Eder, G. Sauer and R. Wiechert, Angew. Chem., Int. Ed. Engl., 1971, 10, 496 CrossRef CAS.
- For examples of organocatalysed synthesis of hydrindane motifs, see: (a) Y. Stöckl, W. Frey, J. Lang, B. Claasen, A. Baro and S. Laschat, Synthesis, 2019, 51, 1123 CrossRef; (b) S. Koshino, E. Kwon and Y. Hayashi, Eur. J. Org. Chem., 2018, 5629 CrossRef CAS; (c) K. S. Halskov, B. S. Donslund, S. Barfüsser and K. A. Jørgensen, Angew. Chem., Int. Ed., 2014, 53, 4137 CrossRef CAS PubMed; (d) P. He, X. Liu, J. Shi, L. Lin and X. Feng, Org. Lett., 2011, 13, 936 CrossRef CAS PubMed; (e) N. T. Vo, R. D. M. Pace, F. O'Hara and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 404 CrossRef CAS PubMed; (f) Y. Hayashi, H. Sekizawa, J. Yamaguchi and H. Gotoh, J. Org. Chem., 2007, 72, 6493 CrossRef CAS PubMed; (g) Y. Hayashi, H. Gotoh, T. Tamura, H. Yamaguchi, R. Masui and M. Shoji, J. Am. Chem. Soc., 2005, 127, 16028 CrossRef CAS PubMed.
- (a) Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212 CrossRef CAS PubMed; (b) M. Marigo, T. C. Wabnitz, D. Fielenbach and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 794 CrossRef CAS PubMed.
- (a) F. G. Calvo-Flores, M. J. Monteagudo-Arrebola, J. A. Dobado and J. Isac-García, Top. Curr. Chem., 2018, 376, 18 CrossRef PubMed; (b) C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927 RSC.
- For recent reviews on flow photoreactors, see: (a) M. Di Filippo, C. Bracken and M. Baumann, Molecules, 2020, 25, 356 CrossRef CAS PubMed; (b) T. H. Rehm, ChemPhotoChem, 2019, 3, 1 CrossRef. For a recent comprehensive review of flow chemistry, see: (c) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob00468e |
| This journal is © The Royal Society of Chemistry 2020 |