
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Site-selective protein modification via disulfide rebridging for fast tetrazine/trans-cyclooctene bioconjugation†
Lujuan
Xu
ab,
Marco
Raabe
 ab,
Maksymilian M.
Zegota
ab,
João C. F.
Nogueira
c,
Vijay
Chudasama
c,
Seah Ling
Kuan
*ab and
Tanja
Weil
*ab
ab,
Maksymilian M.
Zegota
ab,
João C. F.
Nogueira
c,
Vijay
Chudasama
c,
Seah Ling
Kuan
*ab and
Tanja
Weil
*ab
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: weil@mpip-mainz.mpg.de; kuan@mpip-mainz.mpg.de
bInstitute of Inorganic Chemistry I, University of Ulm, Albert-Einstein-Allee 11, 89081 Ulm, Germany
cDepartment of Chemistry, University College London, London, UK
First published on 9th January 2020
Abstract
An inverse electron demand Diels–Alder reaction between tetrazine and trans-cyclooctene (TCO) holds great promise for protein modification and manipulation. Herein, we report the design and synthesis of a tetrazine-based disulfide rebridging reagent, which allows the site-selective installation of a tetrazine group into disulfide-containing peptides and proteins such as the hormone somatostatin (SST) and the antigen binding fragment (Fab) of human immunoglobulin G (IgG). The fast and efficient conjugation of the tetrazine modified proteins with three different TCO-containing substrates to form a set of bioconjugates in a site-selective manner was successfully demonstrated for the first time. Homogeneous, well-defined bioconjugates were obtained underlining the great potential of our method for fast bioconjugation in emerging protein therapeutics. The formed bioconjugates were stable against glutathione and in serum, and they maintained their secondary structure. With this work, we broaden the scope of tetrazine chemistry for site-selective protein modification to prepare well-defined SST and Fab conjugates with preserved structures and good stability under biologically relevant conditions.
Introduction
Peptides and proteins are emerging as powerful treatment options, as exemplified by the application of antibodies and antibody fragments (Fab) in immunotherapy and antibody–drug conjugates in targeted cancer therapy.1 Nevertheless, protein stability, immunogenicity and the time required to engineer recombinant antibodies could limit their development for in vivo studies.2 In this regard, nature has evolved an optimal synthetic factory in the form of posttranslational processes, in which diverse functionalities can be attached to proteins in a site-directed fashion.3 Inspired by this, chemists strive to develop methodologies to impart diverse functionalities to native proteins with similar levels of precision.Site-selective modification of therapeutically relevant peptides and proteins to introduce reactive bioorthogonal handles has emerged for post-modification in an “on-demand” fashion to expand the features and functions of proteins to address the challenges in fundamental biological and medical applications.4 In this manner, protein therapeutics can be synthetically customized to program their properties for envisaged applications.
Over the past decades, several bioorthogonal reactions, including the [3 + 2] azide–alkyne cycloaddition, photoclick 1,3-dipolar cycloaddition and the inverse electron demand Diels–Alder (IEDDA) reactions, have been developed and applied for protein modifications.5 Among these, the IEDDA reaction of 1,2,4,5-tetrazine with trans-cyclooctene (TCO) stands out, providing fast reaction kinetics (rate constant of up to 106 M−1 s−1), excellent orthogonality, catalyst-free conditions and good biocompatibility, and is a widely employed bioorthogonal approach in chemical biology.6 Previously, the IEDDA reaction has been applied for the preparation of antibody conjugates, mainly via statistical modifications using N-hydroxysuccinimide (NHS) ester chemistry for cell imaging,7 nanoparticle functionalization,8 and antibody targeted therapy.6a,9 In the literature, two common approaches to site-selectively modify proteins utilizing the IEDDA reaction are, the introduction of a diene or dienophile by genetic code expansion methods via the incorporation of noncanonical amino acids10 and tag-based posttranslational attachment strategies using enzymes (e.g. lipoic acid protein ligase A).11 Nevertheless, the genetic code expansion method suffers from low yields and tedious synthesis. This in turn limits its scalability and accessibility for further applications, while tag-based posttranslational attachment strategies first require the genetic fusion of a specific peptide sequence (e.g. lipoate acceptor peptide) to the protein of interest.11 A single tetrazine or TCO group has also been introduced site-selectively into peptides or proteins at an unpaired cysteine site via the thiol-maleimide reaction.6a,12
However, very few native proteins contain free cysteine residues.13 Furthermore, thiol–maleimide chemistry easily undergoes the retro-Michael reaction resulting in a maleimide exchange with other reactive thiols present, for example glutathione, under physiological conditions causing off-target effects.4a Therefore, other methods, which can introduce a tetrazine or TCO group in a site-selective fashion into proteins for subsequent IEDDA reactions are highly desirable to expand the scope of their applications. Since many therapeutically relevant peptides and proteins contain at least one disulfide bond close to the protein surface, the disulfide rebridging strategy provides a versatile technique to modify the solvent accessible disulfide bonds on these proteins and peptides to install suitable bioorthogonal tags.13,14 Smith and coworkers presented an elegant approach to introduce the tetrazine group at the disulfide site of proteins/peptides via dichlorotetrazine.15 However, a significantly reduced activity of the protein was reported after the modification. In this context, disulfide modification based on bis-alkylating reagents to form bisthioether conjugates gained wide applications for site-selective protein modifications without compromising their bioactivity. Furthermore, it has been reported that bisthioether conjugates formed by disulfide rebridging are more stable than thiol-maleimide conjugates16 suggesting that it could be a viable strategy for the installation of IEEDA reaction handles. Herein, we present a simple and straightforward method for the site-selective incorporation of a tetrazine group into native peptides and proteins through disulfide rebridging with the allyl sulfone scaffold reported previously.17 In comparison with known bis-sulfone disulfide rebridging reagents, allyl sulfone reagents provide improved reactivity and higher water solubility, which greatly facilitates protein bioconjugations.17 The disulfide rebridging reactions of allyl sulfones proceed in situ without side reactions e.g. with reducing agents such as tris(2-carboxyethyl)-phosphine hydrochloride (TCEP). In comparison, disubstituted maleimide disulfide rebridging reagents, such as 3,4-dibromomaleimide, show side reactions with TCEP, presumably due to the nucleophilic addition of phosphine to the electron-poor alkene moiety.18 The versatility of the method was demonstrated by modifying two model substrates: SST and IgG Fab under mild conditions which yielded a set of well-defined protein conjugates (Fig. 1). The formed protein conjugates showed good stability and maintained their secondary structures after bioconjugation. The reported methodology provides a rapid, robust and straightforward strategy for site-selective protein labeling and expands the scope of using IEDDA chemistry in the functionalization of therapeutically relevant peptides and proteins in a precise manner.
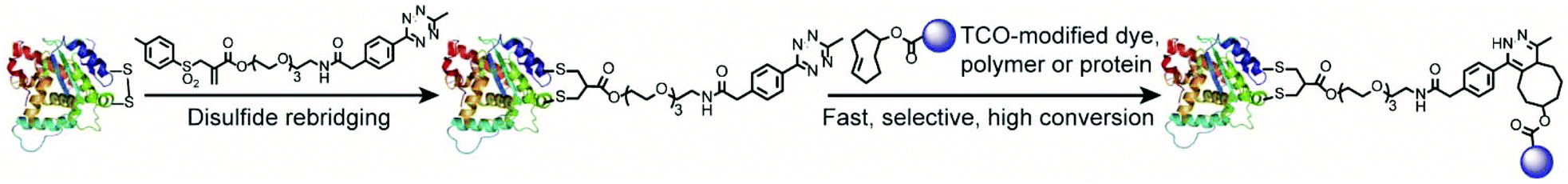 | ||
| Fig. 1 Schematic overview of site-selective incorporation of a reactive tetrazine tag into a solvent-accessible disulfide site of a cell-targeting peptide or antibody fragment. The tetrazine-modified peptide/protein is used for post-functionalization to construct a small set of protein bioconjugates with a (1) dye, (2) polymer and (3) protein in a fast and highly selective manner with high conversions (one isomer is shown as a representation). | ||
Results and discussion
Synthesis and characterization of the tetrazine-based disulfide rebridging reagent
For the site-selective installation of bioorthogonal handles on the proteins, a longer incubation time (usually overnight) is often required.13 Therefore, the diene “6-methyl-1,2,4,5-tetrazine” instead of the dienophile “TCO” was introduced into the protein. This strategy fully utilizes the fast kinetics of the IEDDA reaction but minimizes the inherent isomerization issues of TCO to nonreactive cis-cyclooctene, which occurs at a longer incubation time or in the presence of thiols.4c,19 In this context, a tetrazine-based allylsulfone disulfide rebridging reagent (IC-Tetrazine, Scheme 1) was designed and readily obtained through a four-step synthesis. As shown in Scheme 1, tetraethylene glycol monoamine (1) was protected to afford 2-(2-boc-aminoethoxy)ethanol (2) that underwent condensation with methacryloyl chloride yielding the corresponding methacrylate derivative (3). After a tandem iodosulfonylation–dehydroiodination reaction, the tosyl group was introduced to form allyl-sulfone (4). The tert-butoxycarbonyl group was subsequently removed in an acidic medium to afford the primary amino group, which then reacted with methyl-tetrazine NHS ester to afford the IC-Tetrazine reagent.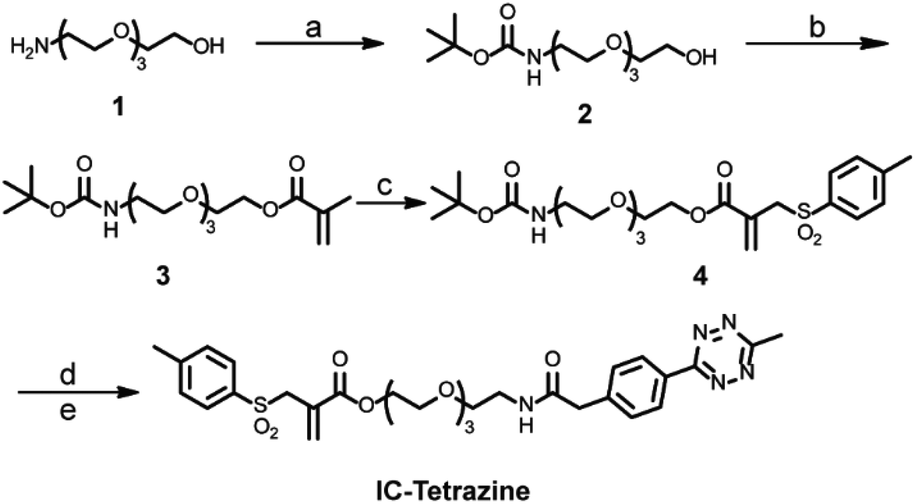 | ||
| Scheme 1 Synthesis of the tetrazine-containing disulfide rebridging reagent (IC-Tetrazine). (a) Di-tert-butyl dicarbonate, CH2Cl2, overnight. (b) Methacryloyl chloride, Et3N, CH2Cl2, 90%. (c) 1. I2, sodium p-toluenesulfinate, CH2Cl2, 3 days. 2. Et3N, CH2Cl2, overnight. 3. Et3N, ethyl acetate, 95 °C, overnight, final yield: 60%. (d) Trifluoroacetic acid (TFA), CH2Cl2, 98%. (e) Methyltetrazine NHS ester, Et3N, CH2Cl2, overnight, 40%. | ||
Site-selective protein modification
Subsequently, two model substrates (somatostatin and human IgG Fab) were selected for site-selective modification with IC-Tetrazine. Somatostatin (SST) regulates the endocrine system and mediates signal transduction via five G protein-coupled receptors (SSTR) that are overexpressed in high levels in various kinds of cancer cells and tumor blood vessels.20 SST conjugates have been widely investigated for targeted drug delivery into SSTR-expressing cancer cells.20,21 IC-Tetrazine was reacted with SST after its single accessible disulfide bond was reduced to generate free thiol groups by the addition of two equivalents of TCEP at room temperature (Fig. 2a). The product (SST-Tetrazine) was purified by high-performance liquid chromatography (HPLC) yielding SST-Tetrazine in 30% yield, which is comparable to that reported in the literature (20% to 40%).13 Characterization by matrix assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF-MS) revealed two signals corresponding to the desired SST-Tetrazine (m/z = 2110.95 [M + H]+, Fig. 2b, the molecular weight of native SST is 1637 g mol−1) and the laser fragmentation species of SST-Tetrazine (m/z = 2041.93, the chemical structure of the fragmentation species is shown in Fig. S3†), respectively. Notably, SST-Tetrazine is stable for more than one year when stored as a solid at −20 °C (Fig. S4†).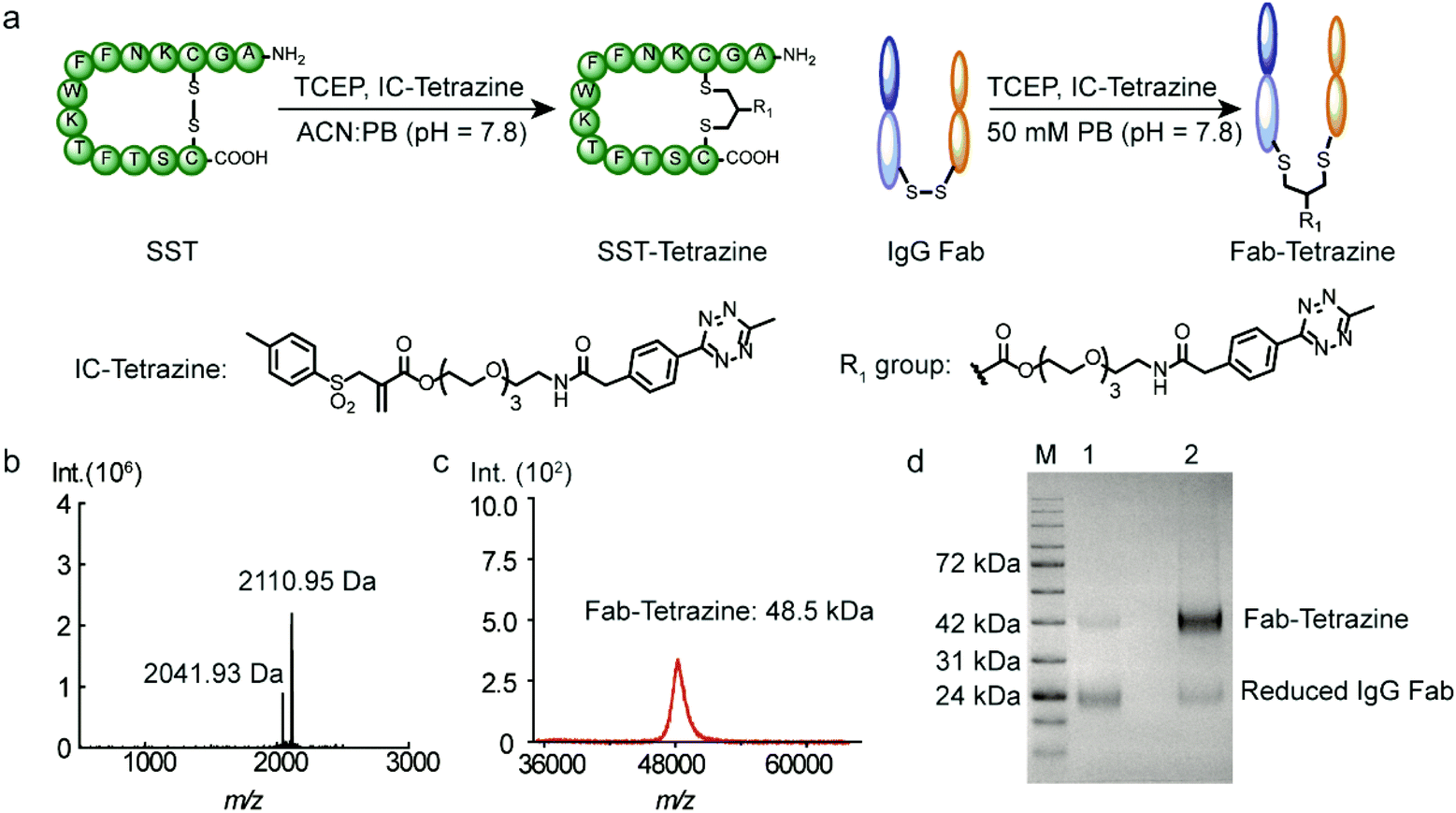 | ||
| Fig. 2 (a) Site-selective modification of SST and IgG Fab with IC-Tetrazine. (b) MALDI-TOF-MS spectrum of SST-Tetrazine (found: 2110.95 [M + H]+, calculated: 2110.95 [M + H]+). The m/z peak at 2041.93 corresponds to the fragmentation of SST-Tetrazine. (c) MALDI-TOF-MS spectrum of Fab-Tetrazine (Fab-Tetrazine: found: 48.5k [M + H]+, calculated: 48.5k [M + H]+). (d) SDS-PAGE analysis of native Fab and Fab-Tetrazine (M: Applichem Protein Marker VI, 1: Fab (native) + 100 eq. TCEP incubation for one hour, and 2: Fab-Tetrazine + 100 eq. TCEP incubation for one hour). | ||
Next, we applied the modification method to an antibody fragment (Fab) from human immunoglobulin G (IgG). Fab fragments have been successfully used for constructing antibody–drug and antibody–nanoparticle conjugates providing several advantages compared to the complete antibody.22 The conjugate retains the antigen-binding region which is crucial for active targeting, but circumvents the non-specific binding of the Fc region of antibodies.22a Since the interchain disulfides are much more exposed to the solvent and less stable relative to the intrachain disulfides that are buried between the two layers of anti-parallel β-sheet structures,23 IgG Fab was incubated with 50 equivalents of TCEP (optimization of the amounts of TCEP is shown in Fig. S5†) to reduce the solvent accessible interchain disulfide bonds and subsequently reacted with IC-Tetrazine overnight (Fig. 2a). The tetrazine modified Fab fragment (Fab-Tetrazine) was purified by ultrafiltration to remove residual TCEP and IC-Tetrazine. The MALDI-TOF-MS analysis of Fab-Tetrazine revealed a molecular weight of 48.5 kDa (Fig. 2c), showing an increase of ∼500 Da compared to that of the native Fab (48.0 kDa, Fig. S5B†) demonstrating its successful site-selective modification. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) of Fab-Tetrazine revealed a single band at ∼48 kDa showing that only a slight reduction occurred after site-selective modification even in the presence of 100 equivalents of TCEP after one hour (Fig. 2d, band 2). In contrast, under the same conditions, the native IgG Fab was almost completely reduced exhibiting a single band appearing at ∼24 kDa on SDS-PAGE (Fig. 2d, band 1). Based on the concentration calculated from gel densitometry (Fig. 2d), about 80% of the Fab fragment was successfully modified. A thiol quantification experiment using the thiol reagent 4,4′-dithiodipyridine was performed and the result showed that about 73% of IgG Fab were successfully modified, which is consistent with the result obtained from gel densitometry (details are shown in the ESI, Fig. S5c†). Taken together, the results clearly indicated that the disulfide bonds in IgG Fab were successfully rebridged using IC-Tetrazine.
Bioconjugation with a chromophore, PEG and enzyme
The versatility of the tetrazine-modified SST and IgG Fab for the construction of well-defined bioconjugates was demonstrated through post-modification with a fluorophore (cyanine-5, Cy5) and a PEG chain. In addition, SST-Tetrazine was also conjugated to a protein enzyme (cytochrome C, CytC) to form a peptide–protein conjugate with high conversion. A TCO-terminated PEG chain of a precise chain length with 12 repeating units (TCO-PEG12) was selected to facilitate the characterization by MALDI-TOF-MS. TCO-PEG12 was incubated with SST-Tetrazine in phosphate buffer (PB, 50 mM, pH = 7.4) and the pink color of the tetrazine group disappeared immediately after mixing, indicating that the reaction took place instantaneously. The bioconjugation reaction was carried out for 30 minutes to ensure completion. Thereafter, the reaction mixture was injected into the HPLC column and a predominant peak of SST-PEG12 was obtained (Fig. S7, ESI†). SST-PEG12 was isolated in nearly quantitative yield (95%) and characterized by MALDI-TOF-MS (Fig. 3b). SST-Tetrazine was also conjugated to TCO-Cy5 under similar conditions yielding the desired conjugate SST-Cy5 in 90% yield. MALDI-TOF-MS showed the m/z signal at 3041.17 (Fig. 3c).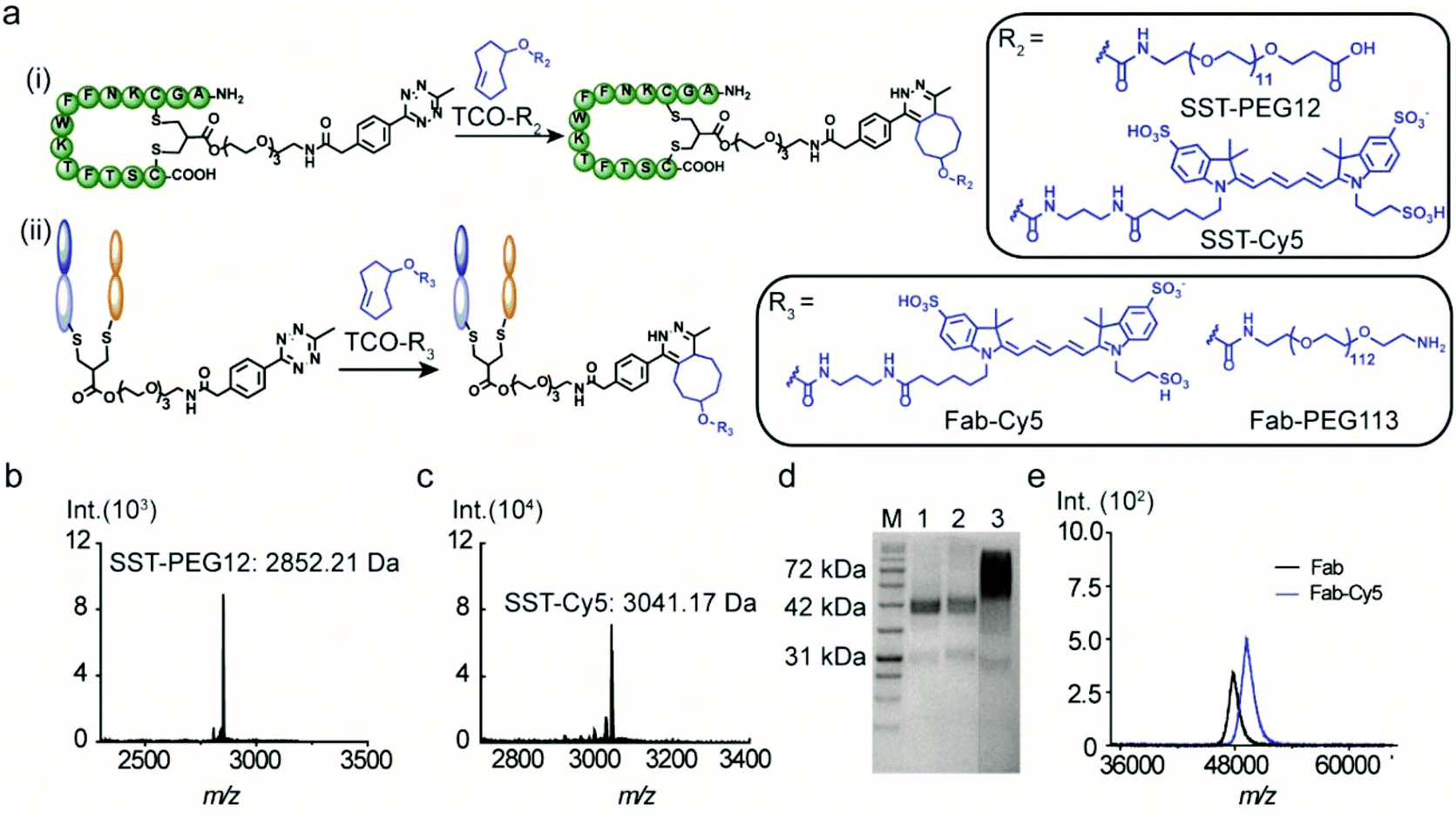 | ||
| Fig. 3 (a) Bioconjugation between (i) SST-Tetrazine with TCO-PEG12 and TCO-Cy5. (ii) Fab-Tetrazine with TCO-Cy5 and TCO-PEG113 via a fast click reaction. (b) MALDI-TOF-MS spectrum of SST-PEG12 (found: 2852.21 [M + H]+, calculated: 2852.28 [M + H]+). (c) MALDI-TOF-MS spectrum of SST-Cy5 (found: 3041.17 [M + H]+, calculated: 3041.28 [M + H]+). Sinapic acid was used as the matrix for all MALDI-TOF-MS measurements. (d) SDS-PAGE analysis of Fab-PEG113 (M: Applichem Protein Marker VI, 1: Fab-Tetrazine, and 2: Fab + TCO-PEG113, 3: Fab-PEG113). (e) MALDI-TOF-MS spectra of Fab-Cy5 (found: 49.5k [M + H]+, calculated: 49.5k [M + H]+) and native Fab (found: 48.0k [M + H]+, calculated: 48.0k [M + H]+). | ||
Due to the much larger molecular weight of Fab-Tetrazine compared to SST-Tetrazine, a longer PEG chain with a molecular weight of 5000 Da containing 113 repeating units on average (TCO-PEG113) was incubated with Fab-Tetrazine in phosphate buffer (pH = 7.4) for 30 minutes to afford Fab-PEG113. The characteristic band of Fab-Tetrazine at ∼48 kDa almost entirely disappeared after PEGylation and a new band emerged, which was broader due to the polydispersity of the PEG chain (Fig. 3d). A control experiment of mixing native Fab and TCO-PEG113 did not show any unspecific absorption on SDS-PAGE (Fig. 3d). MALDI-TOF-MS spectra revealed a signal at 53.5 kDa indicating the successful conjugation (Fig. S11D†). Under similar conditions, the widely applied chromophore Cy5 was also attached to Fab-Tetrazine to afford Fab-Cy5 (molecular weight: 49.5 kDa, Fig. 3e) in 73% yield (the calculation is shown in the ESI†). Successful conjugation was confirmed by an increase in the molecular weight of ∼1.5 kDa in the MALDI-TOF-MS spectra in comparison with those of native Fab (molecular weight: 48 kDa, Fig. 3e).
CytC is a 12 kDa hemeprotein typically located in the mitochondria of viable cells.24 Iso-yeast CytC has a single thiol group on its surface for modification via thiol-maleimide chemistry.25 TCO-PEG3-maleimide was reacted with CytC to introduce the TCO group via thiol-maleimide chemistry affording CytC-PEG3-TCO (Scheme S4†). MALDI-TOF-MS data revealed the successful modification of CytC (Fig. S6, ESI†). In addition, a tetrazine-based dye (tetrazine-5-fluorescein, Tetrazine-FAM, excitation wavelength: 492 nm and emission wavelength: 517 nm) was used to react with CytC-PEG3-TCO to assess the modification yield of CytC. Based on the absorbance of CytC at 280 nm and the FAM dye at 490 nm, about 90% of CytC was successfully modified with the TCO group (experimental details are shown in the ESI†). Thereafter, the targeting peptide SST-Tetrazine was conjugated to CytC-PEG3-TCO by mixing and shaking for 30 minutes.
After ultrafiltration to remove the residual SST-Tetrazine, SST-PEG3-CytC was obtained and characterized by MALDI-TOF-MS and SDS-PAGE. In the MALDI-TOF-MS spectra, the signal at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 315 Da corresponds to the desired SST-PEG3-CytC conjugate (Fig. 4b), whereas the signal for CytC-PEG3-TCO (13233 Da) completely disappeared. On SDS-PAGE, a slight band shift was observed indicating the successful conjugation between SST-Tetrazine and CytC-PEG3-TCO (Fig. 4c). The conjugation of Fab-Tetrazine with CytC-PEG3-TCO was also performed. However, MALDI-Tof-MS data showed almost no conjugation product, presumably due to the steric hindrance of the two bulky macromolecules.
315 Da corresponds to the desired SST-PEG3-CytC conjugate (Fig. 4b), whereas the signal for CytC-PEG3-TCO (13233 Da) completely disappeared. On SDS-PAGE, a slight band shift was observed indicating the successful conjugation between SST-Tetrazine and CytC-PEG3-TCO (Fig. 4c). The conjugation of Fab-Tetrazine with CytC-PEG3-TCO was also performed. However, MALDI-Tof-MS data showed almost no conjugation product, presumably due to the steric hindrance of the two bulky macromolecules.
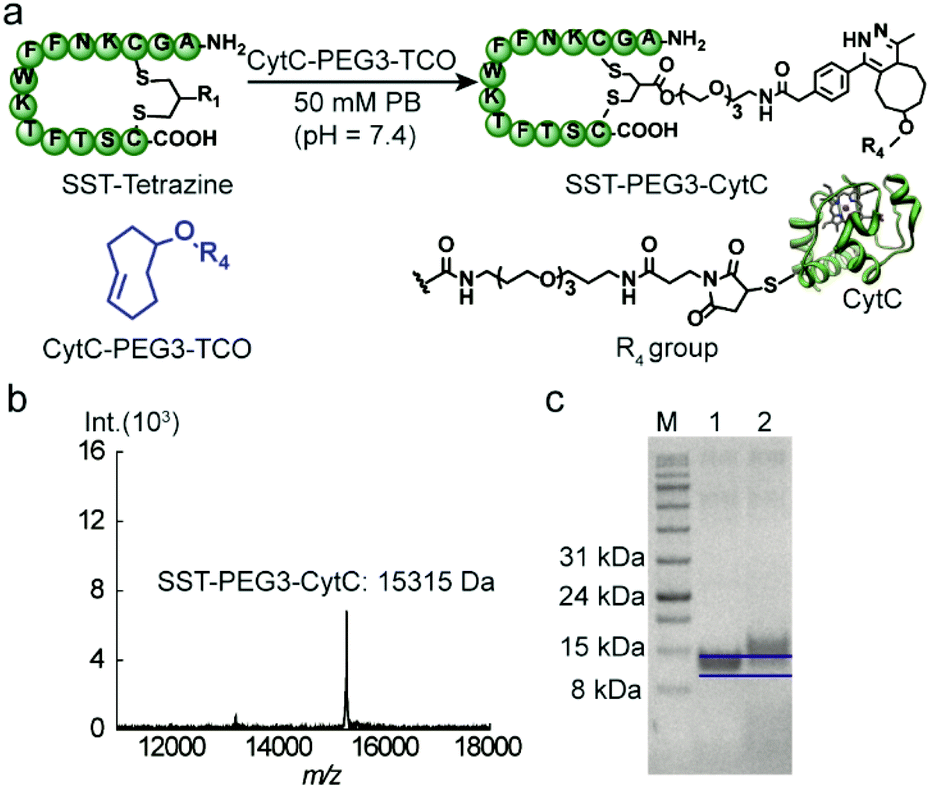 | ||
| Fig. 4 (a) Bioconjugation between SST-Tetrazine and CytC-PEG3-TCO. (b) MALDI-TOF-MS spectrum of SST-PEG3-CytC (found: 15315 [M + H]+, calculated: 15314 [M + H]+) matrix: sinapic acid. (c) SDS-PAGE analysis of SST-PEG3-CytC (M: Applichem Protein Marker VI, 1: CytC-PEG3-TCO, and 2: SST-PEG3-CytC). | ||
Stability studies of SST-Tetrazine, Fab-Tetrazine and Fab-Cy5
The stability of the bioconjugates after disulfide modification is an important consideration for their intended applications in cellular environments. Therefore, the stability of the modified protein with the newly synthesized disulfide rebridging reagent in glutathione (GSH) was investigated using liquid chromatography mass spectrometry (LC-MS). SST-Tetrazine (10 μM) was incubated with two-fold excess of GSH at biologically relevant concentrations (20 μM)26 at 37 °C and SST-Tetrazine and the degradation product (GSH-Tetrazine, 1085 [M + H]+; the chemical structure is shown in Scheme S9†) were identified by simultaneous detection by UV-Vis spectroscopy at an absorption wavelength of 254 nm and selective ion monitoring (SIM). The amount of SST-Tetrazine in each sample was determined as a ratio of the integration of the chromatogram at 254 nm of SST-Tetrazine to the standard (Fmoc-L-phenylalanine) (Fig. 5a and b). LC-MS data showed that SST-Tetrazine remained intact after 24 hours of incubation and no degradation products were detected in the SIM profile (Fig. S12C†). Next, the stability of the Fab conjugates was investigated based on the literature protocol.22b Fab-Tetrazine and Fab-Cy5 were incubated with 1% fetal bovine serum (FBS) at 37 °C and monitored using SDS-PAGE. SDS-PAGE data revealed no change in Fab-Tetrazine after 24 hours, indicating no obvious degradation (Fig. 5c). The stability of Fab-Cy5 was also investigated based on the quantification of the fluorescence of the conjugate (shown in the ESI,† page 15). The data indicated a good stability of Fab-Cy5 after incubation with FBS for 24 hours (Fig. 5d). Taken together, these experiments showed that the resultant peptide and protein bioconjugates formed by the disulfide rebridging strategy and the subsequent IEDDA reaction remained stable under physiological conditions, e.g. in the presence of GSH or in media containing serum proteins.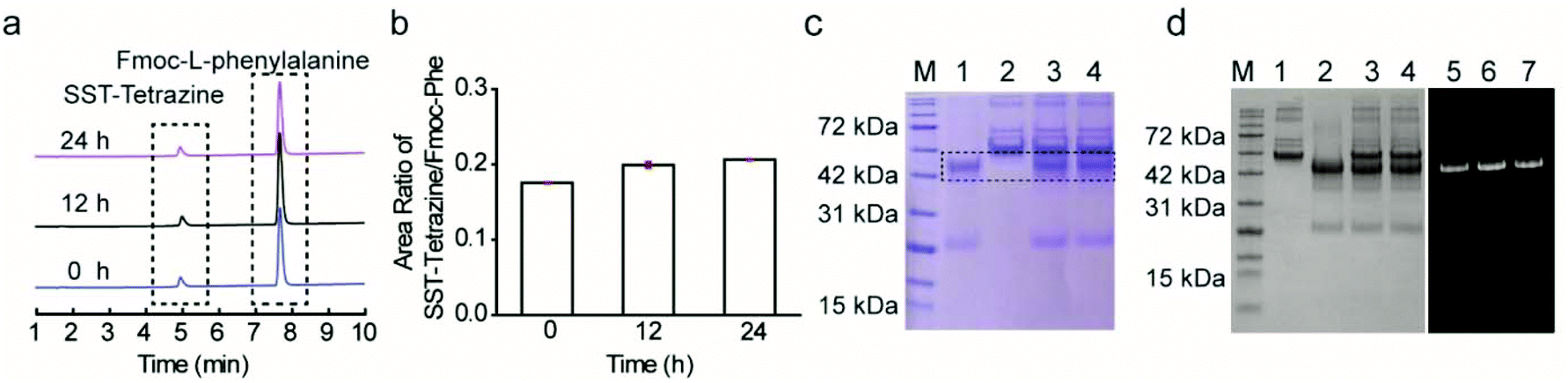 | ||
| Fig. 5 (a) Stability study of SST-Tetrazine via LC-MS. (b) Area ratio of SST-Tetrazine at 254 nm compared to Fmoc-L-phenylalanine (standard) at three time points (n = 3, values are given as mean ± SD). (c) Stability study of Fab-Tetrazine by SDS-PAGE (M: Applichem Protein Marker VI, 1: Fab-Tetrazine, 2: 1% FBS, 3: Fab-Tetrazine + 1% FBS incubated for 12 hours, and 4: Fab-Tetrazine + 1% FBS incubated for 24 hours), (d) Stability study of Fab-Cy5 by SDS-PAGE with (left) and without (right) Coomassie blue (M: Applichem Protein Marker VI, 1: 1% FBS, 2: Fab-Cy5, 3: Fab-Cy5 + 1% FBS incubated for 12 h, 4: Fab-Cy5 + 1% FBS incubated for 24 h, 5: fluorescence of Fab-Cy5, 6: fluorescence of Fab-Cy5 after incubation with 1% FBS for 12 h, and 7: fluorescence of Fab-Cy5 after incubation with 1% FBS for 24 h). | ||
Structural and functional studies
Circular dichroism (CD) is a versatile technique to determine the secondary and tertiary structures of proteins. According to Fig. 6a, SST, SST-Tetrazine and SST-PEG12 showed typical random coil structures with a negative band at 201 nm. The peak at 225 nm of SST is attributed to the presence of a higher degree of polyproline (PPII) conformation.27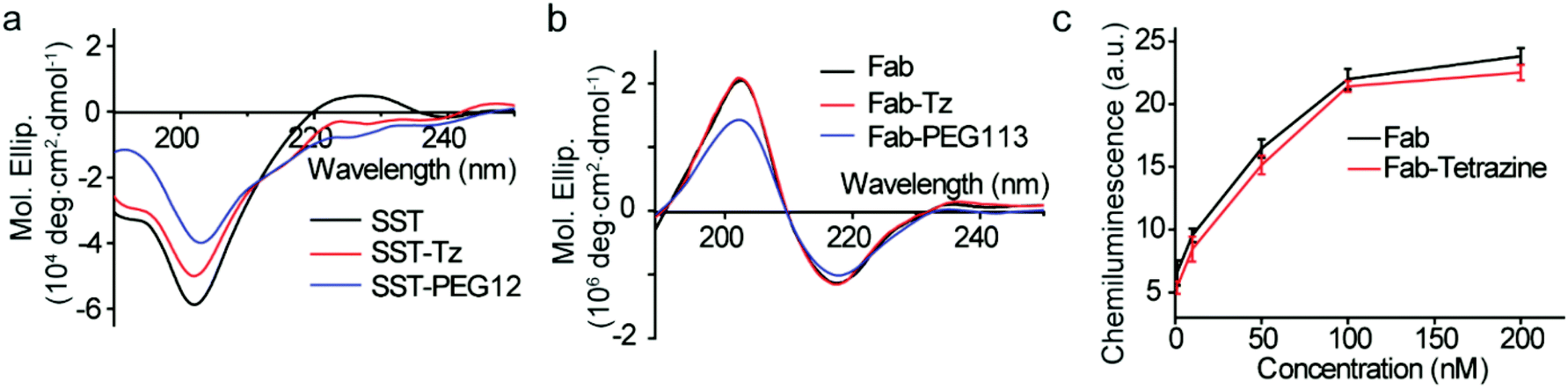 | ||
| Fig. 6 (a) CD spectra of SST, SST-Tetrazine and SST-PEG12 (SST, SST-Tetrazine and SST-PEG12 were diluted to 0.1 mg mL−1 in PBS, the data are the average of three runs). (b) CD spectra of Fab, Fab-Tetrazine and Fab-PEG113 (Fab, Fab-Tetrazine and Fab-PEG113 were measured at 0.1 mg mL−1 in PBS, the data are the average of three runs). (c) ELISA data of Fab and Fab-Tetrazine (five different concentration points: 1 nM, 10 nM, 50 nM, 100 nM, and 200 nM). Data are plotted as mean ± SD (n = 3). | ||
Both native IgG Fab, Fab-Tetrazine and Fab-PEG113 conjugates, possess well-defined antiparallel β-pleated sheets confirmed by the peaks at 218 nm and 202 nm consistent with the literature report,23 which confirmed that the secondary structure of Fab was preserved after modification (Fig. 6b). Next, we assessed whether the modification of IgG Fab affects its function to bind to protein L, a bacterial protein known to interact with Fab fragments of immunoglobulins.28 The binding and recognition of native Fab and Fab-Tetrazine were thus investigated by enzyme-linked immunosorbent assay (ELISA). Different concentrations (1 nM, 10 nM, 50 nM, 100 nM, and 200 nM) of IgG Fab and Fab-Tetrazine were added to the protein L coated well plate and incubated for one hour. After removing the unbound protein, anti-human IgG (Fab specific)-peroxidase antibody was added to the well plate and incubated for one hour. Enhanced chemiluminescence solution was added to the well plate after washing three times with PBS. As shown in Fig. 6c, the binding affinities between IgG Fab and Fab-Tetrazine are comparable, suggesting that the binding of Fab-Tetrazine to protein L was preserved.
Conclusions
Herein, we report a straightforward and broadly applicable bioconjugation method for introducing a single tetrazine group into a targeting peptide and antibody fragment at a pre-defined, distinct site in aqueous media through disulfide modification. The tetrazine functionalized peptide or Fab was further conjugated with a series of TCO-modified functionalities, such as dyes, polymers, or proteins/enzymes yielding a small library of stable bioconjugates within a short reaction time. Such bulky functionalities could only be introduced by a two-step procedure as the disulfide rebridging reaction is greatly limited by the steric demand of the reagent. Bulky substituents attached to the rebridging reagent such as polymers, proteins or drug molecules have only limited access to the reduced disulfide residues resulting in complex product mixtures with large amounts of unreacted starting materials and very low product yield.29 Therefore, the two-step approach presented herein based on the site-selective installation of the tetrazine tag first followed by the IEDDA reaction provides convenient access to compound libraries, in which a broad range of small and bulky TCO-modified functionalities could be attached to the corresponding tetrazine-modified proteins in a convenient, fast, and efficient way.Notably, the modified proteins exhibited good stability under biologically relevant conditions and their secondary structure was preserved after modification. We envision that this method would enable the preparation of the desired bioconjugates “on site”, e.g. in hospitals for the desired applications. In this way, the final conjugate would be applied to the patient immediately after preparation, preventing long term storage, quality control and loss of bioactivity of the final conjugate. The approach presented herein is a valuable addition to the chemical toolbox for site-selective protein labelling and manipulation, offering a fast, robust and straightforward method to be easily adapted in any laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the Max Planck Society, the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Projektnummer 316249678-SFB 1279 (C01), and to the financial support from the European Union's Horizon 2020 Research and Innovation Program under grant agreement no. 667192 (“Hyperdiamond”). LX is grateful to the China Scholarship Council for a scholarship. MMZ and JCFN are grateful to the Marie Curie International Training Network Protein Conjugates for research scholarships. We thank Astrid Heck, Siyuan Xiang and Zhixuan Zhou for their technical support, Nicole Kirsch-Pietz for proofreading and the MPIP mass spectrometry group for the MALDI-TOF-MS measurements. Open Access funding provided by the Max Planck Society.Notes and references
- L. M. Weiner, R. Surana and S. Wang, Nat. Rev. Immunol., 2010, 10, 317 CrossRef CAS PubMed.
- V. Chudasama, A. Maruani and S. Caddick, Nat. Chem., 2016, 8, 114 CrossRef CAS PubMed.
- J. M. Chalker, G. J. Bernardes, Y. A. Lin and B. G. Davis, Chem. – Asian J., 2009, 4, 630–640 CrossRef CAS PubMed.
- (a) N. Krall, F. P. da Cruz, O. Boutureira and G. J. Bernardes, Nat. Chem., 2016, 8, 103–113 CrossRef CAS PubMed; (b) O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed; (c) C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed; (d) G. Zhang, S. Zheng, H. Liu and P. R. Chen, Chem. Soc. Rev., 2015, 44, 3405–3417 RSC; (e) T. B. Nielsen, R. P. Thomsen, M. R. Mortensen, J. Kjems, P. F. Nielsen, T. E. Nielsen, A. L. B. Kodal, E. Cló and K. V. Gothelf, Angew. Chem., Int. Ed., 2019, 58, 9068–9072 CrossRef CAS; (f) J. B. Trads, T. Tørring and K. V. Gothelf, Acc. Chem. Res., 2017, 50, 1367–1374 CrossRef CAS; (g) C. D. Spicer, E. T. Pashuck and M. M. Stevens, Chem. Rev., 2018, 118, 7702–7743 CrossRef CAS PubMed; (h) C. D. Spicer, T. Triemer and B. G. Davis, J. Am. Chem. Soc., 2012, 134, 800–803 CrossRef CAS PubMed; (i) C. D. Spicer, C. Jumeaux, B. Gupta and M. M. Stevens, Chem. Soc. Rev., 2018, 47, 3574–3620 RSC.
- (a) A.-C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131–5142 RSC; (b) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- (a) B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC; (b) A. C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131–5142 RSC.
- (a) N. K. Devaraj, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2008, 19, 2297–2299 CrossRef CAS PubMed; (b) N. K. Devaraj and R. Weissleder, Acc. Chem. Res., 2011, 44, 816–827 CrossRef CAS PubMed; (c) N. K. Devaraj, R. Upadhyay, J. B. Haun, S. A. Hilderbrand and R. Weissleder, Angew. Chem., Int. Ed., 2009, 48, 7013–7016 CrossRef CAS PubMed.
- (a) J. B. Haun, N. K. Devaraj, S. A. Hilderbrand, H. Lee and R. Weissleder, Nat. Nanotechnol., 2010, 5, 660 CrossRef CAS; (b) M. K. Rahim, R. Kota, S. Lee and J. B. Haun, Nanotechnol. Rev., 2013, 2, 215–227 CAS.
- B. Oller-Salvia, G. Kym and J. W. Chin, Angew. Chem., Int. Ed., 2018, 57, 2831–2834 CrossRef CAS PubMed.
- (a) J. L. Seitchik, J. C. Peeler, M. T. Taylor, M. L. Blackman, T. W. Rhoads, R. B. Cooley, C. Refakis, J. M. Fox and R. A. Mehl, J. Am. Chem. Soc., 2012, 134, 2898–2901 CrossRef CAS; (b) K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 10317–10320 CrossRef CAS PubMed; (c) K. Lang, L. Davis, J. Torres-Kolbus, C. Chou, A. Deiters and J. W. Chin, Nat. Chem., 2012, 4, 298 CrossRef CAS PubMed; (d) R. J. Blizzard, D. R. Backus, W. Brown, C. G. Bazewicz, Y. Li and R. A. Mehl, J. Am. Chem. Soc., 2015, 137, 10044–10047 CrossRef CAS PubMed; (e) T. Machida, K. Lang, L. Xue, J. W. Chin and N. Winssinger, Bioconjugate Chem., 2015, 26, 802–806 CrossRef CAS PubMed; (f) Y. Kurra, K. A. Odoi, Y.-J. Lee, Y. Yang, T. Lu, S. E. Wheeler, J. Torres-Kolbus, A. Deiters and W. R. Liu, Bioconjugate Chem., 2014, 25, 1730–1738 CrossRef CAS PubMed.
- (a) M. Baalmann, M. J. Ziegler, P. Werther, J. Wilhelm and R. Wombacher, Bioconjugate Chem., 2019, 30, 1405–1414 CrossRef CAS; (b) D. S. Liu, A. Tangpeerachaikul, R. Selvaraj, M. T. Taylor, J. M. Fox and A. Y. Ting, J. Am. Chem. Soc., 2012, 134, 792–795 CrossRef CAS.
- (a) M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed; (b) C. Canovas, M. Moreau, C. Bernhard, A. Oudot, M. Guillemin, F. Denat and V. Goncalves, Angew. Chem., Int. Ed., 2018, 57, 10646–10650 CrossRef CAS.
- S. L. Kuan, T. Wang and T. Weil, Chem. – Eur. J., 2016, 22, 17112–17129 CrossRef CAS PubMed.
- (a) S. Brocchini, S. Balan, A. Godwin, J.-W. Choi, M. Zloh and S. Shaunak, Nat. Protoc., 2006, 1, 2241–2252 CrossRef CAS PubMed; (b) S. Shaunak, A. Godwin, J.-W. Choi, S. Balan, E. Pedone, D. Vijayarangam, S. Heidelberger, I. Teo, M. Zloh and S. Brocchini, Nat. Chem. Biol., 2006, 2, 312–313 CrossRef CAS PubMed.
- S. P. Brown and A. B. Smith, J. Am. Chem. Soc., 2015, 137, 4034–4037 CrossRef CAS PubMed.
- G. Badescu, P. Bryant, M. Bird, K. Henseleit, J. Swierkosz, V. Parekh, R. Tommasi, E. Pawlisz, K. Jurlewicz, M. Farys, N. Camper, X. Sheng, M. Fisher, R. Grygorash, A. Kyle, A. Abhilash, M. Frigerio, J. Edwards and A. Godwin, Bioconjugate Chem., 2014, 25, 1124–1136 CrossRef CAS PubMed.
- T. Wang, A. Riegger, M. Lamla, S. Wiese, P. Oeckl, M. Otto, Y. Wu, S. Fischer, H. Barth, S. L. Kuan and T. Weil, Chem. Sci., 2016, 7, 3234–3239 RSC.
- (a) F. F. Schumacher, M. Nobles, C. P. Ryan, M. E. B. Smith, A. Tinker, S. Caddick and J. R. Baker, Bioconjugate Chem., 2011, 22, 132–136 CrossRef CAS; (b) M. Henkel, N. Röckendorf and A. Frey, Bioconjugate Chem., 2016, 27, 2260–2265 CrossRef CAS PubMed.
- R. Rossin, S. M. van den Bosch, W. ten Hoeve, M. Carvelli, R. M. Versteegen, J. Lub and M. S. Robillard, Bioconjugate Chem., 2013, 24, 1210–1217 CrossRef CAS PubMed.
- S. L. Kuan, S. Fischer, S. Hafner, T. Wang, T. Syrovets, W. Liu, Y. Tokura, D. Y. W. Ng, A. Riegger, C. Förtsch, D. Jäger, T. F. E. Barth, T. Simmet, H. Barth and T. Weil, Adv. Sci., 2018, 1701036 CrossRef PubMed.
- T. Wang, N. Zabarska, Y. Wu, M. Lamla, S. Fischer, K. Monczak, D. Y. Ng, S. Rau and T. Weil, Chem. Commun., 2015, 51, 12552–12555 RSC.
- (a) D. A. Richards, A. Maruani and V. Chudasama, Chem. Sci., 2017, 8, 63–77 RSC; (b) A. Maruani, M. E. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 6645 CrossRef CAS.
- S. Sun, P. Akkapeddi, M. C. Marques, N. Martinez-Saez, V. M. Torres, C. Cordeiro, O. Boutureira and G. J. L. Bernardes, Org. Biomol. Chem., 2019, 17, 2005–2012 RSC.
- F. R. Salemme, Annu. Rev. Biochem., 1977, 46, 299–330 CrossRef CAS PubMed.
- K. Qu, J. L. Vaughn, A. Sienkiewicz, C. P. Scholes and J. S. Fetrow, Biochemistry, 1997, 36, 2884–2897 CrossRef CAS PubMed.
- T. Wang, D. Y. W. Ng, Y. Wu, J. Thomas, T. TamTran and T. Weil, Chem. Commun., 2014, 50, 1116–1118 RSC.
- (a) Y. Tachibana, G. L. Fletcher, N. Fujitani, S. Tsuda, K. Monde and S.-I. Nishimura, Angew. Chem., Int. Ed., 2004, 43, 856–862 CrossRef CAS; (b) S. Sun, I. Compañón, N. Martínez-Sáez, J. D. Seixas, O. Boutureira, F. Corzana and G. J. L. Bernardes, ChemBioChem, 2018, 19, 48–52 CrossRef CAS PubMed.
- A. C. A. Roque, M. Taipa and C. R. Lowe, J. Chromatogr. A, 2005, 1064, 157–167 CrossRef CAS.
- T. Wang, Y. Wu, S. L. Kuan, O. Dumele, M. Lamla, D. Y. W. Ng, M. Arzt, J. Thomas, J. O. Mueller, C. Barner-Kowollik and T. Weil, Chem. Eur. J., 2015, 21, 228–238 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ob02687h |
| This journal is © The Royal Society of Chemistry 2020 |