
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organocatalytic carbon dioxide fixation to epoxides by perfluorinated 1,3,5-triols catalysts†
Céline
Sperandio
,
Jean
Rodriguez
and
Adrien
Quintard
 *
*
Aix Marseille Univ, CNRS, Centrale Marseille, iSm2, Marseille, France. E-mail: adrien.quintard@univ-amu.fr; Web: http://ism2.univ-amu.fr/fr/annuaire/stereo/quintardadrien
First published on 13th March 2020
Abstract
In order to improve epoxides conversion to carbonates by fixation of CO2 a new type of perfluorinated triol catalysts was developed. These simple acyclic scaffolds of enhanced acidity are efficient for catalysis through selective H-bonding activation of the epoxide. In combination with TBAI as co-catalyst, this useful transformation is performed under only 1 atmosphere of CO2 and between 30 to 80 °C. Both the 1,3,5-triol motif and the perfluorinated side chains are crucial in order to observe this epoxide opening under such mild conditions. In addition, the stereochemistry of the starting epoxide can efficiently be conserved during the carbonate formation.
Designing catalysts with improved activity and/or selectivity is at the heart of modern chemical research. For this purpose, chemists must identify new scaffolds possessing optimized functions and geometry for maximized efficiency in useful catalytic transformations.
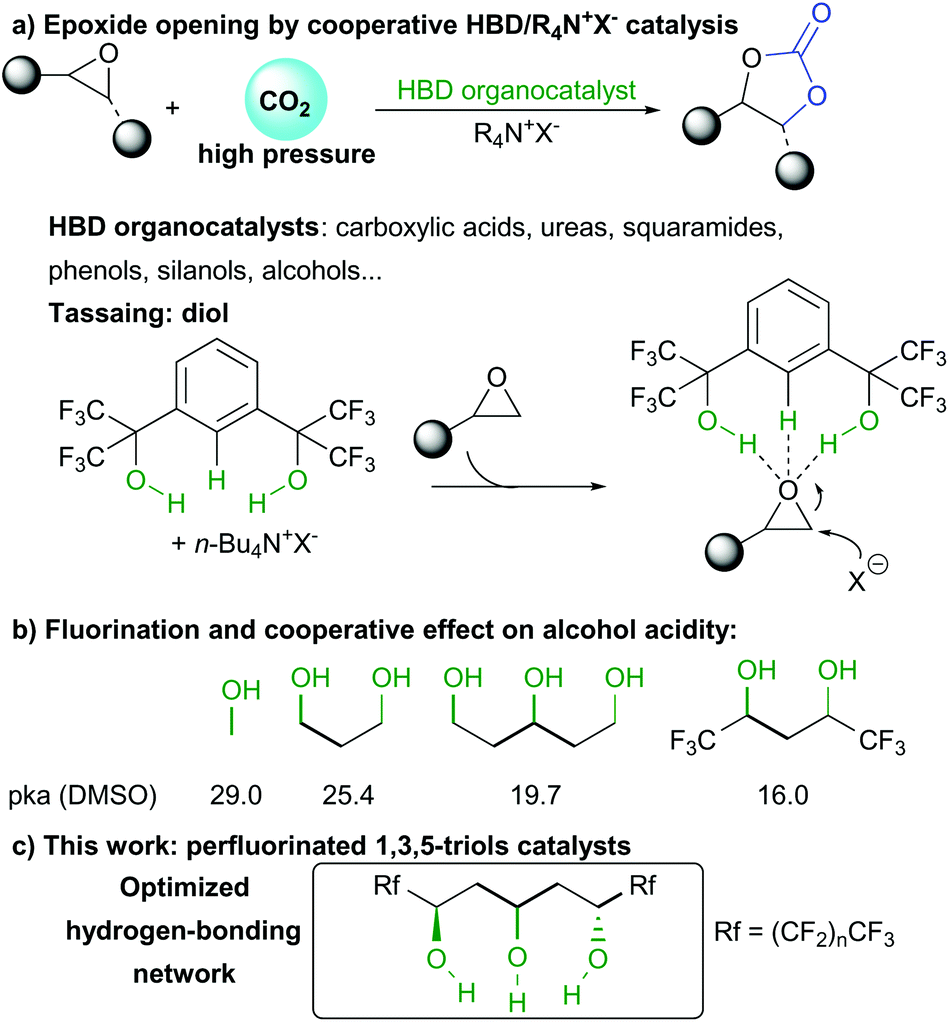
The massive amount of CO2 generated in the last decades have pushed chemists to take advantage of this chemical as an inexpensive and abundant raw material. In this context, CO2 can be used in a circular economy platform either as an energy source through its transformation to fuels such as methanol, or as a source of bulk chemical commodities.1 Among these, cyclic carbonates of great industrial utility can be generated by a fascinating epoxide opening with CO2 (Scheme 1a). These carbonates can be used as alternative solvents or for the production of plastics, pharmaceuticals and other fine chemicals.2 Given this industrial interest, development of new tools enabling the optimum reuse of CO2 for the preparation of these higher value chemicals is vital.
 | ||
| Scheme 1 Organocatalytic epoxide opening with CO2 and proposed catalysts. | ||
In order to activate CO2 towards its appropriate fixation on epoxides, tremendous efforts have been achieved to improve reaction efficiency and decrease costs and hazards.3 Progress in that direction notably imply introduction of inexpensive organocatalysts of improved catalytic activity and avoiding unnecessary toxic metals.4 However, despite the design of better catalysts, the challenge is to introduce alternative catalytic scaffolds enabling CO2 fixation under exquisitely mild conditions. Most notably, simple catalytic structures enabling CO2 activation and fixation at temperatures below 100 °C and atmospheric pressure are highly desirable for greener industrial processes and still constitutes a challenge.5 Given the lack of activity of numerous organocatalysts, a number of systems combining Hydrogen Bond Donor catalysts (HBD) and a quaternary ammonium salt co-catalyst have been developed. The nucleophilic counter anion of the salt acts as a temporary relay, opening the HBD-activated epoxide and serving as leaving group upon ring closure after CO2 fixation.
For such purpose, various HBD catalysts based on different active functions such as carboxylic acids,6 ureas and thioureas,7 squaramides,8 phenols,9 silanols10 or alcohols11 have been described in the last years. Among alcohol HBD, the group of Tassaing extensively studied the structure–activity relationship leading to improved catalytic efficiency (Scheme 1a). They discovered that heavily fluorinated distal 1,5-diols provided the best catalytic activity, even though high CO2 pressure (>10 bars) was required to ensure optimum conversion.12 In depth mechanistic studies indicated that the acidic fluorinated alcohols and the central aromatic proton were involved in the H-bond activation of epoxides. However, DFT calculations clearly indicated that the central aromatic scaffold was not optimum as observed through longer hydrogen bond between the aromatic proton and the Lewis basic oxygen atom.
In addition, in-depth analysis of alcohols acidity by the group of Kass indicated that a H-bonding cooperative effect in 1,3-polyols was responsible for enhanced acidity from a pKa of 29.0 in the case of methanol to 19.7 for pentane-1,3,5-triol (Scheme 1b).13 Furthermore, insertion of CF3 groups also considerably lowered the molecule acidity.14 Given these observations, we hypothesized that a perfluorinated 1,3,5-triol motif we recently discovered15 would provide the requisite H-bonding scaffold geometry for the critical epoxide opening and CO2 activation. In addition, given the known anion coordination properties of such triol scaffolds, further stabilization of anionic intermediates could also facilitate the catalytic cycle (Scheme 1c).
In order to assess the feasibility of the epoxide opening under mild conditions, we tested the catalytic activity of different 1,3,5-triols using styrene oxide 1a and 1 atmosphere of CO2 (Table 1). Under neat conditions, all three perfluorinated triols cat1–3 tested catalyzed the reaction at only 30 °C (entries 1–3). Increasing perfluorinated chains length slightly improved the conversion from 21% to 42% after only 6 hours. This result confirms the importance of the alcohol network acidity over the catalytic activity. Triol catalysts do not suffer from any deactivation and full conversion can be obtained after only 16 hours, highlighting their robustness and excellent activity under mild conditions (entry 4). In the absence of HBD catalyst, TBAI itself scarcely promotes the epoxide opening under those conditions (entry 5). Moreover, the HBD cat3 alone is also poorly efficient providing trace amount of carbonate (entry 6).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Temperature | Time | Conversiona |
| Unless otherwise stated, this reaction was carried with 5 mol% of triol, 5 mol% of TBAI under an atmospheric pressure (balloon) of CO2.a Reaction conversions were obtained by 1H NMR by determining the ratios between 1a and 2a.b In the absence of TBAI. | ||||
| 1 | cat1 | 30 °C | 6 h | 21% |
| 2 | cat2 | 30 °C | 6 h | 36% |
| 3 | cat3 | 30 °C | 6 h | 42% |
| 4 | cat2 | 30 °C | 16 h | >99% |
| 5 | None | 30 °C | 6 h | <5% |
| 6b | cat3 | 30 °C | 6h | <5% |
| 7 | cat1 | 80 °C | 6 h | 75% |
| 8 | cat2 | 80 °C | 6 h | 80% |
| 9 | cat3 | 80 °C | 6 h | 85% |
| 10 | cat2 | 80 °C | 12 h | >99% |
| 11 | cat4 | 80 °C | 6 h | 10% |
| 12 | cat5 | 30 °C | 6 h | <5% |
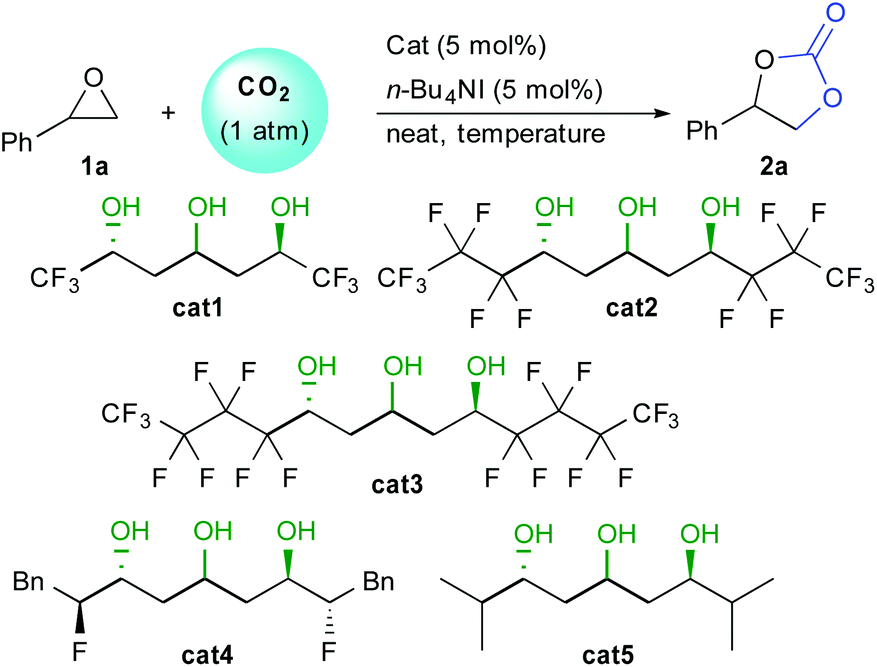
In order to improve kinetics, the reaction was then performed at 80 °C. After 6 hours, good levels of conversion were obtained with cat1–3 (75–85% conversion, entries 7–9), while full conversion was attained after 12 hours (entry 10). To understand the role of the perfluorinated chain, bis-fluorohydrin triol cat4 was also tested under the same conditions.16 Even though this catalyst possessing the same central triol motif had shown good activity as HBD in Friedel–Crafts reactions, here, it provided only 10% conversion after 6 hours. Finally, non-fluorinated triol cat5 did not provide efficient epoxide activation providing only trace amount of 2a (entry 12). These results clearly demonstrate the importance of the enhanced acidity of the triol core for optimum catalytic activity.
With optimal conditions in hand, we then scrutinized the efficiency of the transformation on variously substituted epoxides (Table 2). The remarkable reactivity observed using 5 mol% of organocatalyst cat2 was further confirmed independently of the epoxide structure. In order to ascertain the impact of epoxide nature over reactivity, conversions were determined at different reaction times. Going from styrene oxide 1a to propylene oxide 1b slightly reduced reaction kinetic and full conversion were obtained after 24 h (entries 1–4). Adjacent alcohols and ethers functions were also well tolerated providing carbonates 2c–d with full conversion after 24 hours (entries 5–8). Epichlorohydrin 1e was more reactive providing 82% conversion after only 4 hours and full conversion after 12 hours (entries 9 and 10). Finally, even 1,2-disubstituted epoxides such as 1f, usually recalcitrant to opening could be converted to the corresponding carbonate with full conversion and 89% yield after 48 hours (entry 12). The tolerance of our system towards increased steric hindrance further demonstrates the efficiency of cat2 as HBD.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Time | Conversiona | Yieldb |
| This reaction was carried with 5 mol% of cat2, 5 mol% of TBAI under an atmospheric pressure (balloon) of CO2.a Reaction conversions were obtained by 1H NMR.b Isolated yield. nd = not determined.c Obtained as the cis-diastereomer. | ||||
| 1 | 2a | 6 h | 80% | nd |
| 2 | 2a | 12 h | >99% | 78% |
| 3 | 2b | 4 h | 35% | nd |
| 4 | 2b | 24 h | 99% | 84% |
| 5 | 2c | 4 h | 35% | nd |
| 6 | 2c | 24 h | 99% | 89% |
| 7 | 2d | 4 h | 50% | nd |
| 8 | 2d | 24 h | 99% | 92% |
| 9 | 2e | 4 h | 82% | nd |
| 10 | 2e | 12 h | 99% | 79% |
| 11 | 2f | 4 h | 14% | nd |
| 12 | 2f | 48 h | 99% | 89%c |
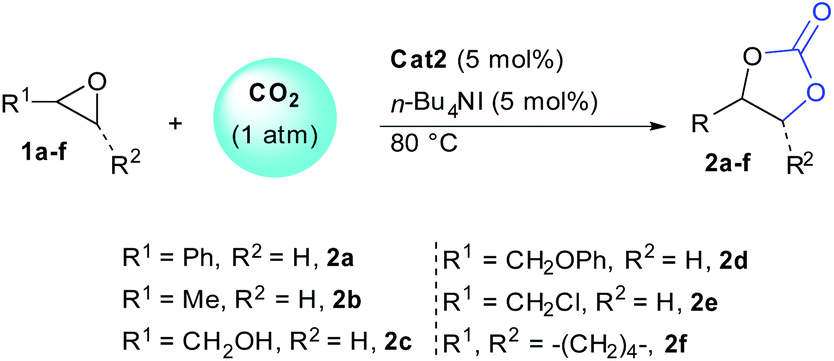
In order to get more insight on the mechanism, several control reactions were then performed (Scheme 2). Using the enantiopure epoxide (R)-1a, the stereochemical information was conserved on both starting epoxide and carbonate, indicating that the epoxide opening must occur preferentially on the primary carbon atom [(a) and (b)]. At full conversion (12 h), the enantioenriched carbonate 2a was obtained in 95% ee (a). On the other hand, starting from a racemic epoxide 1a, the enantiopure cat2* operated without enantioselectivity (c). Finally, at partial or full conversion, no resolution pathways could be observed providing either remaining epoxide or carbonate as racemates [(c) and (d)].
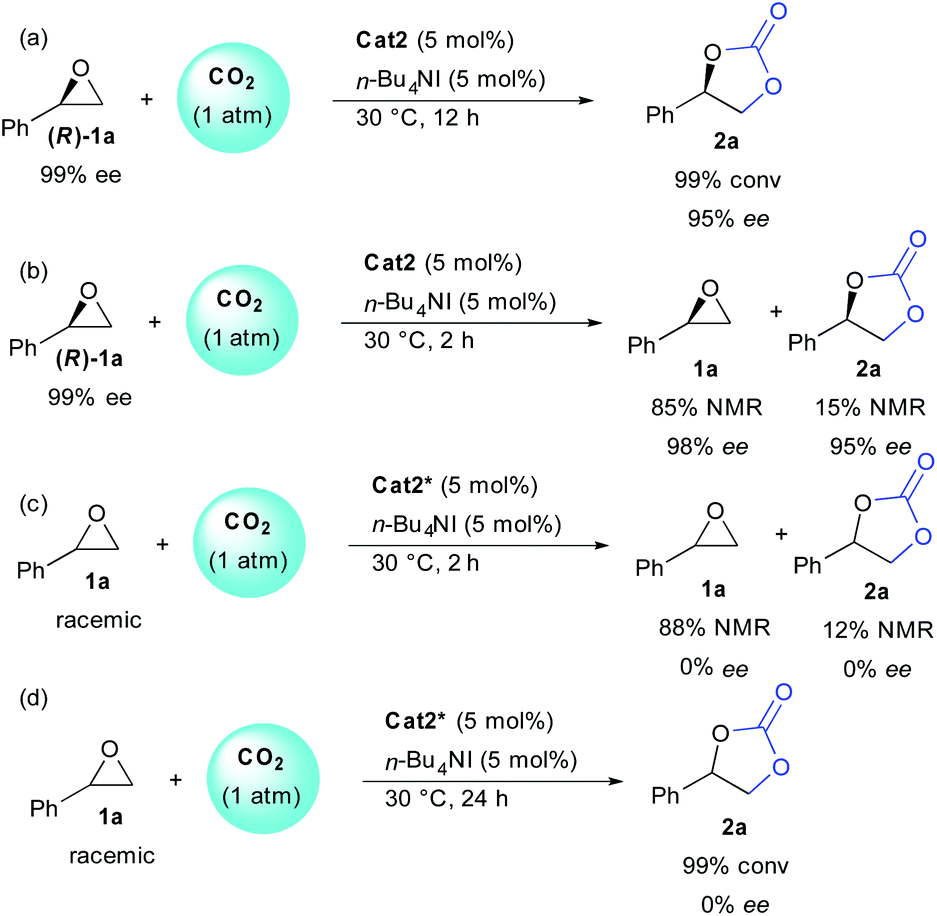 | ||
| Scheme 2 Stereochemical outcome of the epoxide opening. | ||
According to these experiments, the observed higher reactivity of cat2 and from previous mechanistic studies using alcohols as HBD catalysts,7b,11,12 the reaction occurs through the synergistic role of TBAI and the 1,3,5-triol hydrogen bonding network (Scheme 3). According to the catalytic mechanism previously calculated for diols, the epoxide can be activated through simultaneous strong H-bonding interactions.17 Regioselective nucleophilic ring opening by the iodine leads to the triol-stabilized alcoholate B. Given the known anion-binding properties of cat2,15 the triol motif should stabilize the formed anion B. Subsequent addition to CO2 provides iodo-carboxylate C, prone to cyclize forming the carbonate by iodine substitution and catalyst liberation. The improved reactivity of cat2 as compared to previous catalysts is due to the cooperativity of the acyclic triol network enhanced by the presence of the two perfluorinated chains.
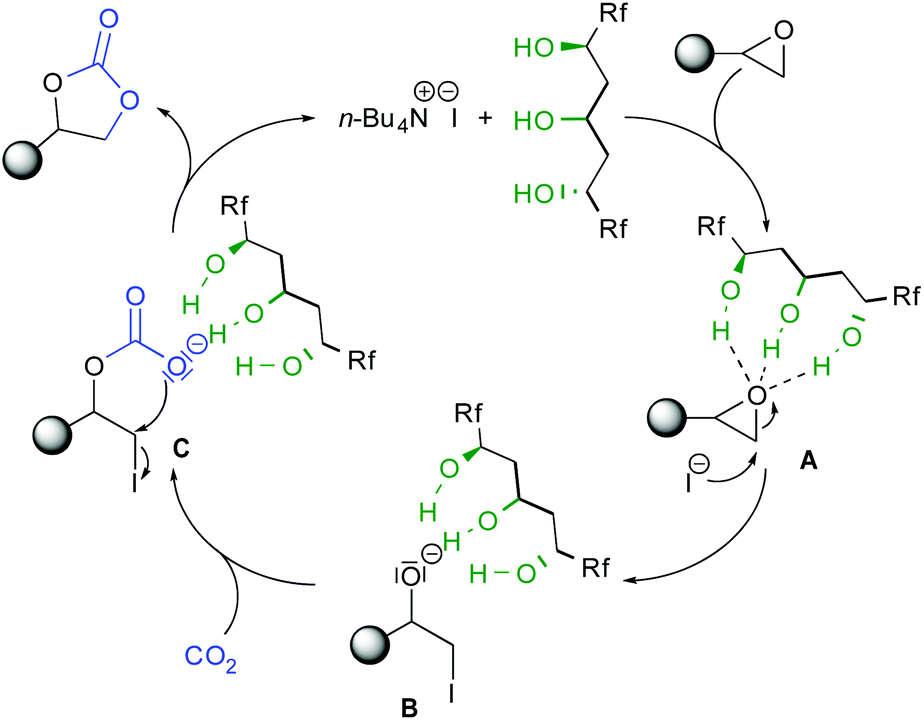 | ||
| Scheme 3 Proposed mechanism for the epoxide opening. | ||
To conclude, we have demonstrated that perfluorinated triol scaffolds of appropriate geometry could be excellent catalysts for the conversion of epoxides to carbonates. In combination with TBAI as synergistic co-catalyst, under moderate temperature (30 to 80 °C) and of utmost importance using only 1 atmosphere of CO2, full conversion to the corresponding carbonates could be observed.
In addition to the improvements it represents for this crucial reaction, this study opens the door to other related use of perfluorinated triols as organocatalysts or to a recyclable version thanks to the fluorous tag present on the catalyst structure.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
A patent on the preparation and properties of perfluorinated 1,3,5-triols has been submitted by Céline Sperandio, Jean Rodriguez and Adrien Quintard. Patent declaration. EP19 305862.5. Submitted 27 June 2019.Acknowledgements
The Centre National de la Recherche Scientifique (CNRS) and the Aix-Marseille Université are warmly acknowledged for financial support. All technical staff from Aix-Marseille Spectropole are acknowledged for their support.Notes and references
- General reviews: (a) A. J. Hunt, E. H. K. Sin, R. Marriott and J. H. Clark, ChemSusChem, 2010, 3, 306 CrossRef CAS PubMed; (b) M. Aresta, A. Dibenedeto and A. Angelini, Chem. Rev., 2014, 114, 1709 CrossRef CAS PubMed; (c) A. Otto, T. Grube, S. Schiebahn and D. Stolten, Energy Environ. Sci., 2015, 8, 3283 RSC; (d) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed; (e) E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840 CrossRef PubMed; (f) D.-H. Lan, N. Fan, Y. Wang, X. Gao, P. Zhang, L. Chen, C.-T. Au and S.-F. Yin, Chin. J. Catal., 2016, 37, 826 CrossRef CAS; (g) J. A. Martens, A. Bogaerts, N. De Kimpe, P. A. Jacobs, G. B. Marin, K. Rabaey, M. Saeys and S. Verhelst, ChemSusChem, 2017, 10, 1039 CrossRef CAS PubMed; (h) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709 CrossRef CAS PubMed.
- (a) C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353 CrossRef; (b) M. Alves, B. Grignard, R. Mereau, C. Jerome, T. Tassaing and C. Detrembleur, Catal. Sci. Technol., 2017, 7, 2651 RSC; (c) A. W. Kleij, M. North and A. Urakawa, ChemSusChem, 2017, 10, 1036 CrossRef CAS PubMed; (d) R. Rajjak Shaikh, S. Pornpraprom and V. D'Elia, ACS Catal., 2018, 8, 419 CrossRef.
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966 RSC.
- (a) G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375 RSC; (b) M. Cokoja, M. E. Wilhelm, M. H. Anthofer, W. A. Herrmann and F. E. Kühn, ChemSusChem, 2015, 8, 2436 CrossRef CAS PubMed; (c) J. Wang, Curr. Green Chem., 2015, 2, 3 CrossRef CAS; (d) M. Alves, B. Grignard, R. Mereau, C. Jerome, T. Tassaing and C. Detrembleur, Catal. Sci. Technol., 2017, 7, 2651 RSC.
- For recent examples of organocatalysts not requiring additional onium salt: (a) X.-F. Liua, Q.-W. Songa, S. Zhanga and L.-N. He, Catal. Today, 2016, 263, 69 CrossRef; (b) X. Wu, C. Chen, Z. Guo, M. North and A. C. Whitwood, ACS Catal., 2019, 9, 1895 CrossRef CAS; (c) R. Sharma, A. Bansal, C. N. Ramachandran and P. Mohanty, Chem. Commun., 2019, 55, 11607 RSC and references cited herein.
- (a) J. Tharun, G. Mathai, A. C. Kathalikkattil, R. Roshan, J.-Y. Kwak and D.-W. Park, Green Chem., 2013, 15, 1673 RSC; (b) N. Liu, Y.-F. Xie, C. Wang, S.-J. Li, D. Wei, M. Li and B. Dai, ACS Catal., 2018, 8, 9945 CrossRef CAS.
- (a) Y.-D. Li, D.-X. Cui, J.-C. Zhu, P. Huang, Z. Tian, Y.-Y. Jiab and P.-A. Wang, Green Chem., 2019, 21, 5231 RSC; (b) Y. Fan, M. Tiffner, J. Schörgenhumer, R. Robiette, M. Waser and S. R. Kass, J. Org. Chem., 2018, 83, 9991 CrossRef CAS PubMed.
- (a) S. Sopeña, E. Martin, E. C. Escudero-Adán and A. W. Kleij, ACS Catal., 2017, 7, 3532 CrossRef; (b) K. Takaishi, T. Okuyama, S. Kadosaki, M. Uchiyama and T. Ema, Org. Lett., 2019, 21, 1397 CrossRef CAS PubMed.
- (a) J. W. Huang and M. Shi, J. Org. Chem., 2003, 68, 6705 CrossRef CAS PubMed; (b) Y. M. Shen, W. L. Duan and M. Shi, Eur. J. Org. Chem., 2004, 3080 CrossRef CAS; (c) C. J. Whiteoak, A. Nova, F. Maseras and A. W. Kleij, ChemSusChem, 2012, 5, 2032 CrossRef CAS PubMed; (d) C. J. Whiteoak, A. H. Henseler, C. Ayats, A. W. Kleij and M. A. Pericàs, Green Chem., 2014, 16, 1552 RSC; (e) S. Liu, N. Suematsu, K. Maruoka and S. Shirakawa, Green Chem., 2016, 18, 4611 RSC; (f) Y. Toda, Y. Komiyama, H. Esaki, K. Fukushima and H. Suga, J. Org. Chem., 2019, 84, 15578 CrossRef CAS PubMed.
- A. M. Hardman-Baldwin and A. E. Mattson, ChemSusChem, 2014, 7, 3275 CrossRef CAS PubMed.
- (a) S. Liang, H. Liu, T. Jiang, J. Song, G. Yang and B. Han, Chem. Commun., 2011, 47, 2131 RSC; (b) R. A. Watile, K. M. Deshmukh, K. P. Dhake and B. M. Bhanage, Catal. Sci. Technol., 2012, 2, 1051 RSC; (c) H. Büttner, K. Lau, A. Spannenberg and T. Werner, ChemCatChem, 2015, 7, 459 CrossRef; (d) M. Wilhelm, M. H. Anthofer, M. Cokoja, I. I. E. Markovits, W. A. Herrmann and F. E. Kühn, ChemSusChem, 2014, 7, 1357 CrossRef CAS PubMed; (e) S. Kaneko and S. Shirakawa, ACS Sustainable Chem. Eng., 2017, 5, 2836 CrossRef CAS; (f) S. Arayachukiat, C. Kongtes, A. Barthel, S. V. C. Vummaleti, A. Poater, S. Wannakao, L. Cavallo and V. D'Elia, ACS Sustainable Chem. Eng., 2017, 5, 6392 CrossRef CAS; (g) Y. A. Rulev, Z. T. Gugkaeva, A. V. Lokutova, V. I. Maleev, A. S. Peregudov, X. Wu, M. North and Y. N. Belokon, ChemSusChem, 2017, 10, 1152 CrossRef CAS PubMed.
- (a) S. Gennen, M. Alves, R. Méreau, T. Tassaing, B. Gilbert, C. Detrembleur, C. Jerome and B. Grignard, ChemSusChem, 2015, 8, 1845 CrossRef CAS PubMed; (b) M. Alves, B. Grignard, S. Gennen, R. Mereau, C. Detrembleur, C. Jerome and T. Tassaing, Catal. Sci. Technol., 2015, 5, 4636 RSC; (c) M. Alves, R. Mereau, B. Grignard, C. Detrembleur, C. Jerome and T. Tassaing, RSC Adv., 2016, 6, 36327 RSC.
- (a) A. Shokri, J. Schmidt, X.-B. Wang and S. R. Kass, J. Am. Chem. Soc., 2012, 134, 16944 CrossRef CAS PubMed; (b) A. Shokri, J. Schmidt, X.-B. Wang and S. R. Kass, J. Am. Chem. Soc., 2012, 134, 2094 CrossRef CAS PubMed; (c) A. Shokri, A. Abedin, A. Fattahi and S. R. Kass, J. Am. Chem. Soc., 2012, 134, 10646 CrossRef CAS PubMed.
- (a) A. Shokri, X.-B. Wang and S. R. Kass, J. Am. Chem. Soc., 2013, 135, 9525 CrossRef CAS PubMed; (b) A. Shokri and S. R. Kass, Chem. Commun., 2013, 49, 11674 RSC.
- C. Sperandio, J. Rodriguez and A. Quintard, Chem. Sci., 2020, 11, 1629 RSC.
- C. Sperandio, G. Quintard, J. V. Naubron, M. Giorgi, M. Yemloul, J.-L. Parrain, J. Rodriguez and A. Quintard, Chem. – Eur. J., 2019, 25, 15098 CrossRef CAS PubMed.
- Alternatively, a cooperative intramolecular H-bonding in the triol can increase the acidity of the catalyst favoring the epoxide activation by a single catalyst (see ref. 14).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob00402b |
| This journal is © The Royal Society of Chemistry 2020 |