
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimple iodoalkyne-based organocatalysts for the activation of carbonyl compounds†
Juan V.
Alegre-Requena
 a,
Alberto
Valero-Tena
ab,
Isaac G.
Sonsona
a,
Santiago
Uriel
b and
Raquel P.
Herrera
*a
a,
Alberto
Valero-Tena
ab,
Isaac G.
Sonsona
a,
Santiago
Uriel
b and
Raquel P.
Herrera
*a
aLaboratorio de Organocatálisis Asimétrica. Dpto de Química Orgánica. Instituto de Síntesis Química y Catálisis Homogénea (ISQCH) CSIC-Universidad de Zaragoza. C/Pedro Cerbuna 12, 50009 Zaragoza, Spain. E-mail: raquelph@unizar.es
bDpto. de Química Orgánica, Escuela de Ingeniería y Arquitectura, Universidad de Zaragoza, 50018 Zaragoza, Spain
First published on 14th January 2020
Abstract
A novel approach for the formation of bisindolylmethane derivatives (BIMs) is described as a proof of concept to evaluate the catalytic capacity of iodoalkynes. The use of these derivatives is reported as an example of simple halogen bond-based organocatalyst. This kind of activation has not been used before for the synthesis of bisindolylmethane derivatives 3. Interestingly, the preparation of 3-(1H-indol-3-yl)-1-phenylbutan-1-one (8) has been also achieved for the first time with an iodoalkyne derivative. We prove the efficiency of this family of new catalysts by developing a simple and easy operational methodology, opening the door to the development of alternative catalysts in the area of halogen bond-based organocatalysts.
Introduction
Along the years, the search for new kinds of interactions or modes of activation has fascinated chemists and has been the focus of research of many scientific groups. Among the variety of non-covalent interactions, where normally hydrogen bonds occupy a privileged place,1 more recently halogen bond (XB) interactions have been making space as an alternative tool.2 Their use as Lewis acid activating Lewis bases has been recently explored in the literature.3 In contrast, during the past two decades, many applications of halogen bonding in fields as diverse as crystal engineering, enantiomer separation, biology, and supramolecular architectures have been reported and reviewed.4–6In comparison, the use of XB in organocatalysis remains underexplored. Since 2008 when Bolm reported an example of perfluoroiodoalkanes as an XB catalyst,7 positively charged iodo heterocycles such as imidazolium,8 1,2,3-triazolium8e,9 and pyridinium9a,10 or neutral halogen bond donors have been used as catalysts in a variety of organic transformations. The most common neutral XB-donor scaffolds typically contain iodopolyfluoroaromatic moieties,11 although N-iodosuccinimide, N-iodosaccharin,12 and tetrabromomethane,13 have also been used.
Halogen atoms bound to carbon, nitrogen or halide atoms show a positive electrostatic potential end-cap, i.e., a σ-hole, that interacts with Lewis bases by halogen bonding.14 Moreover, halogen atoms can be activated to participate in halogen bonding by introducing electron-withdrawing groups into the molecular backbone.
Haloalkynes have a well-established role in synthetic organic chemistry15 but their application as halogen bond donors is less well developed despite their long history.16 Theoretical, statistical and crystallographic studies demonstrate that the sp hybridization of the carbon atom adjacent to the halogen allows the ethynyl-based iodine atom to display a polar σ-hole.17 The maximum positive value of calculated electrostatic potential (Vs,max) of 1,4-bis(iodoethynyl)benzene is 25.2 kcal mol−1 and is similar to those of strong XB donors such as 1,4-diiodo-tetrafluorobenzene (25.9 kcal mol−1), which is one of the most widely used halogen bonding donors. Therefore, haloalkynes can form strong, directional and selective halogen bonds, which makes them suitable for being used as organocatalysts.18,19 Moreover, iodoalkynes are stable towards nucleophiles and elevated temperatures and easily accessible from terminal alkynes,20 which allows to modify the number and arrangement of halogen bond donors, their solubility and their rigidity or flexibility among other characteristics. However, to the best of our knowledge, there is only one example where a perfluorinated iodoethynyl compound has been used as a catalyst.20 Therefore, more contributions in this new field could be of great interest.
Indole is a privileged structural core present in many natural products and biological systems21 and its use in drug discovery has grown over the past decades.22 Consequently, considerable attention has been focused on the development of new synthetic23 and catalytic24 methods, leading to more complex indole structures. Moreover, indole is the structural unit of bisindolylmethane derivatives (BIMs), many of them isolated from marine natural sources.25 This family of indoles discloses a range of biological properties such as antibacterial, antifungal, antimicrobial, anti-inflammatory or antitumor, among others.26
Inspired in our own experience in the field of organocatalysis27 using hydrogen bond-based catalysts, we focused our attention on the activation of aldehydes for the production of bisindoles28 using iodoalkyne derivatives (non-perfluorinated) as promising catalysts for the first time in the literature.
Results and discussion
Recently, several mild and convenient methods have been developed to prepare haloalkynes, most of them from terminal alkynes.9a,15 Among them, we have used the electrophilic iodation of terminal alkynes with N-iodosuccinimide (NIS) and an Ag(I) catalyst29 due to the mild reaction conditions required, high efficiency, and simple manipulation. Organocatalysts I–IV were obtained with yields in the range of 63 to 82% from the corresponding terminal alkynes (Scheme 1).18 The precursors to synthesise I to III are commercially available and the precursor for IV was directly prepared through esterification reaction of 1,3,5-benzenecarbonyl trichloride with 2-propyn-1-ol.30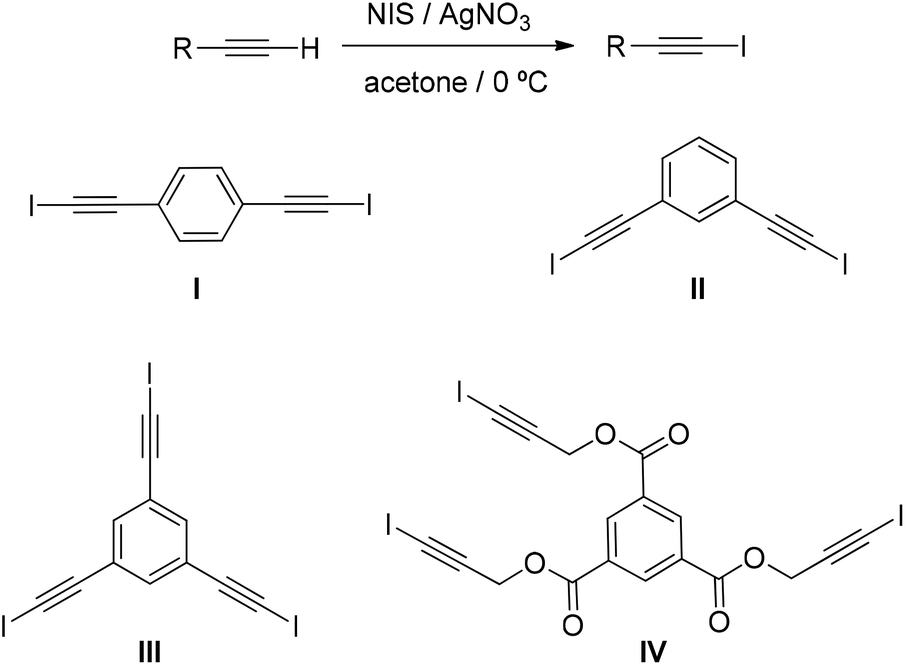 | ||
| Scheme 1 Synthesis of iodoalkynes I–IV. | ||
In order to further evaluate the viability of our work hypothesis, the efficiency of halogen bond-based organocatalysts I–IV was first tested in the model reaction between indole (2a) and 3-nitrobenzaldehyde (1a). Interestingly, the synthesis of bisindole 3aa was successful in all cases. First, the activity of catalysts I–IV was evaluated in CH2Cl2. Although the reaction worked well in all cases (Table S1,† entries 1–4), it was noted that small loss of solvent through evaporation could be accelerating the reaction and the results could be misleading. Therefore, in order to prevent this problem, toluene was tested as a solvent. In this case, catalyst I successfully activated bisindole 3aa formation better than the other three catalysts (Table 1, entry 1).
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Time (days) | Yieldb (%) |
a Experimental conditions: To a mixture of catalyst I–IV (0.03 mmol) and aldehyde 1a (0.1 mmol) in toluene (250 μL), indole (2a) (0.4 mmol, 2 equiv.) was further added at room temperature. After the reaction time, adduct 3aa was isolated by column chromatography (hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AcOEt 8:2).
b Isolated yield. :AcOEt 8:2).
b Isolated yield.
|
|||
| 1 | I | 2 | 98 |
| 2 | II | 3 | 89 |
| 3 | III | 5 | 92 |
| 4 | IV | 2 | 54 |

Although other more polar solvents were also tested, such as acetonitrile, THF, dioxane and ethyl acetate (Table S1,† entries 5–8), they led to reaction quenching or lower yields. This finding is not surprising since polar solvents typically disrupt the activating catalyst⋯substrate XB interactions easier than nonpolar solvents such as toluene. Using toluene and catalyst I in the model reaction, an optimization of different reaction conditions was carried out (Table 2).
|
|
||||
|---|---|---|---|---|
| Entry | I (mol%) | Indole 2a (mmol)/equiv. | Time (days) | Yieldb (%) |
|
a Experimental conditions: to a mixture of catalyst I (30–0 mol%) and aldehyde 1a (0.1 mmol) in toluene (0.5–0.25 mL), indole (2a) (0.4–0.2 mmol) was further added at room temperature. After the reaction time, adduct 3aa was isolated by column chromatography (hexane:AcOEt 8:2).
b Isolated yield.
c 0.25 mL of toluene.
d No reaction observed.
|
||||
| 1 | 30 | 0.4/2 | 2 | 98 |
| 2 | 30 | 0.3/1.5 | 2 | 98 |
| 3 | 30 | 0.2/1 | 2 | 47 |
| 4 | 30c | 0.2/1 | 2 | 74 |
| 5 | 20 | 0.4/2 | 2 | 98 |
| 6 | 20c | 0.4/2 | 2 | 98 |
| 7 | 20c | 0.3/1.5 | 2 | 98 |
| 8 | 20c | 0.2/1 | 2 | 35 |
| 9 | 10 | 0.4/2 | 5 | 77 |
| 10 | — | 0.4/2 | 2 | n.r.d |
Reactions were stopped after two days to allow for a better comparison of the yields obtained; however, reactions using 2 and 1.5 equivalents of indole 2a (0.4 mmol or 0.3 mmol) with 20–30 mol% of catalyst were probably completed before the reaction time employed (Table 2, entries 1, 2, 5, 6 and 7). The results showed that 20 and 30 mol% of catalyst lead to similar yields while 10 mol% of catalyst affords lower yields. Also, raising the amount of indole 2a from one to two equivalents (Table 2, entries 6 and 8) and increasing concentration (Table 2, entries 3 and 4) have great positive impacts on the yields obtained. The best conditions found included 1.5 equivalents of indole 2a and 20 mol% of catalyst in 250 μL of toluene (Table 2, entry 7). It is worth noting that in the absence of catalyst I the reaction did not work (Table 2, entry 10), which supports the role of I as the catalyst of this process. In all cases, the crudes of the reactions are very clean and only the excess of indole and the final product appear in the NMR spectra.
Using the best reaction conditions, the scope of this methodology was evaluated employing diverse commercially available aldehydes and indoles. This methodology was successfully applied to produce a great number of substituted bisindoles 3 with very good results (Table 3).
|
|
||||
|---|---|---|---|---|
| Entry | Aldehyde | Indole | Product | Yieldb (%) |
|
a Experimental conditions: to a mixture of catalyst I (20 mol%) and aldehyde 1b–k (0.1 mmol) in toluene (250 μL), indole 2a–c (0.3 mmol) was further added at room temperature. After two days of reaction time, adducts 3 were isolated by column chromatography (hexane:AcOEt 8:2).
b Isolated yield.
c After 3 days of reaction.
d 30 mol% of I.
|
||||
| 1 | 4-NO2Ph (1b) | H (2a) | 3ba | 98 |
| 2 | 4-CNPh (1c) | H (2a) | 3ca | 95 |
| 3c | 4-ClPh (1d) | H (2a) | 3da | 95 |
| 4c | 4-BrPh (1e) | H (2a) | 3ea | 85 |
| 5 | 3-ClPh (1f) | H (2a) | 3fa | 89 |
| 6 | 3-BrPh (1g) | H (2a) | 3ga | 90 |
| 7d | Ph (1h) | H (2a) | 3ha | 98 |
| 8d | 4-MePh (1i) | H (2a) | 3ia | 85 |
| 9 | 2-Phenylethyl (1j) | H (2a) | 3ja | 95 |
| 10d | 2-Furyl (1k) | H (2a) | 3ka | 81 |
| 11d | Ph (1h) | 2-Me (2b) | 3hb | 98 |
| 12d | Ph (1h) | 5-OMe (2c) | 3hc | 54 |
As reported in Table 3, high yields were achieved in a reasonable reaction time using a representative spectrum of aldehydes 1b–k. Non-activated aldehydes 1h,i (without an electron withdrawing group in their skeleton, entries 7 and 8) or heteroaromatic aldehyde 1k (entry 10) showed a lower reactivity and a 30 mol% of catalyst I was necessary to achieve high yields in the same reaction time. In contrast, activated aldehydes 1b–g (entries 1–6) or aliphatic 1j (entry 9) afforded good levels of reactivity in all cases. Surprisingly, aldehydes 1d,e (entries 3 and 4) required longer reaction times (72 h), although with good yields. Activated indole 2b (entry 11) showed high reactivity with aldehyde 1h while indole 2c only led to a moderate yield (entry 12).
A plausible mechanism is proposed assuming the same route as that previously reported by us when using AgOTf as a Lewis acid (Scheme 2).28 Hence, catalyst I would activate the first addition of one molecule of indole 2 to aldehyde 1 through halogen bonding (4).31 Unstable intermediate 5 would promote the elimination of a molecule of H2O to give azafulvene derivative 6.32 Finally, intermediate 6 would undergo a further addition of a second molecule of indole 2 to produce the final observed product 3.
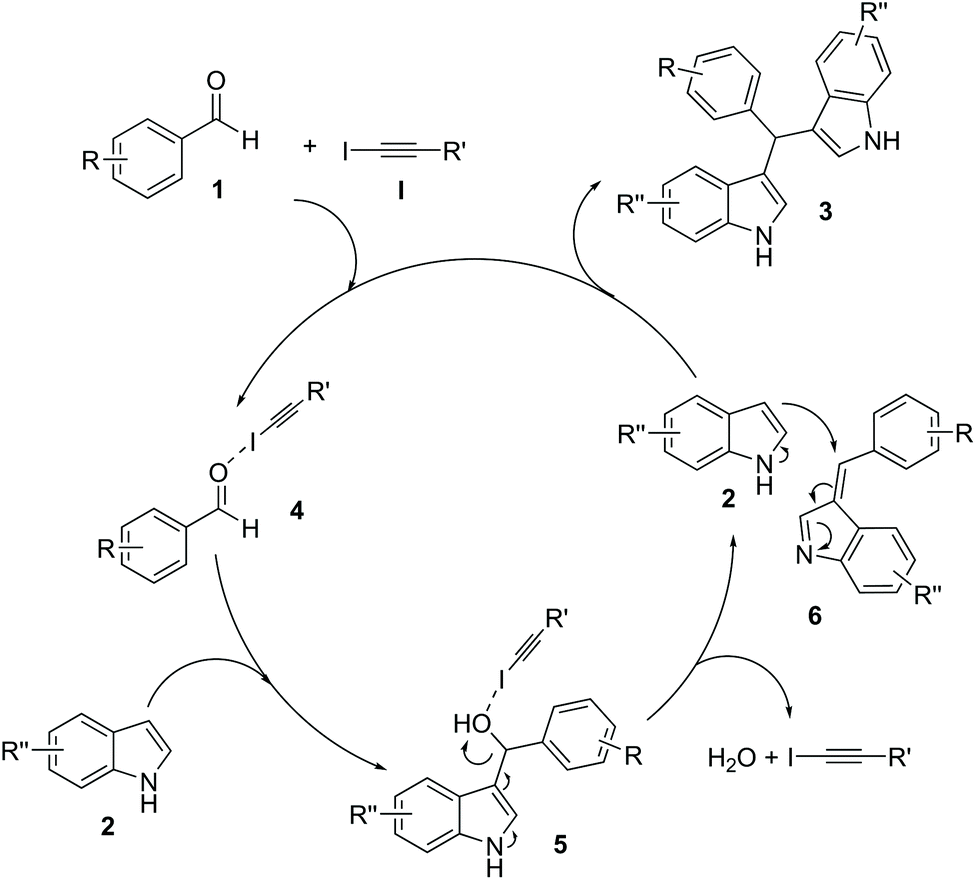 | ||
| Scheme 2 Plausible reaction mechanism. | ||
In order to expand the utility of catalyst I, an additional model reaction between trans-1-pheny-2-buten-1-one (7) and indole (2a) has been also explored as described in Table 4.8c,33
|
|
|||
|---|---|---|---|
| Entry | I (mol%) | Solvent | Yieldb (%) |
|
a Experimental conditions: to a mixture of catalyst I (0–20 mol%) and trans-crotonophenone (7) (0.1 mmol) in the corresponding solvent (0.25 mL), indole 2a (0.2 mmol) was further added at room temperature. After 48 h of reaction, product 8 was isolated by column chromatography (hexane:AcOEt 8:2).
b Isolated yield.
c No reaction observed.
|
|||
| 1 | — | MeOH | n.r.c |
| 2 | 10 | CH3CN | n.r.c |
| 3 | 10 | Toluene | n.r.c |
| 4 | 10 | MeOH | 37 |
| 5 | 20 | MeOH | 82 |
Interestingly, we have also observed reactivity using catalyst I in the Michael addition reaction depicted in Table 4 with MeOH as solvent at room temperature (entries 4 and 5). It is remarkable that the process did not work in other less polar solvents such as toluene or in polar aprotic solvents such as acetonitrile (entries 2 and 3, respectively). Since it is remarkable that even with the presence of protic solvents, such as MeOH, the halogen–bond interaction is formed, in order to be sure that the reaction was not promoted by MeOH itself, the background of this Michael addition was also examined. In the absence of catalyst, the formation of the final product 8 was not observed at the same reaction time (entry 1). This reaction shows the importance of our catalyst promoting also this benchmark reaction with a simpler iodine-based catalyst in comparison with those catalysts previously used to promote this reaction.8c,33 Moreover, Breugst and co-workers found that an iodoalkyne-based catalyst did not promote the latter reaction.33d Therefore, this proves that structural variations in the aromatic ring could be pivotal to promote the process. The efficiency of catalyst I to activate other α,β-unsaturated ketone derivatives is being explored in this moment in our lab.
Computational study of the I⋯O halogen bond
We have computationally analysed the impact that the I⋯O halogen bond created between catalyst I and the initial aldehydes has on the reaction. Initially, the I⋯O interaction is compared with the H⋯O interaction created by an analogous hydrogenated catalyst (IH) that does not contain iodine atoms (Fig. 1).34 | ||
| Fig. 1 Optimised complexes of benzaldehyde (1h) with catalyst I (I⋯1h) or IH (H⋯1h) along with the bonding energies (BE) created by the I⋯O and H⋯O interactions. Negative values in the BE values correspond to attractive interactions. Calculated at the ωB97X-D/Def2-QZVPP(SMD)//ωB97X-D/Def2-TZVP(SMD) level of theory.35 | ||
The distance and angle of the calculated halogen bonding are in accordance with those determined in the crystal structure of II⋯acetone halogen bonding complex.18 The results suggest that iodine atoms create stronger interactions with the O atom of benzaldehyde compared to H atoms (more negative BE when catalyst I is used). Analogous to the electron flow in hydrogen bonds, iodine atoms take electron density from O atoms in halogen bonds.2 Therefore, this I⋯O interaction could activate the carbonyl group of the aldehyde molecule towards a subsequent indole attack. In order to verify this, we calculated the electrostatic potentials at nuclei (EPN) of the carbonyl C atom of benzaldehyde and complexes I⋯1h and IH⋯1h (Table 5).
| Entry | System | Relative EPN (kcal mol−1) | Experimental yield (%) |
|---|---|---|---|
| 1 | 1h | 0.0 | 0 |
| 2 | IH⋯1h | 4.5 | 0 |
| 3 | I⋯1h | 11.1 | 98 |
EPN values have previously been correlated to the reactivity of different functional groups, including carbonyl groups.36 As the EPN value of the carbonyl C atom becomes higher, the electrophilicity and reactivity of the carbonyl group increases. As seen in Table 5, benzaldehyde 1h shows the lowest EPN value of the three values calculated (Table 5, entry 1) and suggests that the carbonyl group of benzaldehyde is the least reactive carbonyl group of the carbonyl groups studied. The results also indicate that the I⋯O interaction created by catalyst I (Table 5, entry 3) activates the carbonyl group in a greater extent than the H⋯O interaction created by its hydrogenated analogous IH (Table 5, entry 2). As expected, the reaction was performed experimentally using catalyst IH and no reaction was observed after two days, while catalyst I showed a 98% yield after two days under the same reaction conditions (Table 3, entry 7).
Furthermore, in order to ensure that the I⋯O interaction was generated, NCIPLOT37 was employed to analyse the noncovalent interactions (NCI) generated in the I⋯1h complex (Fig. 2). The results suggest that the I⋯O halogen is the only relevant intermolecular NCI created in the I⋯1h complex and, therefore, the only NCI that activates the carbonyl group towards the nucleophilic attack of indole.
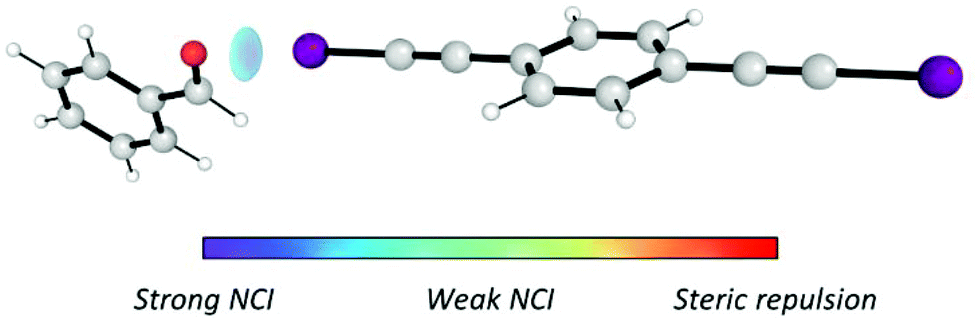 | ||
| Fig. 2 I⋯O halogen bond created between catalyst I and benzaldehyde (1h). Only attractive NCI are shown. | ||
Preliminary kinetic studies performed with the aim of knowing the order of reaction of the aldehyde seem to provide a more complex mechanism of reaction. These studies suggest that more than one molecule of aldehyde is involved in the process. Since at this stage the role of more than one molecule of aldehyde could not be clear, more computational calculations and experimental studies are ongoing in our lab in order to understand this interesting aspect.
Conclusions
A novel approach for the formation of bisindolylmethane derivatives (BIMs) is described as a proof of concept to evaluate the catalytic ability of iodoalkyne organocatalysts. The use of these derivatives is reported as an example of simple halogen bond-based organocatalyst. This kind of activation has not been used before for the synthesis of bisindolylmethane derivatives. We prove the efficiency of this family of new catalysts by developing a simple and easy operational methodology, opening the door to the development of alternative catalysts in the area of halogen bond-based catalysts. Additional kinetic studies and computational calculations are ongoing in our lab in order to shed light to the mechanism of this process.Experimental section
General experimental methods and instrumentation
Purification of reaction products was carried out either by flash chromatography using silical-gel (0.063–0.200 mm). Analytical thin layer chromatography was performed on 0.25 mm silica-gel 60-F plates. ESI ionization method and mass analyser type MicroTof-Q were used for the ESI measurements. 1H and 13C{1H}-APT NMR were recorded at room temperature on a BRUKER AVANCE 400 spectrometer (1H, 300 or 400 MHz; 13C, 75 or 100.6 MHz) in CDCl3, CD3COCD3 or CD3CN as solvent. Chemical shifts were reported in the δ scale relative to residual CHCl3 (7.28 ppm), CH3COCH3 (2.05 ppm) and CH3CN (1.94 ppm) for 1H NMR and to the central line of CHCl3 (77.16 ppm), CH3COCH3 (29.84 ppm) and CH3CN (1.32 ppm) for 13C{1H}-APT NMR. Tri(Prop-2-ynyl)benzene-1,3,5-tricarboxylate was synthesized according to the literature procedure.30 All other reagents were obtained from commercial sources and used without prior purification.All commercially available solvents and reagents were used as received. 1H- and 13C{1H}-APT NMR spectra for compounds 3aa,383ba,393ca,403da,413ea,403fa,383ha,383ia,383ka,423hb,433hc44 and 833d are consistent with values previously reported in the literature.
General procedure for electrophilic iodation of terminal alkynes
2 mmol of the corresponding ethynyl derivative was dissolved in acetone (30 mL) followed by the addition of AgNO3 (0.1 mol/mol C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H). The reaction was kept for 30 min in an ice bath, in the dark, then N-iodosuccinimide (1.2 mol/mol CC–H) was added slowly. The reaction was stirred overnight at room temperature. The crude was filtered over Celite, the solvent was removed in vacuo, and the residue was dissolved in CH2Cl2 (30 mL) and washed with NaHCO3 (10%) (3 × 20 mL). The organic fraction was dried over MgSO4, filtered and the solvent was evaporated. The pure products were recovered as yellow solids (63–82%).
C–H). The reaction was kept for 30 min in an ice bath, in the dark, then N-iodosuccinimide (1.2 mol/mol CC–H) was added slowly. The reaction was stirred overnight at room temperature. The crude was filtered over Celite, the solvent was removed in vacuo, and the residue was dissolved in CH2Cl2 (30 mL) and washed with NaHCO3 (10%) (3 × 20 mL). The organic fraction was dried over MgSO4, filtered and the solvent was evaporated. The pure products were recovered as yellow solids (63–82%).
Representative procedure for synthesis of bis(indolyl)methanes (3)
To a mixture of catalyst I (20 mol%, 7.6 mg) and aldehyde 1a–k (0.1 mmol) in toluene (250 μL), indole 2a–c (0.3 mmol) was further added in a test tube at room temperature. After two days of reaction time, adducts 3 was isolated by column chromatography (hexane:AcOEt 8:2). The yields are given in Table 3.
Representative procedure for synthesis of 3-(1H-indol-3-yl)-1-phenylbutan-1-one (8)
To a mixture of catalyst I (20 mol%, 7.6 mg) and ketone 7 (0.1 mmol) in MeOH (250 μL), indole 2a (0.2 mmol) was further added in a test tube at room temperature. After two days of reaction time, adduct 8 was isolated by chromatography (hexane:AcOEt 9:1).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a 2018 Leonardo Grant for Researchers and Cultural Creators, BBVA Foundation. Authors also thank the Ministerio de Economía, Industria y Competitividad (MINECO/FEDER CTQ2017-88091-P and PGC2018-093761-B-C31), and DGA-FSE (E7_17R and E47_17R) for financial support of this research. All the calculations were performed in the Trueno cluster facility of SGAI-CSIC and the Extreme Science and Engineering Discovery Environment (XSEDE) through allocation CHE190111. J.V.A.-R. thanks Dr Robert S. Paton (Colorado State University) for his support with NCIPLOT and PyMOL. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).Notes and references
- Hydrogen Bonding in Organic Synthesis, ed. P. M. Pihko, Wiley-VCH, Weinheim, 2009 Search PubMed.
- (a) S. Scheiner, Acc. Chem. Res., 2013, 46, 280–288 CrossRef CAS PubMed; (b) Halogen Bonding I, ed. P. Metrangolo and G. Resnati, Springer, Heidelberg, 2015 Search PubMed; (c) Halogen Bonding II, ed. P. Metrangolo and G. Resnati, Springer, Heidelberg, 2015 Search PubMed; (d) D. Bulfield and S. M. Huber, Chem. – Eur. J., 2016, 22, 14434–14450 CrossRef CAS PubMed; (e) S. Schindler and S. M. Huber, Top. Curr. Chem., 2015, 359, 167–203 CrossRef CAS PubMed; (f) S. Benz, A. I. Poblador-Bahamonde, N. Low-Ders and S. Matile, Angew. Chem., Int. Ed., 2018, 57, 5408–5412 CrossRef CAS PubMed.
- (a) S. H. Jungbauer, S. Schindler, F. Kniep, S. M. Walter, L. Rout and S. M. Huber, Synlett, 2013, 2624–2628 CAS; (b) M. Breugst, D. von der Heiden and J. Schmauck, Synthesis, 2017, 49, 3224–3236 CrossRef CAS; (c) R. Tepper and U. S. Schubert, Angew. Chem., Int. Ed., 2018, 57, 6004–6016 CrossRef CAS PubMed.
- For selected reviews, see: (a) P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS PubMed; (b) H.-J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924–3977 CrossRef CAS PubMed; (c) M. Fourmigué, Curr. Opin. Solid State Mater. Sci., 2009, 13, 36–45 CrossRef; (d) A. C. Legon, Phys. Chem. Chem. Phys., 2010, 12, 7736–7747 RSC; (e) A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS PubMed; (f) H. Wang, H. K. Bisoyi, A. M. Urbas, T. J. Bunning and Q. Li, Chem. – Eur. J., 2019, 25, 1369–1378 CrossRef CAS PubMed.
- For selected reviews, see: (a) Y. Lu, Y. Wang and W. Zhu, Phys. Chem. Chem. Phys., 2010, 12, 4543–4551 RSC; (b) R. Wilcken, M. O. Zimmermann, A. Lange, A. C. Joerger and F. M. Boeckler, J. Med. Chem., 2013, 56, 1363–1388 CrossRef CAS PubMed; (c) M. C. Ford and P. S. Ho, J. Med. Chem., 2016, 59, 1655–1670 CrossRef CAS PubMed; (d) L. Mendez, G. Henriquez, S. Sirimulla and M. Narayan, Molecules, 2017, 22, 1397, DOI:10.3390/molecules22091397.
- (a) T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC; (b) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed; (c) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed; (d) B. Li, S. Q. Zang, L. Y. Wang and T. C. W. Mak, Coord. Chem. Rev., 2016, 308, 1–21 CrossRef CAS.
- A. Bruckmann, M. Pena and C. Bolm, Synlett, 2008, 900–902 CAS.
- (a) M. Saito, Y. Kobayashi, S. Tsuzuki and Y. Takemoto, Angew. Chem., Int. Ed., 2017, 56, 7653–7657 CrossRef CAS PubMed; (b) W. He, Y.-C. Ge and C.-H. Tan, Org. Lett., 2014, 16, 3244–3247 CrossRef CAS PubMed; (c) J.-P. Gliese, S. H. Jungbauer and S. M. Huber, Chem. Commun., 2017, 53, 12052–12055 RSC; (d) S. H. Jungbauer, S. M. Walter, S. Schindler, L. Rout, F. Kniep and S. M. Huber, Chem. Commun., 2014, 50, 6281–6284 RSC; (e) S. H. Jungbauer and S. M. Huber, J. Am. Chem. Soc., 2015, 137, 12110–12120 CrossRef CAS PubMed.
- (a) F. Kniep, L. Rout, S. M. Walter, H. K. V. Bensch, S. H. Jungbauer, E. Herdtweck and S. M. Huber, Chem. Commun., 2012, 48, 9299–9301 RSC; (b) A. Dreger, E. Engelage, B. Mallick, P. D. Beer and S. M. Huber, Chem. Commun., 2018, 54, 4013–4016 RSC; (c) R. Haraguchi, S. Hoshino, M. Sakai, S. Tanazawa, Y. Morita, T. Komatsu and S. Fukuzawa, Chem. Commun., 2018, 54, 10320–10323 RSC.
- Y.-C. Chan and Y.-Y. Yeung, Angew. Chem., Int. Ed., 2018, 57, 3483–3487 CrossRef CAS PubMed.
- (a) S. M. Walter, S. H. Jungbauer, F. Kniep, S. Schindler, E. Herdtweck and S. M. Huber, J. Fluor. Chem., 2013, 150, 14–20 CrossRef CAS; (b) F. Kniep, S. H. Jungbauer, Q. Zhang, S. M. Walter, S. Schindler, I. Schnapperelle, E. Herdtweck and S. M. Huber, Angew. Chem., Int. Ed., 2013, 52, 7028–7032 CrossRef CAS PubMed.
- N. Tsuji, Y. Kobayashi and Y. Takemoto, Chem. Commun., 2014, 50, 13691–13694 RSC.
- (a) C. Huo, M. Wu, F. Chen, X. Jia, Y. Yuan and H. Xie, Chem. Commun., 2015, 51, 4708–4711 RSC; (b) I. Kazi, S. Guha and G. Sekar, Org. Lett., 2017, 19, 1244–1247 CrossRef CAS PubMed.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CAS.
- W. Wu and H. Jiang, Acc. Chem. Res., 2014, 47, 2483–2504 CrossRef CAS PubMed.
- (a) A. Sun, J. W. Lauher and N. S. Goroff, Science, 2006, 312, 1030–1033 CrossRef CAS PubMed; (b) H. M. Yamamoto, Y. Kosaka, R. Maeda, J. I. Yamaura, A. Nakao, T. Nakamura and R. Kato, ACS Nano, 2008, 2, 143–155 CrossRef CAS PubMed; (c) C. Perkins, S. Libri, H. Adams and L. Brammer, CrystEngComm, 2012, 14, 3033–3038 RSC; (d) P. M. J. Szell, S. Zablotny and D. L. Bryce, Nat. Commun., 2019, 10, 916 CrossRef PubMed; (e) M. D. Perera and C. B. Aakeröy, New J. Chem., 2019, 43, 8311–8314 RSC.
- (a) C. B. Aakeröy, M. Baldrighi, J. Desper, P. Metrangolo and G. Resnati, Chem. – Eur. J., 2013, 19, 16240–16247 CrossRef PubMed; (b) L. González, S. Graus, R. M. Tejedor, P. López, J. Elguero, J. L. Serrano and S. Uriel, CrystEngComm, 2018, 20, 3167–3170 RSC; (c) S. T. Nguyen, T. L. Ellington, K. E. Allen, J. D. Gorden, A. L. Rheingold, G. S. Tschumper, N. I. Hammer and D. L. Watkins, Cryst. Growth Des., 2018, 18, 3244–3254 CrossRef CAS.
- L. González, N. Gimeno, R. M. Tejedor, V. Polo, M. B. Ros, S. Uriel and J. L. Serrano, Chem. Mater., 2013, 25, 4503–4510 CrossRef.
- L. González, R. M. Tejedor, E. Royo, B. Gaspar, J. Munariz, A. Chanthapally, J. Serrano, J. J. Vittal and S. Uriel, Cryst. Growth Des., 2017, 17, 6212–6223 CrossRef.
- A. Matsuzawa, S. Takeuchi and K. Sugita, Chem. - Asian J., 2016, 11, 2863–2866 CrossRef CAS PubMed.
- (a) P. Ruiz-Sanchis, S. A. Savina, F. Albericio and M. Álvarez, Chem. – Eur. J., 2011, 17, 1388–1408 CrossRef CAS PubMed; (b) R. Vicente, Org. Biomol. Chem., 2011, 9, 6469–6480 RSC.
- (a) M. Lounasmaa and A. Tolvanen, Nat. Prod. Rep., 2000, 17, 175–191 RSC; (b) S. Hibino and T. Choshi, Nat. Prod. Rep., 2001, 18, 66–87 RSC; (c) Y.-J. Wu, Top. Heterocycl. Chem., 2010, 26, 1–29 CAS.
- (a) Indole Ring Synthesis: From Natural Products to Drug Discovery, ed. G. W. Gribble, John Wiley & Sons Ltd. 2016 Search PubMed; (b) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875–2911 CrossRef CAS PubMed.
- (a) M. Bandini, A. Melloni, S. Tommasi and A. Umani-Ronchi, Synlett, 2005, 1199–1222 CrossRef CAS; (b) E. Marqués-López, A. Diez-Martinez, P. Merino and R. P. Herrera, Curr. Org. Chem., 2009, 13, 1585–1609 CrossRef; (c) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608–9644 CrossRef CAS PubMed; (d) V. Terrasson, R. M. de Figueiredo and J. M. Campagne, Eur. J. Org. Chem., 2010, 2635–2655 CrossRef CAS; (e) M. Zeng and S.-L. You, Synlett, 2010, 1289–1301 CAS; (f) P. Chauhan and S. S. Chimni, RSC Adv., 2012, 2, 6117–6134 RSC.
- (a) U. Pindur and T. Lemster, Curr. Med. Chem., 2001, 8, 1681–1698 CrossRef CAS PubMed; (b) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489–4497 CrossRef CAS PubMed.
- (a) S. Safe, S. Papineni and S. Chintharlapalli, Cancer Lett., 2008, 269, 326–338 CrossRef CAS PubMed; (b) M. C. Bell, P. Crowley-Nowick, H. L. Bradlow, D. W. Sepkovic, D. Schmidt-Grimminger, P. Howell, E. J. Mayeaux, A. Tucker, E. A. Turbat-Herrera and J. M. Mathis, Gynecol. Oncol., 2000, 78, 123–129 CrossRef CAS PubMed; (c) C. Hong, G. L. Firestone and L. F. Bjeldanes, Biochem. Pharmacol., 2002, 63, 1085–1097 CrossRef CAS PubMed; (d) H. T. Le, C. M. Schaldach, G. L. Firestone and L. F. Bjeldanes, J. Biol. Chem., 2003, 278, 21136–21145 CrossRef CAS PubMed; (e) V. Fernández-Moreira, C. Val-Campillo, I. Ospino, R. P. Herrera, I. Marzo, A. Laguna and M. C. Gimeno, Dalton Trans., 2019, 48, 3098–3108 RSC.
- (a) G. Dessole, R. P. Herrera and A. Ricci, Synlett, 2004, 2374–2378 CAS; (b) R. P. Herrera, V. Sgarzani, L. Bernardi and A. Ricci, Angew. Chem., Int. Ed., 2005, 44, 6576–6579 CrossRef CAS PubMed; (c) E. Marqués-López and R. P. Herrera, in New Strategies in Chemical Synthesis and Catalysi, ed. B. Pignataro. Wiley-VCH, Weinheim; 2012, pp 175–199; (d) D. Roca-López, E. Marqués-López, A. Alcaine, P. Merino and R. P. Herrera, Org. Biomol. Chem., 2014, 12, 4503–4510 RSC; (e) V. Juste-Navarro, E. Marqués-López and R. P. Herrera, Asian J. Org. Chem., 2015, 4, 884–889 CrossRef CAS.
- J. Beltrá, M. C. Gimeno and R. P. Herrera, Beilstein J. Org. Chem., 2014, 10, 2206–2214 CrossRef PubMed.
- (a) T. Nishikawa, S. Shibuya and S. Hosokawa, Synlett, 1994, 485–486 CrossRef CAS; (b) K. Bouchmella, B. Boury, S. G. Dutremez and A. Van Der Lee, Chem. – Eur. J., 2007, 13, 6130–6138 CrossRef CAS PubMed.
- G. Chen, J. Kumar, A. Gregory and M. H. Stenzel, Chem. Commun., 2009, 6291–6293 RSC.
- V. Nemec, L. Fotović, T. Vitasović and D. Cinčić, CrystEngComm, 2019, 21, 3251–3255 RSC.
- (a) A. H. Cook and J. R. Majer, J. Chem. Soc., 1944, 486–487 RSC; (b) A. H. Cook and J. R. Majer, J. Chem. Soc., 1944, 488–489 RSC.
- For other examples of this reaction activated with halogen bond-based catalysts, see also: (a) B. K. Banik, M. Fernandez and C. Alvarez, Tetrahedron Lett., 2005, 46, 2479–2482 CrossRef CAS; (b) M. Breugst, E. Detmar and D. von der Heiden, ACS Catal., 2016, 6, 3203–3212 CrossRef CAS; (c) D. von der Heiden, S. Bozkus, M. Klussmann and M. Breugst, J. Org. Chem., 2017, 82, 4037–4043 CrossRef CAS PubMed; (d) D. von der Heiden, E. Detmar, R. Kuchta and M. Breugst, Synlett, 2018, 29, 1307–1313 CrossRef CAS; (e) R. A. Squitieri, K. P. Fitzpatrick, A. A. Jaworski and K. A. Scheidt, Chem. – Eur. J., 2019, 25, 10069–10073 CrossRef CAS PubMed.
- IH is the commercially available compound 1,4-diethynylbenzene.
- Calculations were performed using Gaussian 16: M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- (a) B. Galabov, S. Ilieva, B. Hadjieva, Y. Atanasov and H. F. Schaefer III, J. Phys. Chem. A, 2008, 112, 6700–6707 CrossRef CAS PubMed; (b) B. Galabov, S. Ilieva, G. Koleva, W. D. Allen, H. F. Schaefer, III and P. von R. Schleyer, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 37–55 CAS.
- NCIPLOT Version 3.0. (a) E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed; (b) J. Contreras-Garcia, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef CAS PubMed.
- H.-E. Qu, C. Xiao, N. Wang, K.-H. Yu, Q.-S. Hu and L.-X. Liu, Molecules, 2011, 16, 3855–3868 CrossRef CAS PubMed.
- R. Martínez, A. Espinosa, A. Tárraga and P. Molina, Tetrahedron, 2008, 64, 2184–2191 CrossRef.
- L.-T. An, F.-Q. Ding, J.-P. Zou, X.-H. Lu and L.-L. Zhang, Chin. J. Chem., 2007, 25, 822–827 CrossRef CAS.
- C. J. Magesh, R. Nagarajan, M. Karthik and P. T. Perumal, Appl. Catal., A, 2004, 266, 1–10 CrossRef CAS.
- A. Khalafi-Nezhad, A. Parhami, A. Zare, A. R. M. Zare, A. Hasaninejad and F. Panahi, Synthesis, 2008, 617–621 CrossRef CAS.
- M. L. Deb and P. J. Bhuyan, Synthesis, 2008, 2891–2898 CAS.
- R. Ramachandiran, D. Muralidharan and P. T. Perumal, Tetrahedron Lett., 2011, 52, 3579–3583 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ob02688f |
| This journal is © The Royal Society of Chemistry 2020 |