
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective catalytic synthesis of α-aryl-α-SCF3-β2,2-amino acids†
Andreas
Eitzinger
a,
Jean-François
Brière
 b,
Dominique
Cahard
*b and
Mario
Waser
*a
b,
Dominique
Cahard
*b and
Mario
Waser
*a
aJohannes Kepler University Linz, Institute of Organic Chemistry, Altenbergerstraße 69, 4040 Linz, Austria. E-mail: mario.waser@jku.at
bCNRS, UMR 6014 COBRA, Normandie Univ, UNIROUEN, INSA Rouen, 76000 Rouen, France. E-mail: dominique.cahard@univ-rouen.fr
First published on 26th December 2019
Abstract
We herein report a novel entry towards chiral α-SCF3-β2,2-amino acids by carrying out the ammonium salt-catalyzed α-trifluoromethylthiolation of isoxazolidin-5-ones. This approach allowed for high enantioselectivities and high yields and the obtained heterocycles proved to be versatile platforms to access other targets of potential interest.
While nature employs only twenty-two genetically encoded proteinogenic amino acids (AAs) as vital building blocks for life, a plethora of chemically synthesized unnatural AAs exists. In principle, the later offer infinite tools to expand the genetic code and to investigate protein's structure and function. Of particular importance, fluorinated amino acids (FAAs) fall within the realm of specific investigations related to structural and functional learning of peptidic compounds.1 The introduction of a single fluorine atom or a CF3 motif are common approaches to obtain FAAs,2 whereas the supply of alternative F-containing motifs remains much less-explored (Scheme 1A). The incorporation of the highly lipophilic trifluoromethylthio group CF3S into amino acids (and subsequent peptides) should be beneficial for the elucidation of structure and function, as well as for pharmaceutical developments. Indeed, as analogues of the sulfur-containing methionine and cysteine, SCF3-derivatives should help to escape the hydrophobic core where thiol residues are usually buried while ensuring protection against oxidation. Furthermore, thanks to the power of 19F NMR techniques and some recently developed 18F-radiolabeling strategies of the CF3S group, the site-specific installation of trifluoromethylthio amino acids (SCF3-AAs) to probe local events in peptides is a powerful tool to investigate structure and function in a complementary fashion.3
 | ||
| Scheme 1 (A) Overview of FAAs and SCF3-AAs; (B) targeted asymmetric α-SCF3-β2,2-AA syntheses starting from α-aryl-isoxazolidin-5-ones 1. | ||
The common strategy to access SCF3-AAs, relies on the trifluoromethylation of thiols4 or disulfides.5 As for the direct introduction of the whole CF3S motif in AAs, rare examples include regioselective trifluoromethylthiolation of unactivated C–H bonds of amino esters,6 electrophilic α-trifluoromethylthiolation of glycine Schiff bases,7a aminotrifluoromethylthiolation of α,β-unsaturated carbonyl compounds7b and nucleophilic trifluoromethylthiolation of a variety of cyclic sulfamidates based on a stereospecific SN2-type process (i.e. this strategy developed by one of us gives access to an enantiopure L-threonine derivative where the OH-group is replaced for a CF3S-group).8 However, to the best of our knowledge, no enantioselective approach for the synthesis of chiral SCF3-AAs was reported until now.9
The lack of enantioselective catalytic procedures to access novel SCF3-AAs led us to study the construction of β2,2-AAs featuring the CF3S motif at the tetrasubstituted non-racemizable stereocenter.10 β-AA have attracted considerable attention over the last years,11 and more recently the easily accessible α-substituted isoxazolidin-5-ones 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 (as masked β2-AAs) have gained significant interest as precursors for β2,2-AA derivatives.9,13–15 Especially, the use of asymmetric phase-transfer catalysis (PTC), by means of chiral quaternary ammonium salt ion pairing catalysts,16 allowed highly enantioselective α-functionalization reactions of 1 towards α,α-disubstituted isoxazolidin-5-ones 2, albeit with a limited number of electrophiles so far (compounds 1 and 2 in Scheme 1B show the specific substitution patterns we wish to investigate herein but several other derivatives have been reported recently). Importantly, this masked β2,2-AA derivatives can then undergo facile reductive ring-opening reactions to give β2,2-AA derivatives14,15 or may be directly incorporated into peptides by means of the so-called KAHA ligation technique.14d,15a,17
12 (as masked β2-AAs) have gained significant interest as precursors for β2,2-AA derivatives.9,13–15 Especially, the use of asymmetric phase-transfer catalysis (PTC), by means of chiral quaternary ammonium salt ion pairing catalysts,16 allowed highly enantioselective α-functionalization reactions of 1 towards α,α-disubstituted isoxazolidin-5-ones 2, albeit with a limited number of electrophiles so far (compounds 1 and 2 in Scheme 1B show the specific substitution patterns we wish to investigate herein but several other derivatives have been reported recently). Importantly, this masked β2,2-AA derivatives can then undergo facile reductive ring-opening reactions to give β2,2-AA derivatives14,15 or may be directly incorporated into peptides by means of the so-called KAHA ligation technique.14d,15a,17
Based on our recent progress in the utilization of compounds 1 to access masked chiral β2,2-AAs 2 under chiral phase-transfer catalysis,14 we now became interested in introducing an asymmetric protocol to access novel α-SCF3-α-aryl-substituted targets 2 by carrying out the electrophilic trifluoromethylthiolation of α-aryl-1 under chiral ammonium salt catalysis (Scheme 1B). The hereby obtained masked SCF3-β2,2-AAs 2 represent a unique entry to SCF3-β2,2-AAs, dipeptides or the corresponding SO2CF3-analogs.
We started our investigations by carrying out the α-trifluoromethylthiolation of the phenyl-substituted 1a with the established electrophilic CF3S-transfer reagents I–IV18 using Maruoka's catalysts A19 (Table 1 gives an overview of the most significant results of a broad and systematic screening). It should be noted that other chiral ammonium salt catalysts were tried as well, but in analogy to our recent observations,14 none of these quaternary ammonium salts led to useful enantioselectivities.20
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. (mol%) | [CF3S]+ (eq.) | Solv. | Base (eq.) | Yieldb [%] | e.r.c (S:R)d |
| a All reactions were run for 20 h at room temperature using 0.1 mmol 1a in the indicated solvent (0.1 M with respect to 1a) with the given reagents and catalysts unless otherwise stated. b Isolated yields. c Determined by HPLC using a chiral stationary phase. d Absolute configuration was determined by X-ray diffraction analysis of single crystals of enantioenriched (S)-2a.21 e 0.05 M with respect to 1a. f Run at −20 °C. g After recrystallization. h Added with a syringe pump over 5 h. i 1 mmol scale. | ||||||
| 1 | A2 (5) | I (3) | Et2O | Cs2CO3 (2) | 80 | 82:18 |
| 2 | A2 (5) | II (3) | Et2O | Cs2CO3 (1.1) | 70 | 75:25 |
| 3 | A2 (5) | III (3) | Et2O | Cs2CO3 (1.1) | 0 | — |
| 4 | A2 (5) | IV (3) | Et2O | Cs2CO3 (1.1) | 0 | — |
| 5 | A1 (5) | I (1.5) | Et2O | Cs2CO3 (1.1) | 77 | 90:10 |
| 6 | A1 (5) | I (1.2) | Et2O | K2CO3 (0.2) | 89 | 89:11 |
| 7 | A1 (5) | I (1.2) | iPr2O | K2CO3 (0.2) | 85 | 87:13 |
| 8 | A1 (5) | I (1.2) | THF | K2CO3 (0.2) | 66 | 76:24 |
| 9 | A1 (5) | I (1.2) | CH2Cl2 | K2CO3 (0.2) | 53 | 75:25 |
| 10 | A1 (5) | I (1.2) | PhCH3 | K2CO3 (0.2) | 56 | 84:16 |
| 11 | A1 (5) | I (1.2) | Et2O | KOAc (0.2) | 25 | 90:10 |
| 12 | A1 (5) | I (1.2) | Et2O | NaOPh (0.2) | 71 | 89:11 |
| 13 | A1 (5) | I (1.2) | Et2O | K2HPO4 (0.2) | 88 | 90:10 |
| 14 | A1 (5) | I (1.05) | Et2Oe | K2HPO4 (0.2) | 82 | 91:9 |
| 15f | A1 (5) | I (1.05) | Et2Oe | K2HPO4 (0.2) | 90 | 93:7 (98:2)g |
| 16f | A1 (5) | I (1.05)h | Et2Oe | K2HPO4 (0.2) | 90 | 95:5 |
| 17f,i | A1 (3) | I (1.05) | Et2Oe | K2HPO4 (0.2) | 90 | 93:7 |

First experiments with catalyst A2 and the succinimide-based reagent I allowed for a promising initial enantiomeric ratio of 82:18 already (entry 1). In addition, reagent I performs slightly better than the phthalimide-based II, while neither the saccharin-derived III nor the cumyl O-SCF3 reagent IV did allow for any product formation at all (entries 1–4). Using the catalyst A1, substituted by the 3,4,5-trifluorophenyl pendant at 3,3′-positions, a slight increase in enantioselectivity could be achieved, combined with lowering the excess of reagent I (entry 5). Even more interestingly, the reaction allowed the use of catalytic amounts of base, without affecting the enantioselectivity (entry 6). The evaluation of different solvents showed that ethers in general furnished higher selectivities than aromatic or chlorinated solvents (entries 6–10). As Et2O performed slightly better than other ethers, the base-optimization was performed in Et2O subsequently (entries 11–13). Noteworthy, the nature of the base did not affect the enantioselectivity significantly, contrary to the yield.
As the base can be used catalytically in these reactions, which is rather similar to our recent observations for asymmetric α-sulfanylation reactions of compounds 1,14a it can be rationalized that the in situ formed K-succinimide may also serve as a base during these reactions (thus rationalizing why different bases resulted in more or less the same enantioselectivities). Then, we carried out one reaction using a catalytic amount of K-phthalimide under the final optimized conditions (vide infra) and the observed selectivity and yield were exactly the same, thus substantiating this mechanistic proposal.
As K2HPO4 gave somewhat higher yields than the other bases (entry 13), we carried out the final optimization with this base in Et2O. It turned out that lowering the reaction temperature to −20 °C, with a slightly higher dilution and slow addition of I provided enantioselectivities up to 95:5 (entries 15 and 16). To our delight, product 2a can be further enantioenriched by recrystallization and can also easily be accessed on 1 mmol scale with 3 mol% of catalyst A1 in high yield and enantioselectivity (entry 17) (lower catalyst loadings then lead to significantly reduced conversions). The absolute configuration of product 2a was unambiguously assigned by single crystal X-ray analysis21 and the sense of enantioinduction is in line with our previous observations using Maruoka catalysts A with pronucleophiles 1.14
With high yielding and highly enantioselective catalytic conditions at hand, we next investigated the application scope of this approach for a variety of variously substituted α-aryl-derivatives 1 (Scheme 2). For ease of experimental operation, we carried out the trifluoromethylthiolation reactions under the conditions given in entry 15 (Table 1) avoiding the electrophile addition via syringe pump. As outlined in Scheme 2, a variety of novel masked α-SCF3-α-aryl-β2,2-AAs 2a–2k were readily obtained in high yields and with satisfying enantiomeric ratios, ranging from 91:9 to 95:5 e.r., regardless of the nature of the aryl moiety.
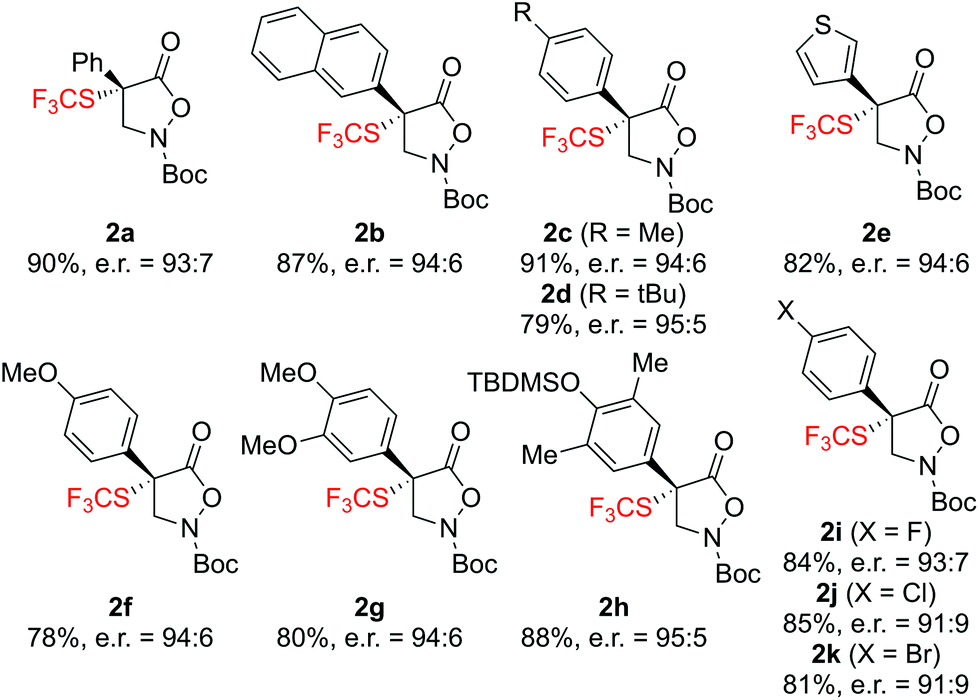 | ||
| Scheme 2 Application scope (all reactions were carried out on 0.1 mmol scale under the conditions given in Table 1, entry 15). | ||
To demonstrate the versatility of the herein accessed heterocyclic platform 2, we also carried out the transformations of compound 2a as depicted in Scheme 3. The N–O-bond could be easily cleaved under Pd-catalyzed hydrogenation conditions to afford the free carboxylic acid 4 in literally quantitative yield.12,14 In addition, the CF3S-group could be oxidized to the corresponding sulfone (giving product 5) straightforwardly with NaIO4 under RuCl3-catalysis.22 Furthermore, compound 2a can directly be engaged into amide bond forming reactions by carrying out a nucleophilic ring opening reaction with benzylic amines to give compounds 6. On the other hand, after a facile N-Boc deprotection of 2a, the KAHA ligation17 with reagent 8 occurred to give access to the corresponding dipeptide 7 under previously described aqueous-conditions.14d,15a All these performed transformations furnished high yields under robust reaction conditions, thus substantiating the synthetic versatility of our SCF3-containing heterocyclic platform 2.
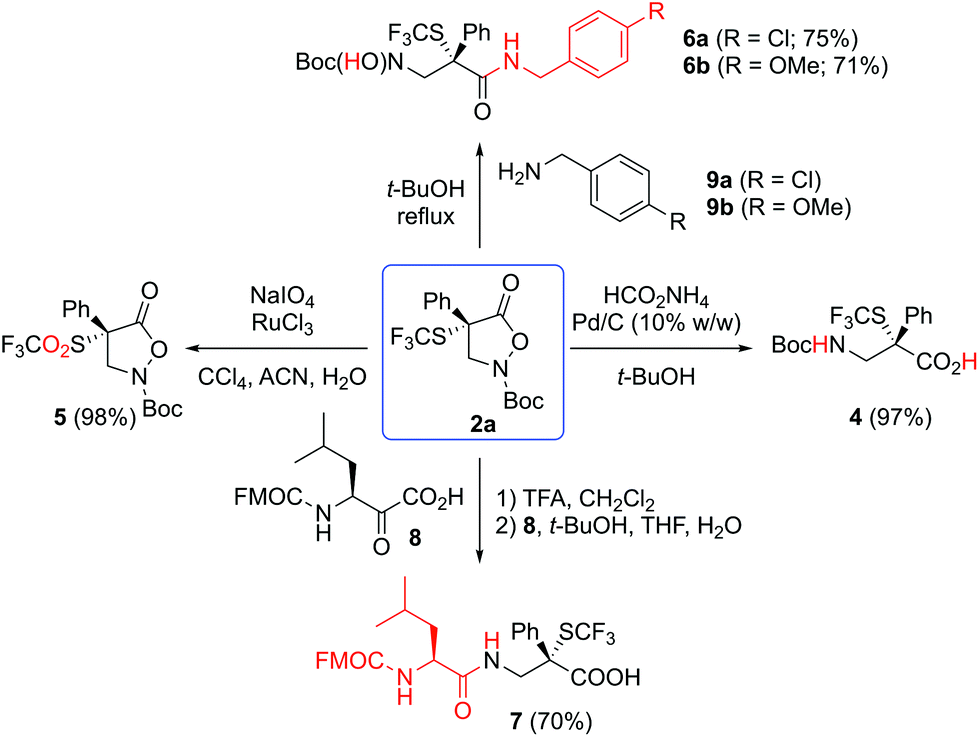 | ||
| Scheme 3 Further transformations of compound 2a. | ||
Conclusions
We have shown that α-aryl-isoxazolidin-5-ones 1 can undergo α-trifluoromethylthiolation reactions with high enantiomeric excesses and excellent yields under asymmetric phase-transfer catalysis conditions by using the commercially available Maruoka catalyst A1.9 These α-heterofunctionalization reactions perform under operationally simple conditions and could easily be carried out on 1 mmol scale as well. In addition, the hereby accessed masked α-SCF3-β2,2-AAs 2 can be utilized to obtain unprotected α-SCF3-β2,2-AAs, amides, dipeptides and the corresponding SO2CF3-analogs either.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was generously supported by the Austrian Science Funds (FWF): Project No. P30237 (financed by the Austrian National Foundation for Research, Technology and Development and Research Department of the State of Upper Austria). The used NMR spectrometers were acquired in collaboration with the University of South Bohemia (CZ) with financial support from the European Union through the EFRE INTERREG IV ETC-AT-CZ program (project M00146, “RERI-uasb”). We are grateful to Markus Himmelsbach (JKU Linz) for support with HRMS analysis and to Kirill Faust (JKU Linz) for support with X-ray analysis. This work has also been partially supported by INSA Rouen of Normandie, Rouen University of Normandie, the Centre National de la Recherche Scientifique (CNRS), EFRD, and Labex SynOrg (ANR-11-LABX-0029), and by Region Normandie (CRUNCh network).Notes and references
- (a) J. Moschner, V. Stulberg, R. Fernandes, S. Huhmann, J. Leppkes and B. Koksch, Chem. Rev., 2019, 119, 10718 CrossRef CAS PubMed; (b) A. A. Berger, J. Völler, N. Budisa and B. Koksch, Acc. Chem. Res., 2017, 50, 2093 CrossRef CAS PubMed; (c) E. N. G. Marsh and Y. Suzuki, ACS Chem. Biol., 2014, 9, 1242 CrossRef CAS PubMed.
- (a) R. Smits, C. D. Cadicamo, K. Burger and B. Koksch, Chem. Soc. Rev., 2008, 37, 1727 RSC; (b) J. L. Acena, A. E. Sorochinsky and V. A. Soloshonok, Synthesis, 2012, 44, 1591 CrossRef CAS.
- (a) S. Liu, H. Ma, Z. Zhang, L. Lin, G. Yuan, X. Tang, D. Nie, S. Jiang, G. Yang and G. Tang, Theranostics, 2019, 9, 1144 CrossRef CAS PubMed; (b) S. Verhoog, C. W. Kee, Y. Wang, T. Khotavivattana, T. C. Wilson, V. Kersemans, S. Smart, M. Tredwell, B. G. Davis and V. Gouverneur, J. Am. Chem. Soc., 2018, 140, 1572 CrossRef CAS PubMed; (c) J. Wu, Q. Zhao, T. Wilson, S. Verhoog, V. Gouverneur, L. Lu and Q. Shen, Angew. Chem., Int. Ed., 2019, 58, 2413 CrossRef CAS PubMed; (d) E. Carbonnel, T. Besset, T. Poisson, D. Labar, X. Pannecoucke and P. Jubault, Chem. Commun., 2017, 53, 5706 RSC; (e) T. Khotavivattana, S. Verhoog, M. Tredwell, L. Pfeifer, S. Calderwood, K. Wheelhouse, T. Lee Collier and V. Gouverneur, Angew. Chem., Int. Ed., 2015, 54, 9991 CrossRef CAS PubMed.
- (a) M. E. Houston and J. F. Honek, J. Chem. Soc., Chem. Commun., 1989, 761 RSC; (b) V. Soloshonok, V. Kukhar, Y. Pustovit and V. Nazaretyan, Synlett, 1992, 657 CrossRef CAS; (c) C. Bottecchia, X. J. Wei, K. P. Kuijpers, V. Hessel and T. Noel, J. Org. Chem., 2016, 81, 7301 CrossRef CAS PubMed; (d) I. Kieltsch, P. Eisenberger and A. Togni, Angew. Chem., Int. Ed., 2007, 46, 754 CrossRef CAS PubMed; (e) S. Capone, I. Kieltsch, O. Flogel, G. Lelais, A. Togni and D. Seebach, Helv. Chim. Acta, 2008, 91, 2035 CrossRef CAS.
- (a) T. Umemoto and A. Ando, Bull. Chem. Soc. Jpn., 1986, 59, 447 CrossRef CAS; (b) B. Langlois, D. Montègre and N. Roidot, J. Fluorine Chem., 1994, 68, 63 CrossRef CAS; (c) H. Yasui, T. Yamamoto, E. Tokunaga and N. Shibata, J. Fluorine Chem., 2011, 132, 186 CrossRef CAS; (d) C. Gadais, N. Saraiva-Rosa, E. Chelain, J. Pytkowicz and T. Brigaud, Eur. J. Org. Chem., 2017, 246 CrossRef CAS; (e) T. Billard, N. Roques and B. R. Langlois, J. Org. Chem., 1999, 64, 3813 CrossRef CAS.
- (a) S. Guo, X. Zhang and P. Tang, Angew. Chem., Int. Ed., 2015, 54, 4065 CrossRef CAS PubMed; (b) S. Mukherjee, B. Maji, A. Tlahuext-Aca and F. Glorius, J. Am. Chem. Soc., 2016, 138, 16200 CrossRef CAS PubMed.
- (a) S. Alazet, L. Zimmer and T. Billard, Chem. – Eur. J., 2014, 20, 8589 CrossRef CAS PubMed; (b) Q. Xiao, Q. He, J. Li and J. Wang, Org. Lett., 2015, 17, 6090 CrossRef CAS PubMed.
- J. L. Zeng, H. Chachignon, J. A. Ma and D. Cahard, Org. Lett., 2017, 19, 1974 CrossRef CAS PubMed.
- During the submission of this article Della Sala, Aleman and co-workers independently reported the chiral ammonium salt-catalyzed α-trifluoromethylthiolation of isoxazolidin-5-ones 1 essentially with α-benzyl pendants in good to moderate yields and with high enantioselectivities. They also reported two α-aryl derivatives, which however performed less selectively. Under our conditions α-benzyl-derivatives 1 did not react, but, as a complementary approach, we succeeded in the elaboration of α-aryl-containing 1 with high yields and enantiomeric ratios: V. Capaccio, M. Sicignano, R. I. Rodriguez, G. Della Sala and J. Aleman, Org. Lett., 2020, 22(1), 219–223 CrossRef CAS PubMed.
- M. A. Hardy, H. Chachignon and D. Cahard, Asian J. Org. Chem., 2019, 8, 591 CrossRef CAS.
- (a) R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219 CrossRef CAS PubMed; (b) G. Lelais and D. Seebach, Pept. Sci., 2004, 76, 206 CrossRef CAS PubMed; (c) M.-I. Aguilar, A. W. Purcell, R. Devi, R. Lew, J. Rossjohn, A. I. Smith and P. Perlmutter, Org. Biomol. Chem., 2007, 5, 2884 RSC; (d) D. Seebach and J. Gardiner, Acc. Chem. Res., 2008, 41, 1366 CrossRef CAS PubMed; (e) Y.-D. Wu, W. Han, D.-P. Wang, Y. Gao and Y.-L. Zhao, Acc. Chem. Res., 2008, 41, 1418 CrossRef CAS PubMed.
- (a) T. Tite, M. Sabbah, V. Levacher and J.-F. Brière, Chem. Commun., 2013, 49, 11569 RSC; (b) N. Lespes, E. Pair, C. Maganga, M. Bretier, V. Tognetti, L. Joubert, V. Levacher, M. Hubert-Roux, C. Afonso, C. Loutelier-Bourhis and J.-F. Brière, Chem. – Eur. J., 2018, 24, 4086 CrossRef CAS PubMed.
- For review on catalytic asymmetric synthesis of β2-amino acid derivatives, see: H. Noda and M. Shibasaki, Eur. J. Org. Chem., 2019 DOI:10.1002/ejoc.201901596.
- (a) T. Cadart, C. Berthonneau, V. Levacher, S. Perrio and J.-F. Brière, Chem. – Eur. J., 2016, 22, 15261 CrossRef CAS PubMed; (b) T. Cadart, V. Levacher, S. Perrio and J.-F. Brière, Adv. Synth. Catal., 2018, 360, 1499 CrossRef CAS; (c) V. Capaccio, K. Zielke, A. Eitzinger, A. Massa, L. Palombi, K. Faust and M. Waser, Org. Chem. Front., 2018, 5, 3336 RSC; (d) A. Eitzinger, M. Winter, J. Schörgenhumer and M. Waser, Chem. Commun., 2020 10.1039/c9cc09239k.
- (a) J.-S. Yu, H. Noda and M. Shibasaki, Angew. Chem., Int. Ed., 2018, 57, 818 CrossRef CAS PubMed; (b) M. N. de Oliveira, S. Arseniyadis and J. Cossy, Chem. – Eur. J., 2018, 24, 4810 CrossRef PubMed; (c) J.-S. Yu, H. Noda and M. Shibasaki, Chem. – Eur. J., 2018, 24, 15796 CrossRef CAS PubMed; (d) F. Amemiya, H. Noda and M. Shibasaki, Chem. Pharm. Bull., 2019, 67, 1046 CrossRef PubMed.
- (a) J.-F. Brière, S. Oudeyer, V. Dalla and V. Levacher, Chem. Soc. Rev., 2012, 41, 1696 RSC; (b) S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 4312–4348 CrossRef CAS PubMed; (c) J. Tan and N. Yasuda, Org. Process Res. Dev., 2016, 20, 129 CrossRef; (d) L. Zong and C.-H. Tan, Acc. Chem. Res., 2017, 50, 842 CrossRef CAS PubMed; (e) J. Schörgenhumer, M. Tiffner and M. Waser, Beilstein J. Org. Chem., 2017, 13, 1753 CrossRef PubMed.
- (a) J. W. Bode, R. M. Fox and K. D. Baucom, Angew. Chem., Int. Ed., 2006, 45, 1248 CrossRef CAS PubMed; (b) T. G. Wucherpfennig, V. R. Pattabiraman, F. R. P. Limberg, J. Ruiz-Rodriguez and J. W. Bode, Angew. Chem., Int. Ed., 2014, 53, 12248 CrossRef CAS PubMed.
- H. Chachignon and D. Cahard, Chin. J. Chem., 2016, 34, 445 CrossRef CAS.
- (a) T. Ooi, M. Kameda and K. Maruoka, J. Am. Chem. Soc., 2003, 125, 5139 CrossRef CAS PubMed; (b) R. He, S. Shirakawa and K. Maruoka, J. Am. Chem. Soc., 2009, 131, 16620 CrossRef CAS PubMed.
- Further details can be found in the ESI.†.
- CCDC 1914375 contains the supplementary crystallographic data for 2a.†.
- W. Su, Tetrahedron Lett., 1994, 35, 4955 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and analytical data. CCDC 1914375. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ob02666e |
| This journal is © The Royal Society of Chemistry 2020 |