
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSequential conjugation methods based on triazole formation and related reactions using azides
Suguru
Yoshida

Laboratory of Chemical Bioscience, Institute of Biomaterials and Bioengineering, Tokyo Medical and Dental University (TMDU), 2-3-10 Kanda-Surugadai, Chiyoda-ku, Tokyo 101-0062, Japan. E-mail: s-yoshida.cb@tmd.ac.jp
First published on 23rd January 2020
Abstract
The recent remarkable progress in azide chemistry has realized sequential conjugation methods with selective 1,2,3-triazole formation. On the basis of the diverse reactivities of azides and azidophiles, including terminal alkynes and cyclooctynes, various selective reactions to furnish triazoles and a wide range of platform molecules, such as diynes, diazides, triynes, and triazides, have been developed so far for bis- and tris(triazole) syntheses. This review highlights recent transformations involving selective triazole formation, allowing the efficient preparation of unsymmetric bis- and tris(triazole)s using diverse platform molecules.
 Suguru Yoshida | Suguru Yoshida received his Ph.D. in 2009 from Kyoto University under the supervision of Prof. Koichiro Oshima. After a postdoctoral fellowship in the group of Prof. Katsuhiko Tomooka at Kyushu University and the group of Prof. Marcus Tius at the University of Hawaii in Manoa, he became Assistant Professor at Tokyo Medical and Dental University (TMDU), Japan, working with Prof. Takamitsu Hosoya in 2010 and was promoted to Associate Professor in 2015. His research interests include new methods in synthetic organic chemistry. He received the Chemical Society of Japan Award for Young Chemists (2017) and the Thieme Chemistry Journal Award (2019). |
Introduction
Azides are recognized as reliable compounds to conjugate with several types of azidophiles, such as terminal alkynes and cycloalkynes.1 In particular, copper-catalyzed azide–alkyne cycloaddition (CuAAC) and strain-promoted azide–alkyne cycloaddition (SPAAC) have been utilized as “click reactions” for connecting two molecules in broad disciplines, including the pharmaceutical sciences, chemical biology, and materials science (Fig. 1).2–6 These reliable methods for forming a 1,2,3-triazole ring have enabled the preparation of a wide range of compounds involving functionalized proteins. | ||
| Fig. 1 Triazole formation by cycloaddition of azides with terminal alkynes or cyclooctyne. | ||
In 2002, Sharpless's group and Meldal's group independently reported that a catalytic amount of copper(I) salt efficiently facilitated the [3 + 2] cycloaddition reaction between azides and terminal alkynes.4a,b This catalytic reaction realizes the selective synthesis of a wide range of 1,4-triazoles, leaving diverse functional groups unreacted. When cyclooctynes were treated with azides, the triazole formation took place smoothly without copper catalysis.5 The SPAAC reaction reported by Bertozzi and coworkers in 2004 served in the chemical modification of proteins.6a Various cycloalkynes have so far been developed for efficient conjugation with azides.5,6
The diversity of synthesizable triazoles has been expanded by the development of sequential triazole formation and related reactions. Sequential reactions using platform molecules, such as diynes, triynes, diazides, and triazides, have allowed for the modular synthesis of bis- and tris(triazole)s from simple modules.
This review summarizes recent sequential conjugation methods based on triazole formation and related chemistry using azides. In particular, various platform compounds bearing two or more clickable moieties are highlighted in terms of their azido- or alkyne-type selectivities.
Multi(triazole) syntheses via selective reactions of diynes or related compounds
One-pot bis(triazole) synthesis using a peptide platform with two types of alkynyl groups, i.e. an ethynyl group and a trimethylsilyl-protected alkyne moiety, was developed by Aucagne and Leigh in 2006 (Fig. 2A).7 Indeed, the copper-catalyzed reaction of diyne 1 with azide 2 proceeded selectively at the terminal alkyne moiety. The subsequent Ag(I)-mediated desilylprotonation and chemoselective triazole formation catalyzed by copper catalysis with azide 3 provided bis(triazole) 4 in high yield. The unsymmetrical bis(triazole) synthesis through selective triazole formation using 1-trimethylsilyl-1,3-butadiyne (5) was reported by Fiandanese and coworkers in 2009 (Fig. 2B).8 The Cu(II) catalyzed azide–alkyne cycloaddition between diyne 5 and benzyl azide (6) and subsequent Cu(I)-promoted triazole formation with n-decyl azide (7) in the presence of tetrabutylammonium fluoride yielded bis(triazole) 8 in good yield. In 2010, Aizpurua, Fratila, and coworkers reported the efficient preparation of bis(triazole) 13 by selective desilylprotonation of 1,4-bis(trimethylsilyl)-1,3-butadiyne (9) with methyllithium followed by a first CuAAC reaction with azide 10, desilylprotonation with cesium fluoride, and a second CuAAC reaction with azide 12 (Fig. 2C).9 Simpson et al. also developed a bis(triazole) synthesis from TIPS-protected 1,3-diyne 14 in a one-pot manner (Fig. 2D).10 | ||
| Fig. 2 Bis(triazole) syntheses by two CuAAC reactions of diynes. (A) Aucagne and Leigh's work. (B) Fiandanese's work. (C) Aizpurua and Fratila's work. (D) Simpson's work. NaAsc = sodium ascorbate; TBTA = tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine. | ||
Sequential triple-triazole-formation methods have been accomplished through selective desilylprotonation (Fig. 3). For example, the synthesis of tris(triazole) 17 using triazacyclophane-scaffold 16 was achieved by Liskamp et al. in 2014 (Fig. 3A).11 Indeed, a first CuAAC reaction with an azide followed by Ag(I)-mediated selective deprotection of the TES group proceeded efficiently. Then, a second CuAAC, desilylprotonation of the TIPS group with TBAF, and a third CuAAC resulted in the convergent synthesis of tris(triazole) 17 bearing three cyclic peptide moieties. In 2015, Jiráček and coworkers developed a versatile trifunctional scaffold 18 with three alkynyl groups; an ethynyl group and TES- and TIPS-protected alkynyl moieties, which enabled a solid-phase triple-click synthesis (Fig. 3B).12 Stepwise triple-click functionalization of a peptide-type triyne platform 19 was also accomplished by Vrabel et al. in 2018.13
 | ||
| Fig. 3 Tris(triazole) syntheses using triynes. (A) Liskamp's work. (B) Other triyne platforms for the tris(triazole) syntheses. TG = TentaGel NH2 resin. | ||
Bis(triazole) syntheses using diynes without silyl protective groups have also been achieved through selective triazole formation (Fig. 4). For example, Girard and coworkers reported selective triazole formation of N-propargyl propiolic amide (20) in 2011 (Fig. 4A).14 The remaining terminal alkyne in 22a reacted with azide 6 in the presence of a copper catalyst to yield bis(triazole) 23 in high yield. In 2016, Burley, Watson, and coworkers found that aromatic ynamines showed higher reactivity than simple terminal alkynes in triazole formation catalyzed by Cu(II) (Fig. 4B).15 On the basis of the different reactivities, selective bis(triazole) synthesis was accomplished by selective Cu(II)-catalyzed triazole formation followed by Cu(I)-catalyzed azide–alkyne cycloaddition.
 | ||
| Fig. 4 Bis(triazole) syntheses using diynes. (A) Girard's work. (B) Watson and Burley's work. (C) Tanimoto's work. A-21 = Amberlyst A-21. AMTC = 2-(4-(dimethylamino)methyl-1,2,3-triazol-1-yl)cyclohexan-1-ol. | ||
Tanimoto and coworkers developed a unique azide–alkyne cycloaddition between azides and propargyl alcohols through cationic intermediates (Fig. 4C).16 Since the cationic intermediates generated from diyne 29 and azide 6 can react with azides, bis(triazole) synthesis was accomplished in moderate yield.16a Furthermore, an elegant four-component coupling by sequential triazole formation and subsequent amination has been achieved using diyne 29, azides 6 and 30, and allylamine (Fig. 4C).16b
Since aryne intermediates spontaneously react with azides without catalysis to efficiently provide benzotriazoles,17 we developed an efficient bis(triazole) synthesis using aryne precursors bearing a terminal alkyne moiety (Fig. 5A).18 Indeed, treatment of o-iodoaryl triflate 32 with a silylmethyl Grignard reagent in the presence of azides followed by a CuAAC reaction furnished bis(triazole)s 33a and 33b in good yields. The aryne–azide cycloaddition and following azide–alkyne cycloaddition catalyzed by ruthenium afforded bis(triazole) 34a with a 1,5-triazole moiety.19 Furthermore, the synthesis of tris(triazole) 39 was achieved from o-iodoaryl triflate 35 and azides 36–38 by aryne–azide cycloaddition, a first CuAAC, Ag(I)-mediated desilylprotonation, and a second CuAAC (Fig. 5B).
 | ||
| Fig. 5 Bis- and tris(triazole) syntheses through aryne intermediates. (A) Bis(triazole) synthesis. (B) Tris(triazole) synthesis. | ||
Bis(triazole) synthesis using a platform with an ethynyl group and a cyclooctyne moiety was accomplished (Fig. 6). Dual labeling of biomolecules using diyne 40 through SPAAC and CuAAC was reported by Kele, Wolfbeis, and coworkers in 2009 (Fig. 6A).20 The SPAAC reaction between cyclooctyne 40 and azide 41 took place smoothly without catalysis, and subsequent CuAAC reaction at the remaining ethynyl group efficiently furnished a regioisomeric mixture of bis(triazole)s 43. In 2011, Boons et al. reported that the selective bis(triazole) synthesis allowed for a dendrimer-type multi(triazole) synthesis using platform 44 with ethynyl groups and a cycloalkyne moiety by a SPAAC reaction and following CuAAC reaction efficiently providing multi(triazole) 46 (Fig. 6B).21
 | ||
| Fig. 6 Multi(triazole) syntheses by SPAAC followed by CuAAC. (A) Kele and Wolfbeis's work. (B) Boons's work. DIPEA = N,N-diisopropylethylamine. | ||
In 2014, we reported a novel method to prepare bis(triazole) 49 using diyne 47 through transient protection of the cyclooctyne moiety toward the click reaction with azides (Fig. 7A).22a Indeed, terminal alkyne-selective triazole formation took place smoothly via a copper–cyclooctyne complex A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23 with (MeCN)4CuBF4 and a following CuAAC reaction with azide and removal of the copper salt with aqueous ammonia solution. Since the terminal alkyne-selective click reaction via the transient protection of diyne 47 with (MeCN)4CuBF4 was realized to retain a more reactive cycloalkyne moiety, the azide-to-cycloalkyne switching approach served in the preparation of a wide range of cyclooctynes with diverse functional groups.24 This approach achieved the chemical modification of azido-incorporated proteins with functional azides.22b In 2016, Dudley and coworker developed a novel platform 50 bearing a cyclononyne and terminal alkyne moieties (Fig. 7B).25 The SPAAC reaction of the cyclononyne moiety of 50 with azide 6 took place efficiently with gentle heating. Transient protection of the cycloalkyne moiety by complexation with copper enabled the selective CuAAC reaction of platform 50 at the ethynyl group followed by SPAAC with azide 53 to afford bis(triazole) 54 in good yield.
23 with (MeCN)4CuBF4 and a following CuAAC reaction with azide and removal of the copper salt with aqueous ammonia solution. Since the terminal alkyne-selective click reaction via the transient protection of diyne 47 with (MeCN)4CuBF4 was realized to retain a more reactive cycloalkyne moiety, the azide-to-cycloalkyne switching approach served in the preparation of a wide range of cyclooctynes with diverse functional groups.24 This approach achieved the chemical modification of azido-incorporated proteins with functional azides.22b In 2016, Dudley and coworker developed a novel platform 50 bearing a cyclononyne and terminal alkyne moieties (Fig. 7B).25 The SPAAC reaction of the cyclononyne moiety of 50 with azide 6 took place efficiently with gentle heating. Transient protection of the cycloalkyne moiety by complexation with copper enabled the selective CuAAC reaction of platform 50 at the ethynyl group followed by SPAAC with azide 53 to afford bis(triazole) 54 in good yield.
 | ||
| Fig. 7 Bis(triazole) syntheses through transient protection of cycloalkynes. (A) Our work. (B) Dudley's work. | ||
Bis(triazole) synthesis using platform 56 with a dicobalt-protected cycloheptyne moiety and an ethynyl group was also achieved by Fouquet, Hermange, and coworker in 2019 (Fig. 8A).26 Platform 56 was prepared through the Nicholas reaction27 of dicobalt-protected alkyne 55. Then, the generation of cycloheptyne by deprotection with trimethylamine N-oxide and SPAAC reaction with benzyl azide took place to afford triazole 57 in moderate yield without damaging the ethynyl group. Bis(triazole) 58 was synthesized by the CuAAC reaction of 57.
 | ||
| Fig. 8 Bis(triazole) syntheses using platforms 56 and 59. (A) Fouquet and Hermange's work. (B) Popik's work. | ||
Based on the photo-triggered click chemistry developed by Popik, Boons, and coworkers,28a bis(triazole) synthesis by sequential SPAAC reactions was accomplished using platform 59 by Popik et al. in 2014 (Fig. 8B).28b Indeed, the first SPAAC reaction of diyne 59 followed by photoirradiated removal of carbon monoxide to generate dibenzo-fused cyclooctyne and a subsequent SPAAC reaction efficiently provided bis(triazole) 60.
In 2010, the double-click reaction of Sondheimer–Wang diyne 6129 was accomplished by Hosoya, Kii, and coworkers (Fig. 9A).30 The double-click reaction served in the chemical modification of azido proteins with diyne 61 and azides, but selective bis(triazole) synthesis was not easy.31 In 2016, Popik et al. succeeded in a selective bis(triazole) synthesis via the mono-cyclopropene formation of diyne 61 followed by SPAAC reaction with butyl azide and a further SPAAC reaction with benzyl azide through the generation of a cycloalkyne moiety (Fig. 9B).28c
 | ||
| Fig. 9 Double-click reactions using diyne 61. (A) Hosoya and Kii's work. (B) Popik's work. | ||
Bis(triazole) synthesis and a thiol–ene reaction using platform 65 enabled the assembly of three modules by reliable conjugation methods (Fig. 10).32 In 2012, Beal and coworkers developed platform 65 with cyclooctyne and terminal alkyne moieties and an acetylthio group. Sequential SPAAC and CuAAC reactions efficiently yielded bis(triazole) 67. Then, removal of the acetyl group followed by a thiol–ene reaction using a maleimide conjugated with bovine serum albumin (BSA) resulted in the dual-modification of the BSA protein.
 | ||
| Fig. 10 Three-component coupling using platform 65. | ||
Multi(triazole) syntheses using platforms with both azido and alkyne moieties
Platform compounds bearing both azido and alkyne moieties have also served in the preparation of bis(triazole)s and tris(triazole)s (Fig. 11). For example, Kaliappan et al. reported that the CuAAC reaction of platform 69 with methyl propiolate (70) and a following CuAAC reaction with p-tolyl azide (71) furnished bis(triazole) 72 in moderate yield by virtue of the higher clickability of methyl propiolate than that of the ethynyl group of platform 69 (Fig. 11A).33 Platform 73, with both an azido group and TES- and TIPS-protected alkyne moieties, was developed by Aucagne and coworkers in 2009 (Fig. 11B).34 Indeed, an elegant 5-step transformation was achieved through a first CuAAC with alkyne 74, TES-selective desilylprotonation with silver nitrate, a second CuAAC with azide 75, TIPS-selective desilylprotonation by TBAF, and a third CuAAC with azide 76. | ||
| Fig. 11 Bis(triazole) syntheses using platforms 69 and 73. (A) Kalippan's work. (B) Aucagne's work. | ||
In 2015, Workentin, Gilroy, and coworkers developed platform 80 bearing an azide and dicobalt-protected cyclooctyne moieties (Fig. 12A).35 The protection of bicyclononyne 78 with dicobalt octacarbonyl successfully proceeded to afford quantitative amounts of 79, enabling the formation of carbamate 80 using 2-azidoethylamine without an SPAAC reaction. Subsequent a CuAAC reaction using the azido group followed by removal of the dicobalt moiety and a further SPAAC reaction furnished bis(triazole) 83.
 | ||
| Fig. 12 Bis(triazole) syntheses using platforms 80 and 84. (A) Gilroy and Workentin's work. (B) Wright and Couty's work. DIC = diisopropylcarbodiimide. | ||
Platform compound 84 with an azido group and alkyne precursor moiety has been developed by Wright, Couty, and coworkers (Fig. 12B).36 Indeed, the CuAAC reaction of platform 84 with alkyne 74 followed by alkyne formation by DIC took place efficiently to provide 86, which reacted with azide 87 catalyzed by copper to afford bis(triazole) 88 in excellent yield.
Recently, we reported bis(triazole) syntheses using platform compounds with an azido group and an aryne precursor moiety (Fig. 13). In 2015, platform compound 89 bearing both an azido group and an o-silylaryl triflate moiety for aryne generation was prepared through Ir-catalyzed C–H borylation37,38 of o-silylaryl triflate and following deborylazidation (Fig. 13A).39a Since aliphatic azide 6 showed higher reactivity than azide 89 in cycloaddition with aryne intermediate B, bis(triazole) 91 was efficiently prepared by azide–aryne cycloaddition and subsequent CuAAC with alkyne 90. Platform 92 with both an azido group and an o-iodoaryl triflate moiety was also developed in 2016 (Fig. 13B).39b For instance, a CuAAC reaction with terminal alkynes and aryne–azide cycloaddition with a silylmethyl Grignard reagent as an activator enabled the facile synthesis of bis(triazole)s 93a–93c, leaving various functional groups untouched.
 | ||
| Fig. 13 Bis(triazole) syntheses using platforms 89 and 92. (A) Platform 89 with both an azido group and an o-silylaryl triflate moiety. (B) Platform 92 with both an azido group and an o-iodoaryl triflate moiety. | ||
Multi(triazole) syntheses using multi-azido platforms
Azido-type selective reactions have served in multi(triazole) syntheses. In 2012, Zhu et al. found efficient reactions between 2-picolyl azides and terminal alkynes in the presence of a catalytic amount of copper(II) acetate by virtue of significant enhancement in the clickability of the picolyl azido group facilitated by chelation (Fig. 14).40 On the basis of this remarkable clickability of the 2-picolyl azido group, bis(triazole) 98 was efficiently prepared from diazide 94 by Cu(II)-catalyzed azide–alkyne cycloaddition of 2-picolyl azido group with alkyne 95 followed by Cu(I)-catalyzed cycloaddition of the remaining azido group with alkyne 97. This method allowed for the stepwise click functionalization of alkyne-installed DNA.41 | ||
| Fig. 14 Bis(triazole) synthesis using diazide 94. | ||
Efficient bis(triazole) syntheses using platform compound 100 with an aromatic and an aliphatic azido group were achieved through triazole formation with nucleophilic species (Fig. 15).42–44 In 2011, Belkheira, Pons, Bressy, and coworker succeeded in efficient bis(triazole) synthesis by an organocatalytic azide–ketone [3 + 2]-cycloaddition reaction. Indeed, triazole 102 was obtained selectively by the reaction between ketone 99 and diazide 100 in the presence of a catalytic amount of proline via an enamine intermediate followed by CuAAC reaction of the remaining benzylic azido group (Fig. 15A).43 In 2015, Ramachary et al. reported that benzyl cyanide also reacted with azides to afford triazoles (Fig. 15B). In the case of using diazide 100, triazole formation took place selectively at the aromatic azido group in the presence of cesium carbonate, and bis(triazole) 106 was successfully prepared in good yield by further cycloaddition with benzyl cyanide 105 using potassium tert-butoxide.44
 | ||
| Fig. 15 Bis(triazole) syntheses using diazide 100. (A) Belkheira, Pons, and Bressy's work. (B) Ramachary's work. | ||
In 2014, Delft, Bickelhaupt, and coworkers reported that electron-deficient aromatic azides showed significantly higher reactivity than aliphatic azides in the SPAAC reaction with bicyclo[6.1.0]non-4-yne (BCN).45 On the other hand, the clickability of aliphatic azides was higher than that of aromatic azides in the SPAAC reaction with dibenzo-fused azacyclooctyne. These SPAAC reactions enabled selective bis(triazole) formation using diazide 108 with two-types of cyclooctynes 107 and 109, showcasing the labeling of BCN-installed protein 107 with fluorescent cyclooctyne 109 (Fig. 16).
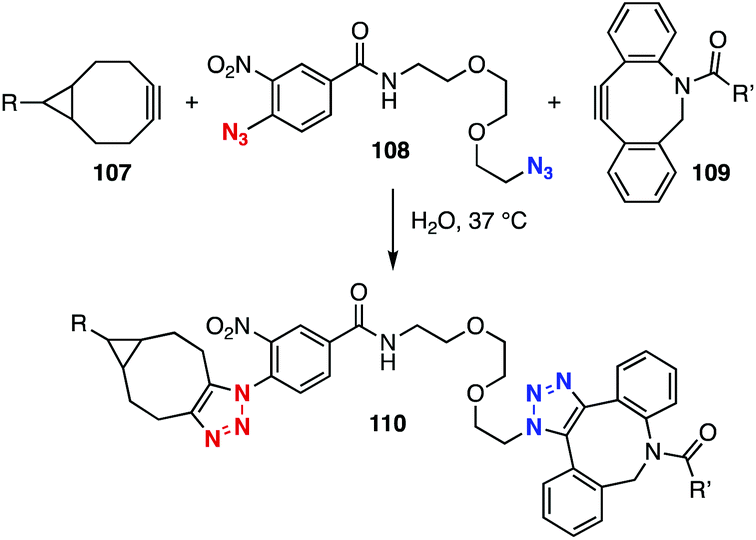 | ||
| Fig. 16 Labeling of BCN-installed protein 107 using diazide 108 and fluorescent cyclooctyne 109. | ||
Bulky tertiary azides also served in selective SPAAC and CuAAC reactions (Fig. 17). In 2016, Koert and coworkers reported that the CuAAC reaction of diazide 113 took place selectively at the primary azido group (Fig. 17A).46 On the basis of this selective CuAAC reaction and cyclooctyne-selective triazole formation using diyne 111, a novel layer-by-layer method was developed. Bis(triazole) synthesis using diazide 116 through two SPAAC reactions was realized by Bickelhaupt, Mikula, and coworkers (Fig. 17B).47 Indeed, the selective SPAAC reaction of diazide 116 at the primary azido group using dibenzo-fused cyclooctyne 117 and a subsequent SPAAC reaction at the remaining bulky azido group with BCN 118 provided bis(triazole) 119 in a quantitative amount.
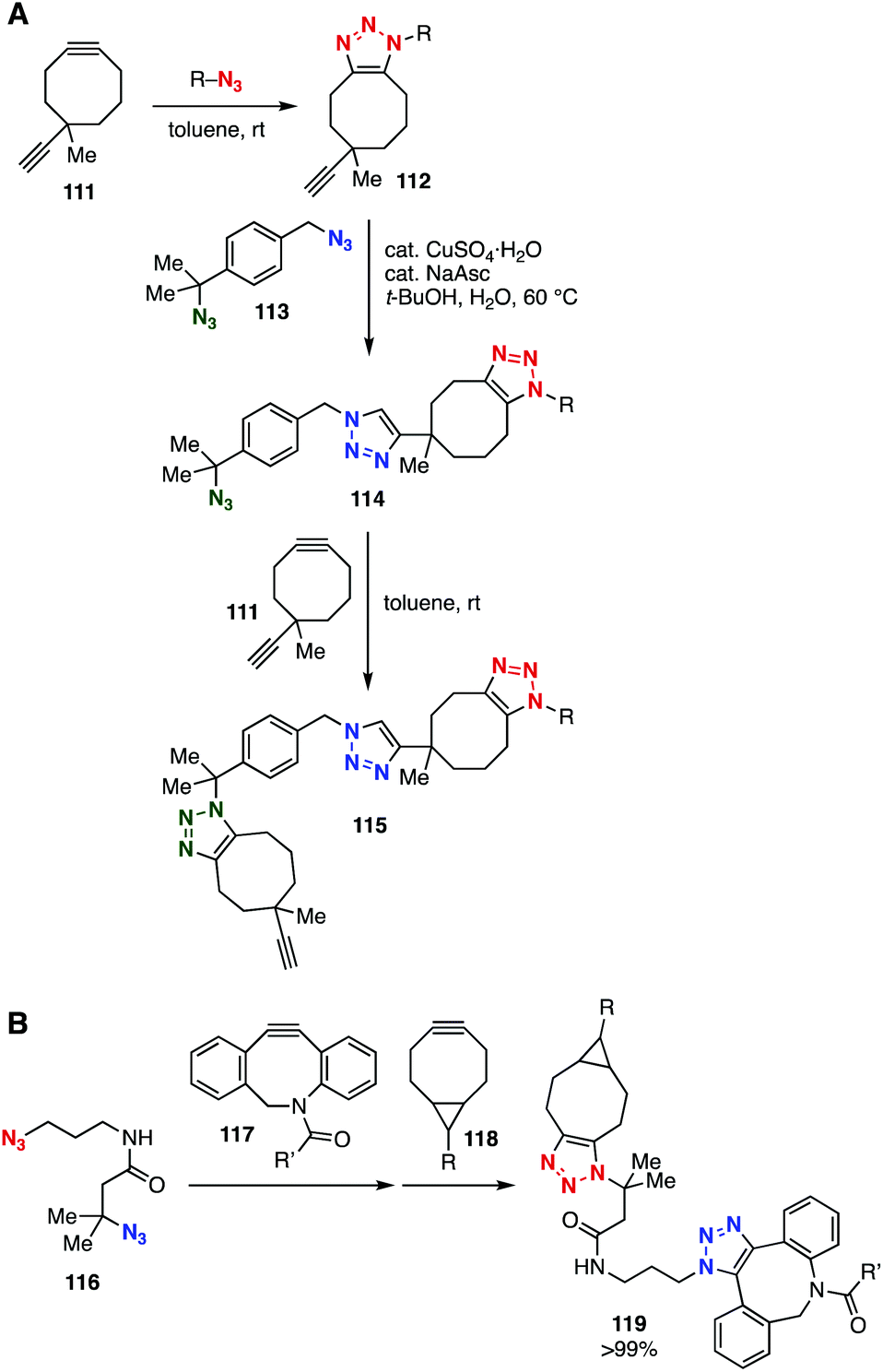 | ||
| Fig. 17 Bis(triazole) syntheses using diazides 113 and 116. (A) Koert's work. (B) Bickelhaupt and Mikula's work. | ||
Remarkable clickability of doubly sterically-hindered aromatic azides also realized selective bis(triazole) formation (Fig. 18).48 In 2011, we found that 2,6-diisopropylphenyl azide showed 76 times higher reactivity than phenyl azide in the SPAAC reaction with dibenzo-fused cyclooctynes due to the steric inhibition of resonance. Thus, a selective SPAAC reaction with cyclooctyne 121 took place efficiently at the doubly sterically hindered aromatic azido group of diazide 120 (Fig. 18A).48a In 2018, a further enhancement in clickability was achieved using 4-amino-2,6-diisopropylphenyl azides, allowing the selective SPAAC reaction of diazide 124 with dibenzo-fused cyclooctyne 121 (Fig. 18B).48b Thus, a following CuAAC reaction with alkyne 125 furnished bis(triazole) 126 in excellent yield.
 | ||
| Fig. 18 Selective triazole formation of diazides 120 and 124. (A) SPAAC reaction between diazide 120 and cyclooctyne 121. (B) Bis(triazole) synthesis using diazide 124. | ||
In 2018, we found three types of selectivities in triazole formation, namely SPAAC, Ru-catalyzed azide–alkyne cycloaddition (RuAAC),19 and base-catalyzed triazole formation with 1,3-dicarbonyl compounds,42c by competitive experiments using an equimolar mixture of 2,6-diisopropylphenyl azide, phenyl azide, and benzyl azide.49 On the basis of these findings, we succeeded in a consecutive tris(triazole) synthesis using triazide platform 127 (Fig. 19). Indeed, selective base-catalyzed triazole formation with 1,3-diketone 128 at the unhindered aromatic azido group followed by selective RuAAC with alkyne 125 at the benzylic azido group and SPAAC reaction with cyclooctyne 121 at the remaining 2,6-diisopropylphenyl azido group efficiently provided tris(triazole) 129a (Fig. 19A). Triple-triazole formation was also achieved by SPAAC reaction with 121 at the doubly sterically-hindered aromatic azido group, aromatic azido-selective base-catalyzed cycloaddition with diketone 128, and CuAAC reaction with alkyne 74. A trifunctional chemical probe 129c as a dual-labeling ligand was efficiently developed by assembling three modules onto triazide platform 127 in three steps (Fig. 19B).
 | ||
| Fig. 19 Tris(triazole) synthesis using triazide 127. (A) Synthesis of 129a and 129b. (B) Dual labeling ligand 129c. | ||
A transient protection method realized selective triazole formation using diazide platform 130 (Fig. 20).50 In 2018, we found that azides were efficiently protected toward SPAAC and CuAAC reactions by phosphazide formation with Amphos (131). Since the formation of phosphazide 132 from diazide 130 selectively proceeded at the aromatic azido group by the addition of an equimolar amount of Amphos (131),51 we accomplished selective SPAAC and CuAAC reactions of diazide 130 with cyclooctyne 78 and alkyne 134, respectively, at the aliphatic azido group through deprotective removal of Amphos by elemental sulfur.
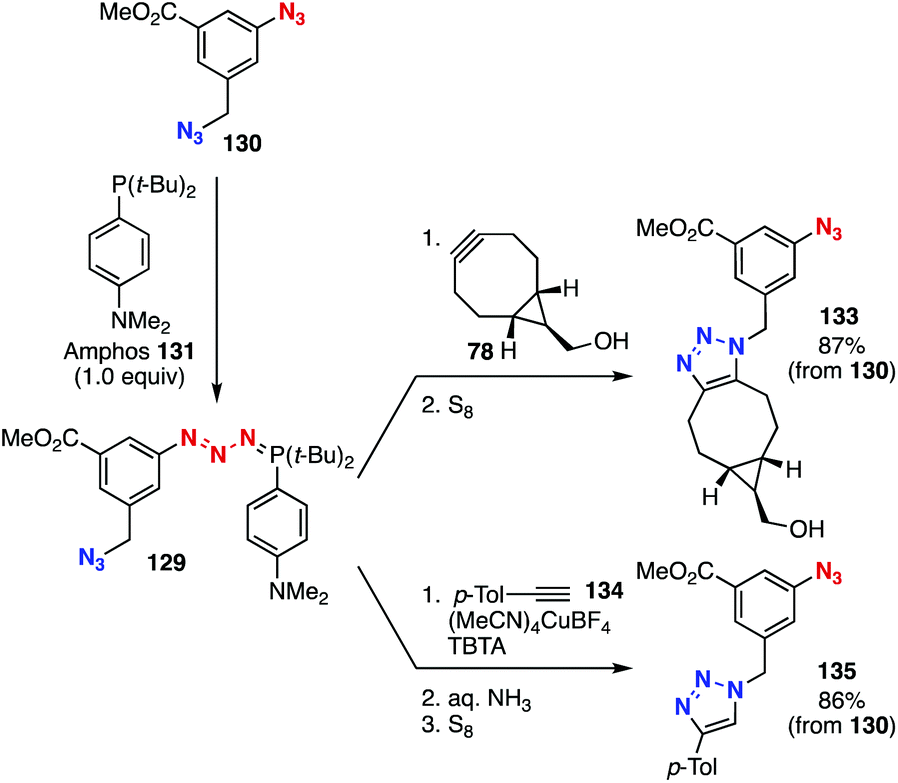 | ||
| Fig. 20 Selective triazole formations through phosphazide formation. | ||
Related sequential conjugation methods based on transformations using azides
Azides show diverse reactivities in conjugation reactions with various azidophiles. In 2015, platform compound 136 with a masked-phosphonite and terminal alkyne moieties was developed by Hackenberger and coworkers (Fig. 21).52,53 Indeed, sequential conjugation was achieved by CuAAC reaction, removal of borane, and reaction with an azide to form a P–N bond.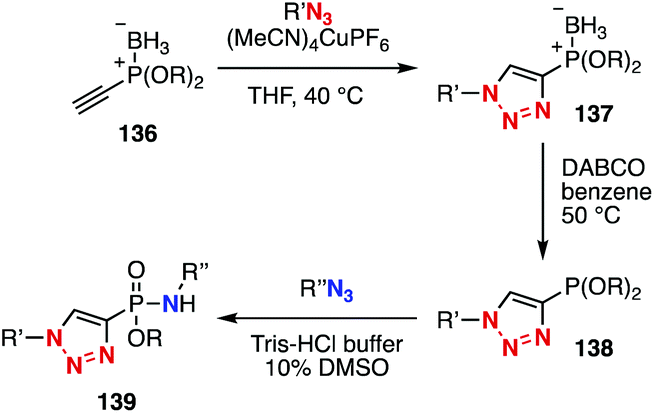 | ||
| Fig. 21 Sequential conjugation using platform 136. | ||
Recently, Yan and Ramstöm's group,54 Yi and Xi's group,55 and our group56 independently reported the Staudinger reaction affording robust aza-ylides. In 2018, we found that the Staudinger reaction between 2,6-dichlorophenyl azide (141) and triphenylphosphine (140) took place smoothly to provide stable aza-ylide 142 even in the presence of cyclooctyne 121, while the SPAAC reaction of benzyl azide (6) with cyclooctyne 121 proceeded faster than that with phosphine 140 (Fig. 22A). We also demonstrated that aza-ylides formed by the Staudinger reaction between 2,6-dichloroaryl azides and triphenylphosphine derivatives showed significant stability in the presence of biomolecules, enabling the chemical modification of azido-incorporated proteins. We also reported a novel sequential conjugation method using diazide 146 with triphenylphosphine (140) and cyclooctyne 121, providing triazole 147 in high yield by three-component coupling (Fig. 22B).
 | ||
| Fig. 22 Selective reactions of azides with phosphine 140 and cyclooctyne 121. (A) Competitive experiments using an equimolar mixture of 140 and 121. (B) Three-component reaction between diazide 146, phosphine 140 and cyclooctyne 121. | ||
In 2019, Workentin et al. developed a sequential conjugation method using platform 148 (Fig. 23A).57 Indeed, the generation of cyclooctyne with an ortho-alkoxycarbonyl-substituted triarylphosphine moiety from 148 enabled the SPAAC reaction with azide 6 followed by a rapid Staudinger reaction with tetrafluorophenyl azide 149, yielding stable aza-ylide 150. In 2019, Yi, Xi, and coworkers developed diazide platform 151 (Fig. 23B).58 Staudinger reaction of diazide 151 with triphenylphosphine derivative 152 proceeded smoothly to afford a stable aza-ylide followed by an SPAAC reaction with cyclooctyne 118, resulting in efficient sequential conjugation.
 | ||
| Fig. 23 Sequential conjugations through stable aza-ylide formation. (A) Workentin's work. (B) Yi and Xi's work. | ||
Elegant sequential conjugations using triazide 154 have been accomplished by Tanimoto and coworkers (Fig. 23). Indeed, selective transformations of azidomethyl groups into diazomethyl groups enabled sequential cycloadditions to efficiently provide 160 (Fig. 24A).59a Furthermore, the selective preparation of oxime was achieved by the transformation of α-azidoacetophenone, leaving two azido groups untouched (Fig. 24B).59b Following the synthesis of triazine 162 with 2-pyridyl aldehyde, diazomethane formation, cycloaddition with acrylic acid ester 163, SPAAC reaction with cyclooctyne 121, and conjugation with trans-cyclooctene 164 at the triazine ring realized efficient three-component assembly.
 | ||
| Fig. 24 Sequential three-component assembly using triazide 154. (A) Transformations via two generations of a diazo group. (B) Transformations via oxime and diazo intermediates. | ||
Conclusions
Complicated multi(triazole)s have been prepared by the consecutive syntheses of simple modules onto platform molecules, such as multiazides, through selective triazole formation. However, three (or more) component coupling still remains challenging due to the limited methods available to control triazole formation. Various methods to synthesize multi(triazole)s would be useful in broad disciplines, including materials science, the pharmaceutical sciences, and chemical biology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number 19K05451; the Naito Foundation; the Japan Agency for Medical Research and Development (AMED) under Grant Numbers JP19am0101098 (Platform Project for Supporting Drug Discovery and Life Science Research, BINDS); and the Cooperative Research Project of Research Center for Biomedical Engineering.Notes and references
- (a) R. Huisgen, Proc. Chem. Soc., 1961, 357 Search PubMed; (b) M. Köhn and R. Breinbauer, Angew. Chem., Int. Ed., 2004, 43, 3106 CrossRef PubMed; (c) C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540 CrossRef CAS PubMed; (d) S. S. van Berkel, M. B. van Eldijk and J. C. M. van Hest, Angew. Chem., Int. Ed., 2011, 50, 8806 CrossRef CAS PubMed; (e) C. I. Schilling, N. Jung, M. Biskup, U. Schepers and S. Bräse, Chem. Soc. Rev., 2011, 40, 4840 RSC; (f) A.-C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131 RSC; (g) Z.-J. Zheng, D. Wang, Z. Xu and L.-W. Xu, Beilstein J. Org. Chem., 2015, 11, 2557 CrossRef CAS PubMed.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS; (b) C. S. McKay and M. G. Finn, Chem. Biol., 2014, 21, 1075 CrossRef CAS PubMed; (c) J. Lahann, Click Chemistry for Biotechnology and Materials Science, John Wiley & Sons, West Sussex, 2009 CrossRef.
- M. Melda and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef PubMed.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (b) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef PubMed.
- (a) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS PubMed; (b) M. F. Debets, C. W. J. van der Doelen, F. P. J. T. Rutjes and F. L. van Delft, ChemBioChem, 2010, 11, 1168 CrossRef CAS PubMed; (c) J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272 RSC; (d) E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666 CrossRef CAS PubMed; (e) S. Arumugam, S. V. Orski, N. E. Mbua, C. McNitt, G.-J. Boons, J. Locklin and V. V. Popik, Pure Appl. Chem., 2013, 85, 1499 CAS; (f) J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delft, Top. Curr. Chem., 2016, 374, 16 CrossRef; (g) S. Yoshida, Bull. Chem. Soc. Jpn., 2018, 91, 1293 CrossRef CAS.
- (a) N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046 CrossRef CAS PubMed; (b) J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793 CrossRef CAS PubMed; (c) J. C. Jewett, E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 3688 CrossRef CAS PubMed; (d) X. Ning, J. Guo, M. A. Wolfert and G.-J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253 CrossRef CAS PubMed; (e) J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. P. J. T. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422 CrossRef CAS PubMed; (f) R. Ni, N. Mitsuda, T. Kashiwagi, K. Igawa and K. Tomooka, Angew. Chem., Int. Ed., 2015, 54, 1190 CrossRef CAS PubMed.
- V. Aucagne and D. A. Leigh, Org. Lett., 2006, 8, 4505 CrossRef CAS PubMed.
- V. Fiandanese, D. Bottalico, G. Marchese, A. Punzi and F. Capuzzolo, Tetrahedron, 2009, 65, 10573 CrossRef CAS.
- J. M. Aizpurua, I. Azcune, R. M. Fratila, E. Balentova, M. Sagartzazu-Aizpurua and J. I. Miranda, Org. Lett., 2010, 12, 1584 CrossRef CAS PubMed.
- B. C. Doak, M. J. Scanlon and J. S. Simpson, Org. Lett., 2011, 13, 537 CrossRef CAS PubMed.
- P. R. Werkhoven, H. van de Langemheen, S. van der Wal, J. A. W. Kruijtzer and R. M. J. Liskamp, J. Pept. Sci., 2014, 20, 235 CrossRef CAS PubMed.
- (a) V. Vaněk, J. Pícha, B. Fabre, M. Buděšínský, M. Lepšík and J. Jiráček, Eur. J. Org. Chem., 2015, 3689 CrossRef; (b) B. Fabre, J. Pícha, V. Vaněk, I. Selicharová, M. Chrudinová, M. Collinsová, L. Žáková, M. Buděšínský and J. Jiráček, ACS Comb. Sci., 2016, 18, 710 CrossRef CAS; (c) B. Fabre, J. Pícha, V. Vaněk, M. Buděšínský and J. Jiráček, Molecules, 2015, 20, 19310 CrossRef CAS PubMed.
- A. Kovalová, R. Pohl and M. Vrabel, Org. Biomol. Chem., 2018, 16, 5960 RSC.
- (a) H. Elamari, F. Meganem, J. Herscovic and C. Girard, Tetrahedron Lett., 2011, 52, 658 CrossRef CAS; (b) H. Elamari, R. Slimi, G. G. Chabot, L. Quentin, D. Scherman and C. Girard, Eur. J. Med. Chem., 2013, 60, 360 CrossRef CAS PubMed.
- (a) M. Z. C. Hatit, J. C. Sadler, L. A. McLean, B. C. Whitehurst, C. P. Seath, L. D. Humphreys, R. J. Young, A. J. B. Watson and G. A. Burley, Org. Lett., 2016, 18, 1694 CrossRef CAS PubMed; (b) M. Z. C. Hatit, C. P. Seath, A. J. B. Watson and G. A. Burley, J. Org. Chem., 2017, 82, 5461 CrossRef CAS PubMed.
- (a) H. Zhang, H. Tanimoto, T. Morimoto, Y. Nishiyama and K. Kakiuchi, Org. Lett., 2013, 15, 5222 CrossRef CAS PubMed; (b) H. Zhang, H. Tanimoto, T. Morimoto, Y. Nishiyama and K. Kakiuchi, Tetrahedron, 2014, 70, 9828 CrossRef CAS.
- F. Shi, J. P. Waldo, Y. Chen and R. C. Larock, Org. Lett., 2008, 10, 2409 CrossRef CAS PubMed.
- S. Yoshida, T. Nonaka, T. Morita and T. Hosoya, Org. Biomol. Chem., 2014, 12, 7489 RSC.
- (a) L. Zhang, X. Chen, P. Xue, H. H. Y. Sun, I. D. Williams, K. B. Sharpless, V. V. Fokin and G. Jia, J. Am. Chem. Soc., 2005, 127, 15998 CrossRef CAS PubMed; (b) B. C. Boren, S. Narayan, L. K. Rasmussen, L. Zhang, H. Zhao, Z. Lin, G. Jia and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 8923 CrossRef CAS PubMed.
- P. Kele, G. Mezö, D. Achatz and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2009, 48, 344 CrossRef CAS PubMed.
- P. A. Ledin, F. Friscourt, J. Guo and G.-J. Boons, Chem. – Eur. J., 2011, 17, 839 CrossRef CAS PubMed.
- (a) S. Yoshida, Y. Hatakeyama, K. Johmoto, H. Uekusa and T. Hosoya, J. Am. Chem. Soc., 2014, 136, 13590 CrossRef CAS PubMed; (b) S. Yoshida, T. Kuribara, H. Ito, T. Meguro, Y. Nishiyama, F. Karaki, Y. Hatakeyama, Y. Koike, I. Kii and T. Hosoya, Chem. Commun., 2019, 55, 3556 RSC.
- (a) G. Wittig and H.-L. Dorsch, Liebigs Ann. Chem., 1968, 711, 46 CrossRef CAS; (b) G. Wittig and S. Fischer, Chem. Ber., 1972, 105, 3542 CrossRef CAS; (c) G. Gröger, U. Behrens and F. Olbrich, Organometallics, 2000, 19, 3354 CrossRef; (d) M. Shelbourne, X. Chen, T. Brown and A. H. El-Sagheer, Chem. Commun., 2011, 47, 6257 RSC; (e) A. Das, C. Dash, M. Yousufuddin, M. A. Celik, G. Frenking and H. V. R. Dias, Angew. Chem., Int. Ed., 2012, 51, 3940 CrossRef CAS PubMed; (f) A. Das, C. Dash, M. A. Celik, M. Yousufuddin, G. Frenking and H. V. R. Dias, Organometallics, 2013, 32, 3135 CrossRef CAS. For a review of metal complexes of cycloalkynes, see: (g) M. A. Bennett and H. P. Schwemlein, Angew. Chem., Int. Ed. Engl., 1989, 28, 1296 CrossRef.
- C. Hansell, Nat. Chem., 2014, 6, 946 CrossRef CAS.
- R. R. Ramsubhag and G. B. Dudley, Org. Biomol. Chem., 2016, 14, 5028 RSC.
- M. Cormier, E. Fouquet and P. Hermange, Org. Chem. Front., 2019, 6, 1114 RSC.
- K. M. Nicholas, Acc. Chem. Res., 1987, 20, 207 CrossRef CAS.
- (a) S. Arumugam and V. V. Popik, J. Org. Chem., 2014, 79, 2702 CrossRef CAS PubMed; (b) D. A. SuttonVladimir and V. Popik, J. Org. Chem., 2016, 81, 8850 CrossRef; (c) D. A. Sutton, S.-H. Yu, R. Steet and V. V. Popik, Chem. Commun., 2016, 52, 553 RSC.
- H. N. C. Wong, P. J. Garratt and F. Sondheimer, J. Am. Chem. Soc., 1974, 96, 5604 CrossRef CAS.
- I. Kii, A. Shiraishi, T. Hiramatsu, T. Matsushita, H. Uekusa, S. Yoshida, M. Yamamoto, A. Kudo, M. Hagiwara and T. Hosoya, Org. Biomol. Chem., 2010, 8, 4051 RSC.
- (a) A. A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G.-J. Boons and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 15769 CrossRef CAS PubMed; (b) F. Xu, L. Peng, K. Shinohara, T. Morita, S. Yoshida, T. Hosoya, A. Orita and J. Otera, J. Org. Chem., 2014, 79, 11592 CrossRef CAS PubMed; (c) M. Tera, Z. H. Taji and N. W. Luedtke, Angew. Chem., Int. Ed., 2018, 57, 15405 CrossRef CAS PubMed.
- D. M. Beal, V. E. Albrow, G. Burslem, L. Hitchen, C. Fernandes, C. Lapthorn, L. R. Roberts, M. D. Selby and L. H. Jones, Org. Biomol. Chem., 2012, 10, 548 RSC.
- K. P. Kaliappan, P. Kalanidhi and S. Mahapatra, Synlett, 2009, 2162 CrossRef CAS.
- I. E. Valverde, A. F. Delmas and V. Aucagne, Tetrahedron, 2009, 65, 7597 CrossRef CAS.
- P. Gobbo, T. Romagnoli, S. M. Barbon, J. T. Price, J. Keir, J. B. Gilroy and M. S. Workentin, Chem. Commun., 2015, 51, 6647 RSC.
- K. Wright, P. Quinodoz, B. Drouillat and F. Couty, Chem. Commun., 2017, 53, 321 RSC.
- (a) C. N. Iverson and M. R. Smith III, J. Am. Chem. Soc., 1999, 121, 7696 CrossRef CAS; (b) J.-Y. Cho, C. N. Iverson and M. R. Smith III, J. Am. Chem. Soc., 2000, 122, 12868 CrossRef CAS; (c) J.-Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka Jr. and M. R. Smith III, Science, 2002, 295, 305 CrossRef CAS PubMed; (d) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390 CrossRef CAS PubMed; (e) T. Ishiyama, J. Takagi, J. F. Hartwig and N. Miyaura, Angew. Chem., Int. Ed., 2002, 41, 3056 CrossRef CAS. For review, see: (f) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed.
- (a) E. Demory, K. Devaraj, A. Orthaber, P. J. Gates and L. T. Pilarski, Angew. Chem., Int. Ed., 2015, 54, 11765 CrossRef CAS PubMed; (b) J. Larsson, E. Demory, K. Devaraj, C. Sollert and L. T. Pilarski, Synlett, 2016, 27, 969 CrossRef CAS.
- (a) S. Yoshida, K. Shimomori, T. Nonaka and T. Hosoya, Chem. Lett., 2015, 44, 1324 CrossRef CAS; (b) S. Yoshida, T. Morita and T. Hosoya, Chem. Lett., 2016, 45, 726 CrossRef CAS.
- (a) G.-C. Kuang, H. A. Michaels, J. T. Simmons, R. J. Clark and L. Zhu, J. Org. Chem., 2010, 75, 6540 CrossRef CAS PubMed; (b) Z. Yuan, G.-C. Kuang, R. J. Clark and L. Zhu, Org. Lett., 2012, 14, 2590 CrossRef CAS.
- (a) S. A. Ingale and F. Seela, J. Org. Chem., 2013, 78, 3394 CrossRef CAS PubMed; (b) S. S. Pujari and F. Seela, J. Org. Chem., 2013, 78, 8545 CrossRef CAS PubMed.
- (a) A. Krasiński, V. V. Fokin and K. B. Sharpless, Org. Lett., 2004, 6, 1237 CrossRef; (b) S. W. Kwok, J. R. Fotsing, R. J. Fraser, V. O. Rodinov and V. V. Fokin, Org. Lett., 2010, 12, 4217 CrossRef CAS PubMed; (c) E. P. J. Ng, Y.-F. Wang, B. W.-Q. Hui, G. Lapointe and S. Chiba, Tetrahedron, 2011, 67, 7728 CrossRef CAS; (d) L. Wu, X. Chen, M. Tang, X. Song, G. Chen, X. Song and Q. Lin, Synlett, 2012, 23, 1529 CrossRef CAS; (e) C. D. Smith and M. F. Greaney, Org. Lett., 2013, 15, 4826 CrossRef CAS PubMed; (f) L. Hong, W. Lin, F. Zhang, R. Liu and X. Zhou, Chem. Commun., 2013, 49, 5589 RSC; (g) A. H. Banday and V. J. Hruby, Synlett, 2014, 25, 1859 CrossRef CAS; (h) A. B. Shashank, S. Karthik, R. Madhavachary and D. B. Ramachary, Chem. – Eur. J., 2014, 20, 16877 CrossRef CAS; (i) M. T. Saraiva, G. P. Costa, N. Seus, R. F. Schumacher, G. Perin, M. W. Paixão, R. Luque and D. Alves, Org. Lett., 2015, 17, 6206 CrossRef CAS PubMed; (j) B. Alcaide, P. Almendros and C. Lázaro-Milla, Chem. Commun., 2015, 51, 6992 RSC.
- (a) M. Belkheira, D. El Abed, J.-M. Pons and C. Bressy, Chem. – Eur. J., 2011, 17, 12917 CrossRef CAS PubMed; (b) M. Belkheira, D. El Abed, J.-M. Pons and C. Bressy, Synthesis, 2018, 50, 4254 CrossRef CAS.
- P. M. Krishna, D. B. Ramachary and S. Peesapatia, RSC Adv., 2015, 5, 62062 RSC.
- J. Dommerholt, O. van Rooijen, A. Borrmann, C. F. Guerra, F. M. Bickelhaupt and F. L. van Delft, Nat. Commun., 2014, 5, 5378 CrossRef CAS.
- N. Münster, P. Nikodemiak and U. Koert, Org. Lett., 2016, 18, 4296 CrossRef PubMed.
- D. Svatunek, N. Houszka, T. A. Hamlin, F. M. Bickelhaupt and H. Mikula, Chem. – Eur. J., 2019, 25, 754 CrossRef CAS PubMed.
- (a) S. Yoshida, A. Shiraishi, K. Kanno, T. Matsushita, K. Johmoto, H. Uekusa and T. Hosoya, Sci. Rep., 2011, 1, 82 CrossRef PubMed; (b) S. Yoshida, J. Tanaka, Y. Nishiyama, Y. Hazama, T. Matsushita and T. Hosoya, Chem. Commun., 2018, 54, 13499 RSC.
- S. Yoshida, K. Kanno, I. Kii, Y. Misawa, M. Hagiwara and T. Hosoya, Chem. Commun., 2018, 54, 3705 RSC.
- T. Meguro, S. Yoshida, K. Igawa, K. Tomooka and T. Hosoya, Org. Lett., 2018, 20, 4126 CrossRef CAS PubMed.
- T. Meguro, S. Yoshida and T. Hosoya, Chem. Lett., 2017, 46, 473 CrossRef CAS.
- M. Robert, J. Vallée, P. Majkut, D. Krause, M. Gerrits and C. P. R. Hackenberger, Chem. – Eur. J., 2015, 21, 970 CrossRef PubMed.
- (a) R. Serwa, I. Wilkening, G. Del Signore, M. Mühlberg, I. Claussnitzer, C. Weise, M. Gerrits and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2009, 48, 8234 CrossRef CAS PubMed; (b) V. Bçhrsch, T. Mathew, M. Zieringer, M. R. J. Valløe, L. M. Artner, J. Dernedde, R. Haag and C. P. R. Hackenberger, Org. Biomol. Chem., 2012, 10, 6211 RSC; (c) N. Nischan, A. Chakrabarti, R. A. Serwa, P. H. M. Bovee-Geurts, R. Brock and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2013, 52, 11920 CrossRef CAS PubMed; (d) N. Nischan, M.-A. Kasper, T. Mathew and C. P. R. Hackenberger, Org. Biomol. Chem., 2016, 14, 7500 RSC; (e) K. D. Siebertz and C. P. R. Hackenberger, Chem. Commun., 2018, 54, 763 RSC; (f) M.-A. Kasper, M. Glanz, A. Oder, P. Schmieder, J. P. von Kries and C. P. R. Hackenberger, Chem. Sci., 2019, 10, 6322 RSC; (g) M.-A. Kasper, M. Glanz, A. Stengl, M. Penkert, S. Klenk, T. Sauer, D. Schumacher, J. Helma, E. Krause, C. Cardoso, H. Leonhardt and C. P. R. Hackenberger, Angew. Chem., 2019, 58, 11625 CrossRef CAS; (h) M.-A. Kasper, A. Stengl, P. Ochtrop, M. Gerlach, T. Stoschek, D. Schumacher, J. Helma, M. Penkert, E. Krause, H. Leonhardt and C. P. R. Hackenberger, Angew. Chem., 2019, 58, 11631 CrossRef CAS.
- M. Sundhoro, S. Jeon, J. Park, O. Ramström and M. Yan, Angew. Chem., Int. Ed., 2017, 56, 12117 CrossRef CAS PubMed.
- (a) Y. Xie, L. Cheng, Y. Gao, X. Cai, X. Yang, L. Yi and Z. Xi, Chem. – Asian J., 2018, 13, 1791 CrossRef CAS PubMed; (b) J. Zhang, Y. Gao, X. Kang, Z. Zhu, Z. Wang, Z. Xi and L. Yi, Org. Biomol. Chem., 2017, 15, 4212 RSC; (c) D. Mac, X. Kanga, Y. Gao, J. Zhua, L. Yi and Z. Xi, Tetrahedron, 2019, 75, 888 CrossRef.
- T. Meguro, N. Terashima, H. Ito, Y. Koike, I. Kii, S. Yoshida and T. Hosoya, Chem. Commun., 2018, 54, 7904 RSC.
- W. Luo, J. Luo, V. V. Popik and M. S. Workentin, Bioconjugate Chem., 2019, 30, 1140 CrossRef CAS PubMed.
- L. Cheng, X. Kang, D. Wang, Y. Gao, L. Yi and Z. Xi, Org. Biomol. Chem., 2019, 17, 5675 RSC.
- (a) T. Yokoi, H. Tanimoto, T. Ueda, T. Morimoto and K. Kakiuchi, J. Org. Chem., 2018, 83, 12103 CrossRef CAS PubMed; (b) T. Yokoi, T. Ueda, H. Tanimoto, T. Morimoto and K. Kakiuchi, Chem. Commun., 2019, 55, 1891 RSC.
| This journal is © The Royal Society of Chemistry 2020 |