
Chimeric γPNA–Invader probes: using intercalator-functionalized oligonucleotides to enhance the DNA-targeting properties of γPNA†

Abstract
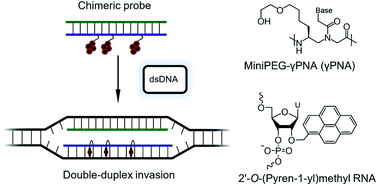
Gamma peptide nucleic acids (γPNAs), i.e., single-stranded PNA strands that are modified at the γ-position with (R)-diethylene glycol, and Invader probes, i.e., DNA duplexes with +1 interstrand zipper arrangements of 2′-O-(pyren-1-yl)methyl-RNA monomers, are two types of nucleic acid mimics that are showing promise for sequence-unrestricted recognition of double-stranded (ds) DNA targets. We recently demonstrated that recognition of dsDNA targets with self-complementary regions is challenging for single-stranded high-affinity probes like γPNAs due to their proclivity for secondary structure formation, but not so for Invader probes, which are engineered to form readily denaturing duplexes irrespective of the target sequence context. In the present study, we describe an approach that mitigates these limitations and improves the dsDNA-recognition properties of γPNAs in partially self-complementary target contexts. Chimeric probes between γPNAs and individual Invader strands are shown to form metastable duplexes that (i) are energetically activated for recognition of complementary mixed-sequence dsDNA target regions, (ii) reduce γPNA dimerization, and (iii) substantially improve the fidelity of the dsDNA-recognition process. Chimeric γPNA–Invader probes are characterized with respect to thermal denaturation properties, thermodynamic parameters associated with duplex formation, UV-Vis and fluorescence trends to establish pyrene binding modes, and dsDNA-recognition properties using DNA hairpin model targets.
- This article is part of the themed collection: Chemical Biology in OBC
 Please wait while we load your content...
Please wait while we load your content...

