
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light-driven photocontrol of the Trp-cage protein fold by a diazocine cross-linker†
Nils
Preußke
 ,
Widukind
Moormann
,
Katrin
Bamberg
,
Matthias
Lipfert
,
Rainer
Herges
* and
Frank D.
Sönnichsen
*
,
Widukind
Moormann
,
Katrin
Bamberg
,
Matthias
Lipfert
,
Rainer
Herges
* and
Frank D.
Sönnichsen
*
Otto-Diels-Institute for Organic Chemistry, Christian-Albrechts-University of Kiel, Otto-Hahn-Platz 4, 24118 Kiel, Germany. E-mail: rherges@oc.uni-kiel.de; fsoennichsen@oc.uni-kiel.de
First published on 24th March 2020
Abstract
Diazocines are characterized by extraordinary photochemical properties rendering them of particular interest for switching the conformation of biomolecules with visible light. Current developments afford synthetic access to unprecedented diazocine derivatives promising particular opportunities in photocontrol of proteins and biological systems. In this work, the well-established approach of photocontrolling the secondary structure of α-helices was exploited using a diazocine to reversibly fold and unfold the tertiary structure of a small protein. The protein of choice was the globulary folded Trp-cage, a widely used model system for the elucidation of protein folding pathways. A specifically designed, short and rigid dicarboxy-functionalized diazocine-based cross-linker was attached to two solvent-exposed side chains at the α-helix of the miniprotein through the use of a primary amine-selective active ester. This cross-linking strategy is orthogonal to the common cysteine-based chemistry. The cross-linked Trp-cage was successfully photoisomerized and exhibited a strong correlation between protein fold and diazocine isomeric state. As determined by NMR spectroscopy, the cis-isomer stabilized the fold, while the trans-isomer led to complete protein unfolding. The successful switching of the protein fold in principle demonstrates the ability to control protein function, as the activity depends on their structural integrity.
Introduction
Biological systems comprise a complex network of biochemical interactions, e.g. signal transduction cascades, of very diverse compounds including small molecules (second messengers, metabolites), peptides (hormones), and proteins (enzymes, receptors). The ability to control the biological activities of peptides or proteins with high spatio-temporal resolution is a powerful tool to elucidate the mechanisms, kinetics and sequence of these biochemical pathways.1–4 Visible light is a perfect trigger as it transfers a desired amount of energy with the required spatial and temporal resolution, does not contaminate the sample, is non-toxic and has a high penetration depth (except through blood-supported tissue). Consequently, irradiation-induced configurational changes of incorporated light-sensitive chromophores have been used to control the three-dimensional structure of peptides and proteins.1–4By far the most frequently used photoswitches are azobenzenes.1–4 Upon irradiation, they undergo reversible isomerization between two states of significantly different geometries (Fig. 1, top). The thermodynamically stable extended trans-configuration can be converted to the shorter, bent cis-isomer with high yields upon irradiation with ultraviolet (UV) light. Due to its planarity, trans-azobenzene exhibits a tendency to aggregate when incorporated into peptides.5,6 Additionally, it may influence condensation and melting of DNA when equipped with positive charges.7–9 Therefore, most peptide or protein switching applications10–19 make use of a water soluble sulfonic acid-substituted azobenzene.20 However, even this cross-linker may lead to aggregation when incorporated into a peptide10 or can unintendedly interfere with the protein fold due to interactions of the sulfonic acid moieties with guanidinium groups of arginine.15
 | ||
| Fig. 1 cis–trans-Isomerization of azobenzene (top) and diazocine (bottom). | ||
Compared to azobenzenes, diazocines show a reversed switchability (Fig. 1, bottom): the ethylene bridge stabilizes the bent and bulky cis-configuration rendering it the thermodynamically stable isomer.21–24trans-Diazocine is not planar, but adopts a twist or chair conformation. It is therefore not prone to π–π-stacking. Diazocines also display improved photochemical properties in organic solvents when compared to azobenzenes.21,22 These include a better separation of absorption bands and exceptionally high quantum yields resulting in higher switching efficiencies, i.e. better isomer ratios in the photostationary states (PSS).21,22,24 Importantly, switching in both directions can be achieved with visible light, which is essential for applications in biological systems. Recent developments in the synthesis of diazocines now render them almost as easily accessible as azobenzenes.25–27
If carefully designed and attached to a protein, the relatively subtle structural change of the molecular switch affects the fold of the much larger biomacromolecule. Photocontrolling the biological function of α-helical peptides by reversibly allowing or preventing formation of their secondary structure is already well-established and has been applied to DNA recognition,10–12,14 protein–protein interactions5,6,12–14,16–19 or RNA binding.15 These examples make use of cross-linking the respective peptide using an azobenzene moiety with both attachment points at the same side of the α-helix. In contrast to azobenzene, a diazocine was applied only once to achieve photocontrol over a simple α-helical peptide in a study by Woolley et al.28
The cis-configuration of an azobenzene or diazocine stabilizes the helical backbone conformation when the cross-linker is connected to residues i, i + 413,19 or i, i + 75,6,10,12–19 while switching to its trans-isomer prevents formation of the secondary structure. In contrast, connected to residues i, i + 11 the trans-cross-linker would stabilize the α-helical fold, which in turn adapts a disordered state upon isomerization to the contracted cis-state of the cross-linker.11,13,28
Either of these approaches is useful to switch single, bioactive helices. A question of greater interest is whether they could be used to control the tertiary structure and thus function of cooperatively folded proteins. α-Helices are central and essential elements of the fold, particularly in small globular proteins or domains. Examples are the apoptosis-regulating Bcl-family of proteins,29,30 the engrailed homeodomain transcription factor,31,32 the KIX domain of CBP,33 the ribonuclease T1,34 the villin headpiece subdomain35 or the Trp-cage.36,37 In these, a solvent-exposed α-helix is of particular importance for establishing a network of tertiary contacts. Reversibly preventing the formation of secondary structure in these domains would be expected to result in their unfolding. This will perturb the integrity of the entire protein or protein complex which can be used as a tool to control their function.
The Trp-cage miniprotein36,37 displays typical protein properties38 despite its length of only 20 residues. It comprises three elements of secondary structure, i.e. an N-terminal α-helix, a short 310-helix and a polyproline II (PPII) helix. The eponymous encapsulation of a central tryptophan residue is driven by hydrophobic interactions and additionally stabilized by hydrogen bonds and a salt bridge. The highly stable and well characterized mutant ‘TC10b’ exhibits characteristics of folding cooperativity including reversible, sigmoidal thermal unfolding curves with a Tm above 55 °C.37 Because of its protein-typical features yet small size the Trp-cage is a well-established model system for the experimental36,37,39–44 and theoretical37,40,45–48 elucidation of protein folding pathways with the aim to deduce general mechanisms applicable to larger proteins.
In this work, the Trp-cage was chosen to evaluate the hypothesis that folding of a surface-exposed α-helix reversibly influences the complete tertiary structure of a protein. Here, we describe the design, synthesis and characterization of a Trp-cage miniprotein conjugated with a diazocine-based cross-linker (Fig. 2). The cross-linker was conjugated with the protein through the use of an active ester which constitutes an adequate alternative to the common cysteine-based chemistry. We set out to reversibly fold and unfold the Trp-cage through visible-light-driven photocontrol of its α-helix. This approach explores the application of diazocines in aqueous environments and their use in achieving photocontrol of proteins.
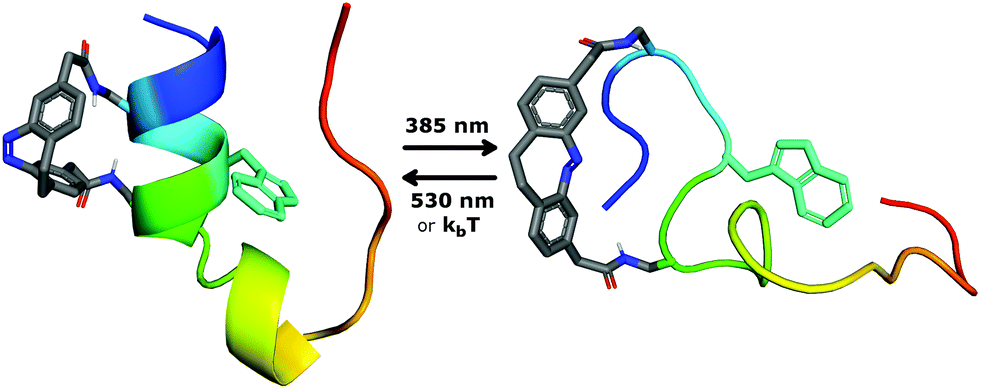 | ||
| Fig. 2 The switchable Trp-cage (switch cage) was designed to exhibit a stable fold in the dark or under irradiation with green light (530 nm) when the diazocine cross-linker is present in its cis-configuration. Upon irradiation with violet light (385 nm) the diazocine is converted to the trans-isomer which prevents formation of the helix and in turn disrupts the tertiary structure. | ||
Results and discussion
The Trp-cage
Due to its small size, most residues in the Trp-cage significantly contribute to the fold stability which limits the options for attaching the cross-linker. Moreover, the short α-helix of the Trp-cage only allows cross-linking to residues i, i + 4 or i, i + 7. Residues 4 and 8 (i, i + 4) of TC10b seemed most suitable for conjugation because they are solvent exposed and contribute relatively little to the fold stability.37 Incidentally, Markiewicz and co-workers came to the same conclusion when investigating the folding dynamics of the Trp-cage using a rigid, non-switchable cross-linker.40In our hands,49 the use of primary-amine-selective active esters in peptide conjugation reactions has proven superior to the usually employed cysteine-based cross-linking chemistry.1,3,28,40 In order to enable selective cross-linking, residues Ala4 and Lys8 of TC10b were mutated to L-2,3-diaminopropionic acid (Dpr, Γ)‡ and the N-terminus was acetylated resulting in a Trp-cage named TC(4,8). Dpr was chosen over lysine because it has a drastically reduced side chain length and flexibility, which is necessary to efficiently couple the geometric properties of the diazocine with the fold state of the Trp-cage.
TC(4,8) was synthesized via Fmoc-based solid phase peptide synthesis. It was confirmed to exhibit the Trp-cage fold as evidenced by CD and two-dimensional (2D) 1H-NMR spectroscopy. However, with a Tm of (31.6 ± 2.7) °C the fold stability of TC(4,8) was reduced compared to TC10b (∼55 °C). This destabilization can be ascribed to the repulsion between the positively charged amines of Dpr4 and Dpr8. It leads to the presence of unfolded by-forms of the Trp-cage (Fig. S5†).
The cross-linking strategy
The diazocine-based cross-linker 1 (Scheme 1) was designed to match the distance between the amino side chains of Dpr4 and Dpr8 (i, i + 4) of the folded Trp-cage. The chosen design is based on an “unfolding-upon-extension/folding-upon-contraction” switching mechanism where the short, contracted cis-diazocine supports the stable fold of the protein. Upon switching to the extended trans-state of cross-linker formation of the Trp-cage fold is prevented (Fig. 2). This concept is in contrast to the diazocine-based photoswitchable α-helix by Woolley and co-workers, which relies on an i, i + 11 approach (folding-upon-extension/unfolding-upon-contraction).28 | ||
| Scheme 1 Comparison of the primary amine-reactive cross-linker (1) presented in this work with the thiol-reactive cross-linker (3) by Woolley et al. Curved arrows indicate rotational degrees of freedom of single bonds of the cross-linker incorporated into a peptide or protein (2 and 4). The dashed arrow indicates reduced rotational freedom due to partial double bond character. | ||
When incorporated into the protein, advantages of cross-linkage 2 over the cysteine-dependent diazocine cross-linkage 4 by Woolley and co-workers28 include reduced length and flexibility (decreased number of rotable bonds) in the cis- as well as in the trans-configuration (Scheme 1). Furthermore, methylene groups in 2 serve as spacers between the diazocine moiety and the amide groups. This should preserve the excellent photochemical properties of the parent diazocine.25
In order to validate the proposed cross-linking strategy, molecular dynamics (MD) simulations of the uncross-linked TC(4,8) and of the hypothetical amidated diazocine 5 (cis-5 and trans-5) were performed (Fig. 3). The distance between the cross-linker attachment points at TC(4,8), i.e. Nγ of Dpr4 and Nγ of Dpr8 was measured during the simulation. Similarly, the cis- and trans-isomer of the amidated diazocine 5 were constructed in silico to assess the accessible N–N′-distance of the cross-linker during the simulations.
 | ||
| Fig. 3 The histograms of the switchable Trp-cage ‘cis-SCa’ (open), the uncross-linked Trp-cage ‘TC(4,8)’ (red), as well as the diazocines cis-5 (gray) and trans-5 (blue) result from an MD simulation over a period of 100 ns for each displayed molecule. Each simulation resulted in 1000 structures from which the indicated N–N′-distances were measured and plotted with a bin-size of 0.1 Å. | ||
The side chain amines (Nγ) of TC(4,8) are always less than 9.8 Å apart. With an accessible distance range of 9.8 Å to 15.2 Å trans-5 does not match this distance. On the other hand, cis-5 displays a large variance in its N–N′-distance covering a range between 3.3 Å and 13.1 Å that largely overlaps with the attachment point distances in TC(4,8). Finally, a model of the peptide-cross-linker-conjugate (cis-SCa, Fig. 3) was constructed and its MD simulation resulted in a narrow distribution of NγDpr4–NγDpr8-distances between 5.6 Å and 7.0 Å. Thus, MD simulations indicated that the trans-cross-linker would prevent a stable fold when conjugated with the Trp-cage while the cis-diazocine supports a stable fold. Importantly, both observations confirm the concept.
The linear five step synthesis of the cross-linker 1 (Scheme 2) was based on a previously published oxidative C–C coupling and reductive azo cyclization.25,26 Initially, the carboxylic acid functional group of 3-nitro-4-methyl-phenylacetic acid (6) was protected with an 4-methyl-2,6,7-trioxa-bicyclo[2.2.2]octane-1-yl (OBO) protecting group. This protection according to Corey and Raju50 started with an esterification with 3-methyl-3-oxetanemethanole (7) followed by a BF3·Et2O-catalysed rearrangement yielding the desired orthoester 9. Potassium butoxide was used as a non-nucleophilic base to selectively deprotonate the toluene α protons of 9. By addition of bromine as an oxidizing agent, the anions were converted into radicals and C–C-bond formation led to the desired dinitro dimer 10. In the ensuing reductive azo cyclization, 10 was reduced with Ba(OH)2/Zn to a hydrazine intermediate and subsequently oxidized with CuCl2 and air. During work-up, the orthoester was hydrolysed to the open ester 11, which was cleaved with Cs2CO3. Without purification, the resulting free carboxylic acid was converted with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS) into the active ester 1.
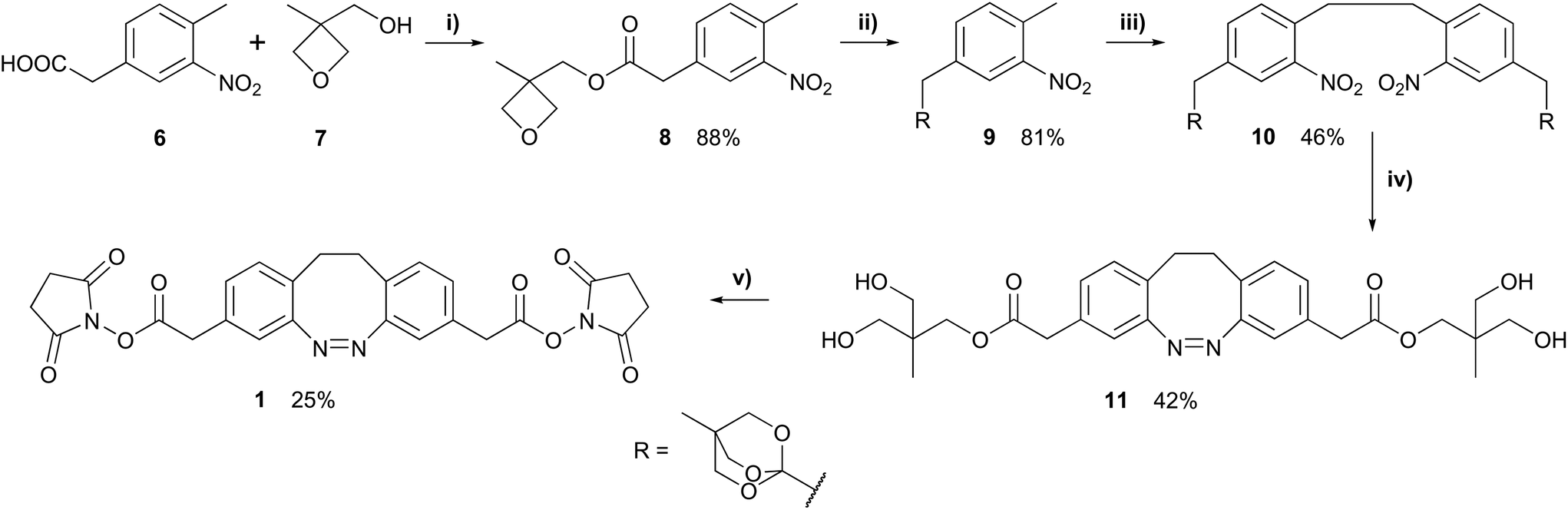 | ||
| Scheme 2 Reaction conditions: (i) N,N′-Dicyclohexylcarbodiimide (DCC), 4-dimethylaminopyridine (DMAP), dichloromethane (DCM); (ii) BF3·Et2O, triethylamine (TEA), DCM; (iii) tBuOK, Br2, tetrahydrofuran (THF); (iv) 1. Zn, Ba(OH)2 H2O/EtOH; 2. CuCl2/O2, NaOH/MeOH; (v) 1. Cs2CO3, MeOH, H2O; 2. 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), N-hydroxysuccinimide (NHS), N,N′-dimethylformamide (DMF). | ||
The PSS of diazocine 11 was determined at 385 nm in MeOH or MeCN at 298 K (Fig. S23†). With 78% and 82% trans-isomer, respectively, 11 largely retains the outstanding properties of the parent diazocine (87% trans-isomer25 at 385 nm in MeCN). In addition to its use in bio- and organic chemistry the dicarboxy-diazocine can therefore be applied as a monomer of polyamides or polyesters in photo- and mechanoresponsive materials.
The folded cis-switch cage
Conjugation of TC(4,8) with cross-linker 1 was carried out in high dilution in dimethylsulfoxide (DMSO) by means of a double syringe pump and resulted in the photo-switchable Trp-cage that we designate ‘switch cage’ (SC; Fig. 2).Dark-adapted cis-SC shows all characteristics of a stable Trp-cage fold as evidenced by NMR chemical shift deviations (CSDs; Table S4†), number and intensity of nuclear Overhauser enhancement (NOE) cross-peaks and CD spectroscopy (Fig. 4). As expected, conjugation with the cis-diazocine markedly increases the stability of the fold reaching a Tm of (48.5 ± 1.4) °C compared to TC(4,8) ((31.6 ± 2.7) °C; Fig. 4a).
 | ||
| Fig. 4 (a) The mean residue molar ellipticity [θ]MR at 222 nm of TC(4,8) (red triangles) and cis-SC (black circles) is plotted over the temperature. A sigmoidal Boltzman function was fitted to each plot to obtain the midpoint of thermal unfolding (melting temperature, Tm). Details regarding the fitted function can be obtained from the ESI.† (b) The CD spectrum of cis-SC (black circles) displays two minima at 206 nm and 222 nm indicative of an α-helix. TC(4,8) (red triangles) appears to have a lower helical propensity as the minimum at 222 nm is less pronounced. Compared to the spectrum of cis-SC, the spectrum of the switch cage after having reached the PSS at 385 nm (blue squares) indicates significantly reduced helical propensity. | ||
The Trp6 indole NH proton (Hε1) is a particularly valuable indicator for the existence of the hydrophobic cluster, as its resonance frequency is not overlapping with other signals. Most importantly, it lies in the centre of the fold and its chemical shift deviation from the random coil chemical shift (∼10.2 ppm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 51,52) correlates with the stability of the Trp-cage. Interestingly, the indole region of the 1H-NMR spectrum indicates two folded species of cis-SC (cis-SCa: 9.728 ppm, cis-SCb: 9.757 ppm; Fig. 5, top). In fact, there is a second set of resonances for the spin systems of many, but not all residues and the diazocine moiety. NMR chemical shifts and NOE cross-peaks confirm that the major (cis-SCa) and the minor (cis-SCb) form both comprise the correctly folded protein and differ only in the orientation of the cross-linker (Fig. 6). The two diastereomers are the result of conjugating the prochiral diazocine moiety with the chiral protein. The cis-cross-linker is incorporated so that either its ethylene bridge (cis-SCa, Fig. 6, left) or its azo-group (cis-SCb, Fig. 6, right) is oriented towards residue Leu7. NOE spectroscopy (NOESY) indicates that the ethylene bridge side of the diazocine is in close vicinity to Hβ2/β3 and Hδ1/δ2 of Leu7 in cis-SCa. Similarly, the orientation of the cross-linker in cis-SCb was also derived from the NOESY data. Notably, cis-SCa is the main form with an approximately 3-fold higher population than its diastereomer cis-SCb.
51,52) correlates with the stability of the Trp-cage. Interestingly, the indole region of the 1H-NMR spectrum indicates two folded species of cis-SC (cis-SCa: 9.728 ppm, cis-SCb: 9.757 ppm; Fig. 5, top). In fact, there is a second set of resonances for the spin systems of many, but not all residues and the diazocine moiety. NMR chemical shifts and NOE cross-peaks confirm that the major (cis-SCa) and the minor (cis-SCb) form both comprise the correctly folded protein and differ only in the orientation of the cross-linker (Fig. 6). The two diastereomers are the result of conjugating the prochiral diazocine moiety with the chiral protein. The cis-cross-linker is incorporated so that either its ethylene bridge (cis-SCa, Fig. 6, left) or its azo-group (cis-SCb, Fig. 6, right) is oriented towards residue Leu7. NOE spectroscopy (NOESY) indicates that the ethylene bridge side of the diazocine is in close vicinity to Hβ2/β3 and Hδ1/δ2 of Leu7 in cis-SCa. Similarly, the orientation of the cross-linker in cis-SCb was also derived from the NOESY data. Notably, cis-SCa is the main form with an approximately 3-fold higher population than its diastereomer cis-SCb.
 | ||
| Fig. 5 The 1D 1H-NMR spectra of cis-SC in its dark adapted state (top) and after having reached the PSS at 385 nm (bottom) are shown. In the expansion of the spectrum at the top the two folded species cis-SCa and cis-SCb are indicated by the two singlets of the Trp6 Hε1 at 9.728 ppm and 9.757 ppm. The expansion of the important indole region between 10.3 ppm and 9.5 ppm clearly shows the rise of an unfolded species indicated by the intense singlet at 10.089 nm (bottom). | ||
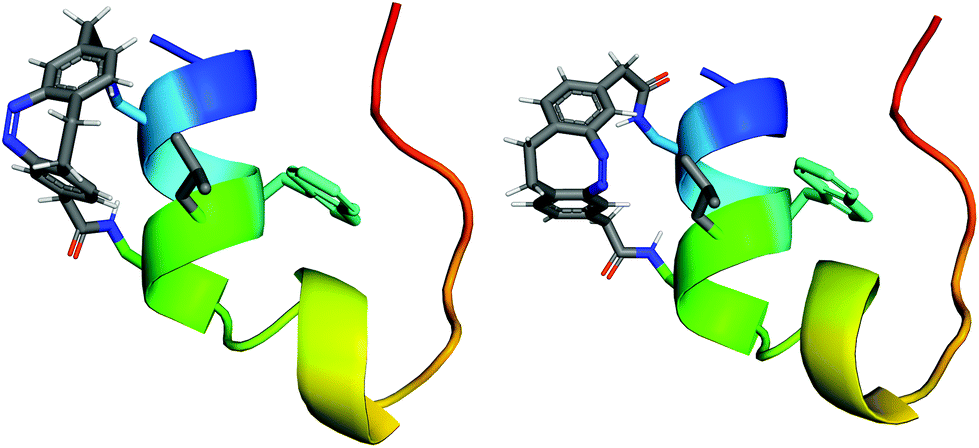 | ||
| Fig. 6 The two different possible orientations of the cross-linker produce the two diasteromers cis-SCa (left) and cis-SCb (right). Structural models of the Trp-cage are shown in cartoon mode with the Trp6 side chain in the center of the molecule. The side chain of Leu7 is shown in grey in stick representation omitting hydrogens for clarity. | ||
Switching from cis-SC to trans-SC
Irradiation with light at the wavelength of 385 nm produces a photo stationary state (PSS) with approximately 46% trans-SC and 54% cis-SC (Table S6†). At first glance, this ratio appears to be low for a diazocine, but few conversion ratios of diazocines in water have been published. For instance, Woolley and colleagues28 provided information on the PSS of their diazocine only in DMSO. In fact, the PSS of the switch cage compares well with the few published PSSs of diazocine derivatives in aqueous solution that range between 16% and 60% of trans-isomer.53–56 Although the conditions differ in pH, buffer and DMSO content, it appears that the PSSs of ethylene bridged diazocines§ in aqueous environment are generally lower than the PSS of the parent diazocine21,22 in organic solvents (>90% trans-species in n-hexane). In addition to the solvent effect, in cis-SC the folding free energy of the Trp-cage has to be overcome in order to unfold the miniprotein upon cis → trans-isomerization which also may lower the PSS.After irradiation with light at 385 nm, new resonances are observed in the NMR spectra (Fig. 5) belonging to the trans-species of the switch cage (trans-SC). It was not possible to assign many atoms of trans-SC, as their resonances overlap with those of cis-SCa and cis-SCb. Moreover, the unfolded nature of trans-SC prohibits the assignment due to overlap of intramolecular proton signals and resulting equivocal inter-residual NOEs. Despite these difficulties it was possible to determine the folding state of trans-SC.
A first evidence, that trans-SC is unfolded and assumes a random coil conformation is provided by the chemical shift of its Trp6 indole NH proton (Hε1) at 10.089 ppm (Fig. 5, bottom). This is very close to the random coil shift (∼10.2 ppm51,52) indicating the absence of the hydrophobic cluster of the folded state. Moreover, at 298 K the Trp6 Hε1 of cis-SCa shows coupling through space to Hβ2, Hβ3, Hγ and the backbone HN of Arg16 as well as Hα and Hδ of Pro18 and Hδ2/δ3 of Pro19 (Fig. 7). In contrast, the Trp6 Hε1 of trans-SC displays only one extremely weak NOE cross-peak (3.498 ppm). Even at 278 K, a temperature that favours folding and which enables long measurements due to slow trans → cis isomerization, no further NOE cross-peaks are observable in the trans-SC NOE spectrum.
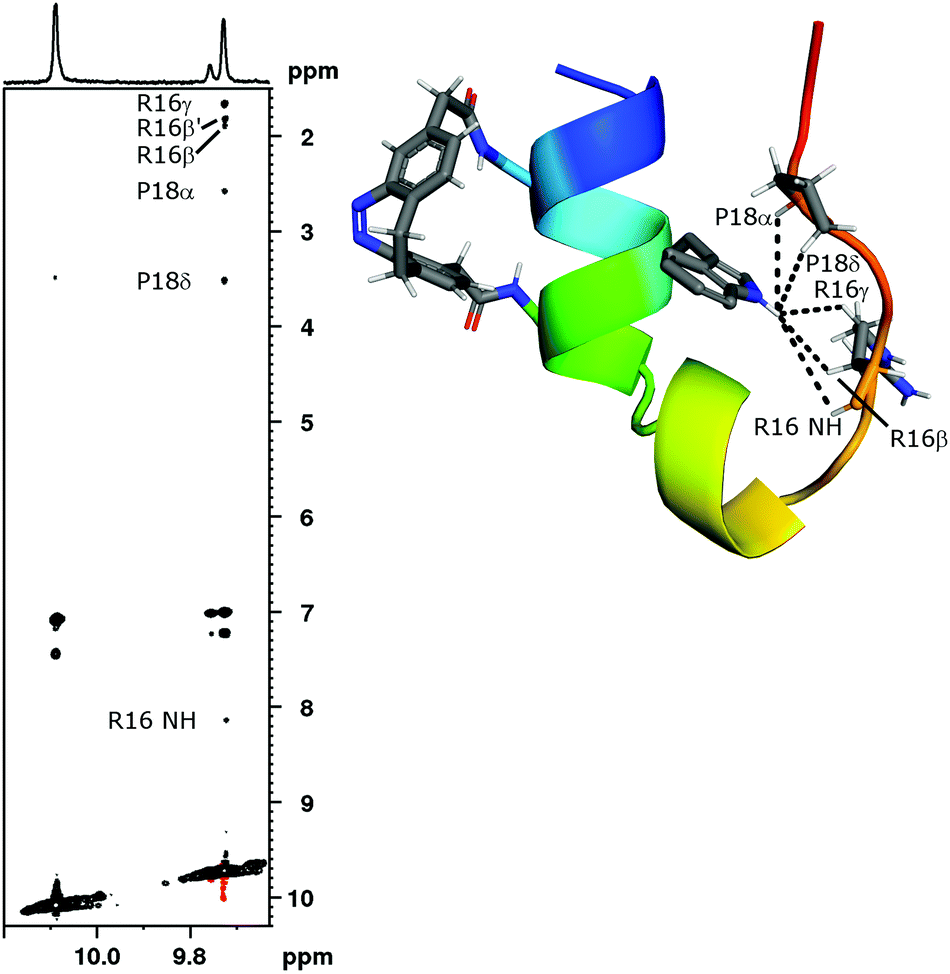 | ||
| Fig. 7 The 1H,1H-NOESY of the switch cage after irradiation to the PSS at 385 nm was recorded at 298 K. The expansion shows the indole region of cis-SCa (F2: 9.728 ppm), cis-SCb (F2: 9.757 ppm) and trans-SC (F2: 10.089 ppm). The six inter-residual cross-peaks in the vertical direction of cis-SCa evidence a stable tertiary structure. The NOE signals highlighted in the expansion are depicted in the model structure of cis-SCa. In contrast to cis-SCa, only one unassigned cross-peak is observed for the indole proton of trans-SC (F2: 10.098 ppm, F1: 3.485 ppm). The NOEs between 7.0 ppm and 7.5 ppm are intra-residual cross-peaks with the neighbouring aromatic protons. For cis-SCb inter-residual NOEs are not visible in this depiction due to low population. | ||
In addition to NMR spectroscopy, CD spectroscopy also provides evidence that trans-SC is completely unfolded. The mean residue molar ellipticity [θMR] at 222 nm in the dark-adapted state amounts to −12017° M−1 m−1, which is reduced to −6725° M−1 m−1 in the PSS at 385 nm (Fig. 4b). This corresponds to a reduction by 44% which is in the same range as the cis → trans-conversion ratio in the PSS indicating complete unfolding of the α-helix and thus absence of secondary and tertiary structure in trans-SC.
The lack of α-helicity as evidenced by CD-spectroscopy, the Trp6 Hε1 chemical shift and the disappearance of inter-residual NOE cross-peaks upon switching to the trans-state unambiguously demonstrate that the switch cage is entirely unfolded in its trans-state. Conversely, the switch cage is nearly quantitatively folded in the presence of the cis-linker. Thus, the folding state of the protein is completely coupled to the isomeric state of the cross-linker.
Switching from trans-SC to cis-SC
Irradiation with light at the wavelength of 530 nm provides almost complete conversion to cis-SC (Fig. S16†). Moreover, the NMR-spectrum of cis-SC recorded immediately after irradiation to its PSS at 530 nm is indistinguishable from that of dark-adapted cis-SC.The thermal trans → cis relaxation process of the switch cage was observed by 1H-NMR spectroscopy and quantified by integration of selected peaks of the protein or the diazocine moiety (Table S5 and Fig. S14, S15†) after irradiation to the PSS at 385 nm. At 298 K in the dark, this process requires 39 h after which trans-SC is not quantifiable anymore (Fig. S16†). It follows first order kinetics with a half-live of (6.2 ± 0.3) h (Table S6†). Within the error of measurement, differences in the determined relaxation rates for diazocine signals and protein signals (Trp6 Hε1) are insignificant. Further, the relaxation of trans-SC to cis-SCa and to cis-SCb is equally fast. Together, this is an additional demonstration of the strict coupling between the isomeric state of the diazocine and the folding state of the Trp-cage.
Throughout the entire study, the same sample was used for all spectroscopic measurements. In consequence, it was subjected to multiple switching cycles without showing any sign of photobleaching or other material fatigue. Furthermore, aggregation was never observed, not even in the PSS at 385 nm, when approximately half of the population of the switch cage was unfolded exposing the hydrophobic core and the trans-diazocine to the solvent. This emphasizes the reversibility of the switching process and the photochemical stability of the switch cage.
The relationship between cis-SCa and cis-SCb
Regardless of the wavelength or intensity of incident light (day-light-adapted, dark-adapted, PSS at 385 nm, PSS at 530 nm), the two diastereomers cis-SCa and cis-SCb have always been observed in a ratio of 3:1. While often indicative of a thermodynamic reaction control, the apparently constant ratio of cis-SCa to cis-SCb in photoreactions appears surprising at first. The parent cis-diazocine 1 is prochiral and converts upon radiation at 385 nm equally fast into two enantiomeric trans-forms, that are believed to comprise an equilibrium of twist- and chair-conformations.57 These forms convert back to cis-diazocine equally well.
In the chiral context of the protein, however, irradiation results in multiple possible cis–trans-isomerizations (Scheme S1†) between diastereomeric states. Unhindered interconversion would lead to the expectation of a 1:1 product ratio of cis-SCa and cis-SCb. The observed uneven cis-SCa/cis-SCb ratio thus suggest that the conversion rates between the states differ between diastereomers implying a form of chiral induction by the protein moiety.
Although it is possible that the conversion from either trans-diastereomer occurs faster to the cis-SCa diastereomer, a chiral induction in the trans-state appears unlikely since the protein moiety is unfolded and should have very limited influence on the conversion kinetics. In the cis-states, however, the environment of the chromophore differs significantly for example with closer protein-diazocine contacts in cis-SCa. These structural differences might slow the photoconversion of cis-SCa sterically or energetically whereas cis-SCb more rapidly isomerizes to either trans-isomer. Accordingly, this chiral induction could lead to the observed bias in the photostationary states.
Conclusions
The present study aimed to reversibly fold and unfold the Trp-cage upon irradiation with visible light of different wavelengths. Since the elongation of the diazocine during cis → trans-isomerization induces unfolding, the switching approach presented in this work is described as “unfolding-upon-extension/folding-upon-contraction”. This goal was achieved through the conjugation of an N-terminally acetylated Trp-cage mutant ‘TC(4,8)’ bearing L-2,3-diaminopropionic acid (Dpr, Γ)‡ in positions 4 and 8. A diazocine-based, primary amine-selective cross-linker (1) was designed to match the distance between the Nγ of both Dpr-residues in its cis-state. Upon cross-linking, the fold of the resulting photoswitchable Tpr-cage (switch cage, SC) is stabilized significantly compared to the uncross-linked peptide. Irradiation with light of the wavelength 385 nm produces a PSS of approximately 46% trans-SC. This isomerization is completely reversed upon irradiation with light at 530 nm or through thermal relaxation. Most importantly, in the trans-state, secondary and tertiary structuring is completely abolished. As predicted, pulling apart only one helical turn between residues 4 and 8 destroys the entire helical structure and in consequence unfolds the hydrophobic cluster.With these features, the switch cage may serve as a valuable tool for the analysis of the Trp-cage folding pathway adding visible light as an alternative trigger to the previously used T-jump39,41 in IR or fluorescence spectroscopy. The reversibility of the switching process enables multiple switching cycles and thus facilitates multiple and repeated observations of folding and unfolding kinetics.
In this study, a dicarboxy-functionalized diazocine was furbished with selective reactivity towards primary amines by introduction of NHS-active ester moieties. This cross-linking strategy adds a valuable alternative to current methods to conjugate the chromophore with the protein. The NHS-ester cross-linking chemistry is orthogonal to the common cysteine-based chemistry.58 When incorporated into the protein (cf.Scheme 1), the presented diazocine cross-linker is shorter and more rigid than the chloroacetamide-functionalized diazocine.28
The active ester cross-linking strategy is especially interesting for synthetic peptides and proteins, as next to lysine several amino group-bearing homologues exist that can be used to tailor length and flexibility of the cyclized product. Particularly short and rigid cross-linkages are obtained when the shortest lysine-homologue Dpr is utilized. The cross-linker 1 may also be incorporated via naturally occurring lysine residues or a free N-terminus, but this would limit its application to “folding-upon-extension/unfolding-upon-contraction”-approaches due to the high flexibility of the lysine side chains.
In conclusion, we were able to reversibly fold and unfold the secondary and tertiary structure of the Trp-cage miniprotein upon irradiation with visible light. Most importantly the present study shows that it is generally possible to reversibly fold and unfold a single, solvent-exposed α-helix in a small, but stable protein resulting in photocontrol of the entire fold. This strengthens our beliefs that this approach is applicable to larger proteins or protein domains.
Experimental
Trp-cage synthesis
TC(4,8) (Ac-DAYΓQWLΓDGGPSSGRPPPS)‡ was synthesized using Fmoc-based solid phase peptide synthesis with a microwave-assisted, semi-automated peptide synthesizer Initiator+ SP Wave (Biotage, Uppsala, Sweden) on Wang-resin (200–400 mesh, 0.86 mmol g−1; 200 mg, 1 eq.; ChemPep, Wellington, FL, USA) at a 0.172 mmol scale. To avoid aspartimide formation during synthesis the dipeptide Fmoc-Asp(OtBu)-(DMB)Gly-OH (Novabiochem, Merck, Darmstadt, Germany) was used. Moreover, the two consecutive serines were coupled as the pseudo-proline dipeptide Fmoc-Ser(tBu)-Ser(psiMe,Mepro)-OH (Novabiochem, Merck, Darmstadt, Germany). In general, the peptide was synthesised using repetitive cycles of coupling, capping (i.e. acetylating unreacted peptide chains) and deprotection. Each coupling, capping or deprotection step was followed by draining the liquid and washing the resin with N,N-dimethylformamide (DMF; 2 ml) four times. Coupling of amino acids was achieved using the corresponding amino acid (860 μmol, 5 eq.), hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU; 320 mg, 843 μmol 4.9 eq.) and N,N-diisopropylethylamine (DIPEA; 300 μl, 1.72 mmol, 10 eq.) dissolved in DMF (2 ml). The reaction solution was heated to 75 °C and vortexed at 600 rpm for 5 min. The coupling step was performed twice (double coupling) for the following amino acids: Fmoc-Ser(tBu)-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Gly-OH, Fmoc-Dpr(Boc)-OH, Fmoc-Gln(Trt)-OH and Fmoc-Ala-OH. (Double) coupling was followed by a capping step using a solution (2 ml) of pyridine (25% v/v), and acetic anhydride (25% v/v) in DMF at 75 °C at 600 rpm for 5 min. Removal of the Fmoc-group was achieved in three consecutive steps (1 min, 4 min, 4 min). Each step used a solution (2 ml) of piperidine (25% v/v) in DMF at 75 °C at 600 rpm which was drained afterwards. After the final deprotection, the peptide was N-terminally acetylated using the capping reagents and conditions. The peptide was cleaved off the resin using a mixture of trifluoroacetic acid (TFA; 90% v/v), m-dimethoxybenzene (DMB; 5% v/v), triisopropylsilane (TIPS; 2.5% v/v) and water (2.5% v/v). The cleavage reaction was completed after gently agitating it for 3 h at room temperature. The peptide was precipitated using cold diethyl ether and centrifuged. The supernatant was decanted. The pellet was dissolved in water and lyophilized.Trp-cage purification
The peptide was purified in four batches by high performance liquid chromatography (HPLC) on a VWR-Hitachi LaChrom Elite instrument (VWR, Darmstadt, Germany) equipped with a semi-preparative PLRP-S column (325 mm × 30 mm, 30 nm pore diameter, 8 μm particle size; Agilent, Santa Clara, CA, USA), a VWR-Hitachi L2400 UV detector (VWR, Darmstadt, Germany) and a Foxy R1 fraction collector (Teledyne ISCO, Lincoln, NE, USA). A flow rate of 6 mL min−1 and the following gradient (solvent A: water, 0.1% formic acid (FA), solvent B: acetonitrile, 0.1% FA) was applied: 0 min: 5% B, 5 min: 5% B, 25 min: 15% B, 55 min: 25% B, 65 min: 95% B, 75 min: 95% B, 80 min: 5% B, 90 min: 5% B. Fractions of 3 ml were collected between 45 min and 75 min. TC(4,8) eluted after retention time of tR = 55.5 min. A dead time of 20 min was observed.Collected fractions were analysed by HPLC-mass spectrometry (HPLC-MS) using the same VWR-Hitachi LaChrom Elite instrument (VWR, Darmstadt, Germany) equipped with a 717 plus autosampler (Waters, Milford, MA, USA), an analytical PLRP-S column (150 mm × 4.6 mm, 30 nm pore diameter, 8 μm particle size; Agilent, Santa Clara, CA, USA), a VWR-Hitachi L2400 UV detector (VWR, Darmstadt, Germany) and an expressionL cms MS device (Advion, Harlow, UK). A flow rate of 1 mL min−1 and the following gradient (solvent A: water, 0.1% FA, solvent B: acetonitrile, 0.1% FA) was applied: 0.0 min: 5% B, 2.5 min: 5% B, 12.5 min: 60% B, 13.5 min: 95% B, 16.0 min: 95% B, 17.0 min: 5% B, 18.5 min: 5% B. A dead time of 2.0 min was observed. TC(4,8) eluted after retention time of tR = 9.8 min. Fractions deemed sufficiently pure were combined and lyophilized. The peptide was obtained as a white fluffy solid (17.8 mg, 8.65 μmol, 5%).
Cross-linker synthesis and purification
Details regarding the cross-linker synthesis, purification and characterization can be found in the ESI.†Cross-linking reaction and purification
DIPEA (33 μL, 190 μmol, 100 eq.) and DMSO (1.5 ml) were added to a 50 ml polypropylene (PP) centrifuge tube equipped with a magnetic stir bar. A solution of TC(4,8) (4.0 mg, 1.9 μmol, 1 eq.) in DMSO (1.5 mL) supplemented with DIPEA (33 μL, 190 μmol, 100 eq.) was transferred into a 2 mL syringe. A second 2 ml syringe was filled with a solution of the NHS-functionalized cross-linker 1 (1 mg, 2.1 μmol, 1.1 eq.) in DMSO (1.81 ml). The two reactants were slowly added to the centrifuge tube, each at a speed of 2 μl min−1 under continuous stirring at room temperature. After complete addition, the reaction was stirred for 2 d. Subsequently, the solution was flash-frozen with liquid nitrogen and lyophilized. The crude product was dissolved in 2.5 ml of water by briefly sonicating it in the centrifuge tube using an ultrasonic bath. The switch cage was purified in ten small batches via HPLC using the VWR-Hitachi LaChrom Elite instrument (VWR, Darmstadt, Germany) equipped with a 717 plus autosampler (Waters, Milford, MA, USA), an analytical PLRP-S column (150 mm × 4.6 mm, 30 nm pore diameter, 8 μm particle size; Agilent, Santa Clara, CA, USA), a VWR-Hitachi L2400 UV detector (VWR, Darmstadt, Germany) and a Foxy R1 fraction collector (Teledyne ISCO, Lincoln, NE, USA). A flow rate of 1 mL min−1 and the following gradient (solvent A: water, 0.1% FA, solvent B: acetonitrile, 0.1% FA) was applied: 0.0 min: 5% B, 2.5 min: 5% B, 10 min: 27% B, 20 min: 29% B, 25 min: 95%B, 29 min: 95% B, 31 min: 5% B, 35 min: 5% B. The switch cage eluted after a retention time of tR = 18.2 min. A dead time of 2.0 min was observed. Fractions of the ten different batches with identical retention times were collected into the same containers. Fractions were analysed using the same HPLC-MS set-up as described in the chapter Trp-cage purification. The switch cage eluted after a retention time of tR = 12.0 min. Fractions deemed sufficiently pure were combined and lyophilized which afforded the switch cage as a yellowish solid (0.8 mg, 330 nmol, 17%).Ultrahigh performance liquid chromatography – high resolution mass spectrometry
TC(4,8) and the switch cage analysed by ultrahigh performance liquid chromatography – high resolution mass spectrometry (UHPLC-HRMS) using an Ultimate 3000 RS instrument coupled to a QExactive Plus Hybrid Quadrupol-Orbitrap MS device (both Thermo Fisher Scientific, Waltham, MA, USA). A Zorbax RRHD Eclipse Plus C18 column with a length of 100 mm and a diameter of 3 mm (Agilent, Santa Clara, CA, USA) was used. The MS was set to detect positive ions and the resolution was set to FWHM = 70000 at m/z = 200. The Full MS method was applied covering an m/z range between 200 and 3000. A flow rate of 0.4 mL min−1 and the following gradient (solvent A: water, 0.1% FA, solvent B: acetonitrile, 0.1% FA) was applied: 0.0 min: 5% B, 1.0 min: 5% B, 9.0 min: 80% B, 9.5 min: 95% B, 11.0 min: 95% B, 11.5 min: 5% B, 13.2 min: 5% B. A dead time of 1.23 min was observed. The TC(4,8) eluted after retention time of tR = 4.92 min. Chromatograms and the corresponding mass spectra of TC(4,8) can be found in the ESI (Fig. S1–S3†). The switch cage eluted after retention time of tR = 6.19 min. Chromatograms and the corresponding mass spectra of the switch cage can be found in the ESI (Fig. S5–S8†).
Molecular modelling and molecular dynamics simulations
All computational studies were performed using the Schrödinger Suite Release 2017-4 (Schrödinger LLC, New York, NY, USA). The amidated cross-linkers cis-5 and trans-5 were built in silico using Maestro (Schrödinger LLC, New York, NY, USA). The structures were geometrically relaxed using MacroModel (Schrödinger LLC, New York, NY, USA) by applying the OPLS3 force field with implicit water. Charges were compensated by the addition of Na+ or Cl−. To avoid isomerization during conformational energy minimization, constraints were applied to the CNNC torsion angle (cis-5: 0°, force constant (FC) = 200 kJ mol−1 Å−2; trans-5: 147°, FC = 300 kJ mol−1 Å−2) and to the NNC bond angles (cis-5: 121°, FC = 200 kJ mol−1 Å−2; trans-5: 112°, FC = 200 kJ mol−1 Å−2).The model of TC(4,8) was built from the first structure of the NMR-derived structure ensemble of TC10b (RCSB PDB accession code 2JOF) using Maestro. Energy minimization was achieved in the same way as for cis-5 and trans-5, but without any constraints.
The model of cis-SC was built from the relaxed model of TC(4,8). The relaxed cis-5 cross-linker was incorporated into the peptide using Maestro resulting in the diastereomer cis-SCa. Relaxation was achieved in the same way as for cis-5 applying the same constraints to the cross-linker.
MD simulations were performed using the Desmond Molecular Dynamics System (D. E. Shaw Research, New York, NY, USA) and set up with the Maestro-Desmond Interoperability Tools (Schrödinger LLC, New York, NY, USA). The molecule was submerged in an orthorhombic box of explicit water complying with the SPC water model using the OPLS3 force field. The size of the box was adjusted to a minimal volume with at least 10 Å distance between the molecule and each side of the box. The temperature was set to 300 K and the pressure was set to 1.01325 bar. MD simulations covered a period of 100 ns with recording intervals of 100 ps (trajectory) and 1.2 ps (energy). The starting geometry of each molecule was the result of the previous minimization using MacroModel. In order to avoid isomerization of the cross-linkers during MD simulations, the central eight-membered ring of the cross-linker was restrained to its starting geometry with a FC of 300 kJ mol−1 Å−2. Distances between atoms were measured and exported using the simulation event analysis panel of the Schrödinger Suite. The structures created with the Schrödinger Suite were exported as PDB files to create the molecule depictions for the figures using the PyMOL Molecular Graphics System Version 1.9.0.0. (Schrödinger LLC, New York, NY, USA).
CD spectroscopy
CD spectra were recorded on a Jasco J-720 spectropolarimeter (Jasco, Pfungstadt, Germany) equipped with a Haake WKL 26 thermostat (Thermo Electron Corporation, now Thermo Fisher Scientific, Waltham, MA, USA) and a Jasco PTC-423S peltier element (Jasco, Pfungstadt, Germany). For all measurements the same Quartz glass cuvette with an optical path length of 1.00 mm was used. All spectra were obtained in water at concentrations of 36 μM. The pH was adjusted to 5.4 ± 0.2 using 0.1 M solutions of NaOH and/or HCl. The concentration of TC(4,8) was determined by the UV absorption of Tyr and Trp according to Gill and von Hippel.59 This method was not applicable to the switch cage because of the unknown extinction coefficient of the diazocine in water. Therefore, the concentration of the switch cage was determined by recording a 1D-1H-NMR spectrum of the sample with a known amount of DSS and correlating the integrals of the DSS signal and the Leu7δ-methyl groups of cis-SCa and cis-SCb, which do not exchange and are not overlapping with other signals.Full spectra are averaged over six measurements and were recorded at 25 °C. In order to obtain the spectra of the switch cage in its PSS, the sample was irradiated at 385 nm for 10 s prior to each of the six measurements.
Thermal unfolding and folding was observed between 5 °C and 90 °C at a wavelength of 222 nm. A heating and cooling rate of 1 °C min−1 was applied. Before starting the temperature gradient, the sample was equilibrated for 5 min at the starting temperature. Two consecutive heating and cooling cycles were averaged for each spectrum. This repetition indicated full reversibility of thermal unfolding and folding of cis-SC and TC(4,8). For determination of the melting point the spectra were fitted with a sigmoidal Boltzmann fit. The fitting results are given in the ESI.†
NMR spectroscopy
The NMR spectra of TC(4,8) and of the switch cage were obtained on a AV III 600 Fourier transform-NMR spectrometer (Bruker, Billerica, MA, USA) at frequencies of 600.1 MHz for 1H-nuclei. The spectrometer was equipped with a triple resonance cryo probe. All samples were dissolved in a solution of 90% H2O and 10% D2O. 4,4-Dimethyl-4-silapentane-1-sulfonic acid (DSS) served as the internal standard for referencing. Water suppression was achieved by excitation sculpting60 using standard bruker pulse sequences.Chemical shifts were assigned using 2D-NMR techniques including 1H,1H-correlated spectroscopy (COSY), 1H,1H-total correlation spectroscopy (TOCSY) and 1H,1H-nuclear Overhauser enhancement spectroscopy (NOESY). Except for the spectra of the switch cage after having reached the PSS at 385 nm, all 2D-NMR spectra were recorded with the following parameters: COSY spectra were recorded with a size of free induction decay (FID) of 4096 (F2) by 1024 (F1) data points and a spectral width of 14.0261 ppm (F2) by 14.0000 ppm (F1). TOCSY data were acquired with a size of FID of 4096 (F2) by 512 (F1), a spectral width of 14.0261 ppm (F2) by 14.0261 ppm (F1) and a mixing time of 60 ms. NOESY data were recorded with a size of FID of 4096 (F2) by 512 (F1), a spectral width of 14.0261 ppm (F2) by 14.0261 ppm (F1) and using a mixing time of 200 ms.
All measurements were performed at 298 K if not stated otherwise. Spectra of TC(4,8) were recorded at concentrations of 3.3 mg ml−1 (1.6 mM) at pH = 5.3 ± 0.1. Each data point of each spectrum was an accumulation of 8 scans. Spectra of the switch cage were recorded at concentrations of 1.2 mg ml−1 (0.5 mM) at pH = 5.5 ± 0.1. Each data point of each spectrum was an accumulation of 16 scans.
In order to obtain spectra of the Trp-cage after having reached its PSS at 385 nm the sample was irradiated for 10 s immediately before each experiment. Due to the relatively fast trans → cis relaxation the 2D-NMR experiments were recorded with a reduced amount of scans and a reduced size of FID in the F1 dimension but otherwise identical parameters as stated above. The COSY, TOCSY and NOESY were recorded with an F1 FID of 512 and 8 scans per data point.
Irradiation
Irradiation was performed immediately prior to the measurements outside the spectrometer (NMR, CD, UV/vis) with samples already inside their container for the respective spectroscopic measurement. Samples were irradiated using light emitting diodes (LEDs) emitting at 385 nm or 530 nm (Sahlmann Photochemical Solutions, Kiel, Germany). Irradiation at 385 nm was performed from four sides by a total of twelve Nichia NCSU034A LEDs with the following specifications: FWHM = 9 nm, P(opt) = 12 × 340 mW. Irradiation at 530 nm was performed from four sides by a total of 16 Luxeon LXML-PM01-0080 LEDs with the following specifications: FWHM = 33 nm, P(opt) = 16 × 200 mW. Regardless of the container and the wavelength the PSS was reached within 10 s of irradiation.Author contributions
F. D. S., M. L., N. P. and W. M. developed the concept. N. P. planned and performed all MD simulations as well as the synthesis and characterization of the Trp-cage and the switch-cage. For his PhD thesis in organic chemistry, W. M. planned and performed the synthesis and characterization of the cross-linker. For his bachelor thesis in biochemistry, W. M. performed the MD-simulations as well as the synthesis and characterization of the Trp-cage and the switch-cage under the guidance of N. P. M. L. and K. B. optimized the cross-linking reaction conditions. N. P. and W. M. prepared the original draft. N. P., W. M., F. D. S. and R. H. revised the manuscript. F. D. S. and R. H. supervised the work. N. P., F. D. S., R. H. and K. B. acquired the funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support by the Deutsche Forschungsgesellschaft (DFG) within the Collaborative Research Center SFB 677, “Function by Switching”. N. P. thanks the Deutsche Bundesstiftung Umwelt (German Federal Environmental Foundation) for a predoctoral fellowship. K. B. thanks the Studienstiftung des deutschen Volkes (German Academic Scholarship Foundation) for a predoctoral fellowship. The authors thank Prof. Joachim Grötzinger for granting access to and use of his CD spectrometer.Notes and references
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40(8), 4422 RSC.
- W. Szymański, J. M. Beierle, H. A. V. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113(8), 6114 CrossRef PubMed.
- R. J. Mart and R. K. Allemann, Chem. Commun., 2016, 52(83), 12262 RSC.
- L. Albert and O. Vázquez, Chem. Commun., 2019, 55(69), 10192 RSC.
- J. R. Kumita, D. G. Flint, G. A. Woolley and O. S. Smart, Faraday Discuss., 2002, 122, 89–103 RSC ; discussion 171–190.
- A. M. Ali, M. W. Forbes and G. A. Woolley, ChemBioChem, 2015, 16(12), 1757 CrossRef CAS PubMed.
- A. Bergen, S. Rudiuk, M. Morel, T. Le Saux, H. Ihmels and D. Baigl, Nano Lett., 2016, 16(1), 773 CrossRef CAS PubMed.
- A. Estévez-Torres, C. Crozatier, A. Diguet, T. Hara, H. Saito, K. Yoshikawa and D. Baigl, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(30), 12219 CrossRef PubMed.
- A.-L. M. Le Ny and C. T. Lee Jr., J. Am. Chem. Soc., 2006, 128(19), 6400 CrossRef CAS PubMed.
- L. Guerrero, O. S. Smart, C. J. Weston, D. C. Burns, G. A. Woolley and R. K. Allemann, Angew. Chem., Int. Ed., 2005, 44(47), 7778 CrossRef CAS PubMed.
- L. Guerrero, O. S. Smart, G. A. Woolley and R. K. Allemann, J. Am. Chem. Soc., 2005, 127(44), 15624 CrossRef CAS PubMed.
- G. A. Woolley, A. S. I. Jaikaran, M. Berezovski, J. P. Calarco, S. N. Krylov, O. S. Smart and J. R. Kumita, Biochemistry, 2006, 45(19), 6075 CrossRef CAS PubMed.
- S. Kneissl, E. J. Loveridge, C. Williams, M. P. Crump and R. K. Allemann, ChemBioChem, 2008, 9(18), 3046 CrossRef CAS PubMed.
- F. Zhang, K. A. Timm, K. M. Arndt and G. A. Woolley, Angew. Chem., Int. Ed., 2010, 49(23), 3943 CrossRef CAS PubMed.
- R. J. Mart, P. Wysoczański, S. Kneissl, A. Ricci, A. Brancale and R. K. Allemann, ChemBioChem, 2012, 13(4), 515 CrossRef CAS PubMed.
- R. J. Mart, R. J. Errington, C. L. Watkins, S. C. Chappell, M. Wiltshire, A. T. Jones, P. J. Smith and R. K. Allemann, Mol. BioSyst., 2013, 9(11), 2597 RSC.
- L. Nevola, A. Martín-Quirós, K. Eckelt, N. Camarero, S. Tosi, A. Llobet, E. Giralt and P. Gorostiza, Angew. Chem., Int. Ed., 2013, 52(30), 7704 CrossRef CAS PubMed.
- A. Martín-Quirós, L. Nevola, K. Eckelt, S. Madurga, P. Gorostiza and E. Giralt, Chem. Biol., 2015, 22(1), 31 CrossRef PubMed.
- B. Jankovic, A. Gulzar, C. Zanobini, O. Bozovic, S. Wolf, G. Stock and P. Hamm, J. Am. Chem. Soc., 2019, 141(27), 10702 CrossRef CAS PubMed.
- Z. Zhang, D. C. Burns, J. R. Kumita, O. S. Smart and G. A. Woolley, Bioconjugate Chem., 2003, 14(4), 824 CrossRef CAS PubMed.
- R. Siewertsen, H. Neumann, B. Buchheim-Stehn, R. Herges, C. Näther, F. Renth and F. Temps, J. Am. Chem. Soc., 2009, 131(43), 15594 CrossRef CAS PubMed.
- R. Siewertsen, J. B. Schönborn, B. Hartke, F. Renth and F. Temps, Phys. Chem. Chem. Phys., 2011, 13(3), 1054 RSC.
- M. Hammerich, C. Schütt, C. Stähler, P. Lentes, F. Röhricht, R. Höppner and R. Herges, J. Am. Chem. Soc., 2016, 138(40), 13111 CrossRef CAS PubMed.
- P. Lentes, E. Stadler, F. Röhricht, A. Brahms, J. Gröbner, F. D. Sönnichsen, G. Gescheidt and R. Herges, J. Am. Chem. Soc., 2019, 141(34), 13592 CrossRef CAS PubMed.
- W. Moormann, D. Langbehn and R. Herges, Beilstein J. Org. Chem., 2019, 15, 727 CrossRef CAS PubMed.
- W. Moormann, D. Langbehn and R. Herges, Synthesis, 2017,(15), 3471 CAS.
- M. S. Maier, K. Hüll, M. Reynders, B. S. Matsuura, P. Leippe, T. Ko, L. Schäffer and D. Trauner, J. Am. Chem. Soc., 2019, 141(43), 17295 CrossRef CAS PubMed.
- S. Samanta, C. Qin, A. J. Lough and G. A. Woolley, Angew. Chem., Int. Ed., 2012, 51(26), 6452 CrossRef CAS PubMed.
- A. M. Petros, A. Medek, D. G. Nettesheim, D. H. Kim, H. S. Yoon, K. Swift, E. D. Matayoshi, T. Oltersdorf and S. W. Fesik, Proc. Natl. Acad. Sci. U. S. A., 2001, 98(6), 3012 CrossRef CAS PubMed.
- S. W. Muchmore, M. Sattler, H. Liang, R. P. Meadows, J. E. Harlan, H. S. Yoon, D. Nettesheim, B. S. Chang, C. B. Thompson, S.-L. Wong, S.-C. Ng and S. W. Fesik, Nature, 1996, 381(6580), 335 CrossRef CAS PubMed.
- C. R. Kissinger, B. Liu, E. Martin-Blanco, T. B. Kornberg and C. O. Pabo, Cell, 1990, 63(3), 579 CrossRef CAS PubMed.
- N. D. Clarke, C. R. Kissinger, J. Desjarlais, G. L. Gilliland and C. O. Pabo, Protein Sci., 1994, 3(10), 1779 CrossRef CAS PubMed.
- I. Radhakrishnan, G. C. Pérez-Alvarado, D. Parker, H. J. Dyson, M. R. Montminy and P. E. Wright, Cell, 1997, 91(6), 741 CrossRef CAS PubMed.
- J. Martinez-Oyanedel, H.-W. Choe, U. Heinemann and W. Saenger, J. Mol. Biol., 1991, 222(2), 335 CrossRef CAS PubMed.
- C. J. McKnight, P. T. Matsudaira and P. S. Kim, Nat. Struct. Biol., 1997, 4(3), 180 CrossRef CAS PubMed.
- J. W. Neidigh, R. M. Fesinmeyer and N. H. Andersen, Nat. Struct. Biol., 2002, 9(6), 425 CrossRef CAS PubMed.
- B. Barua, J. C. Lin, V. D. Williams, P. Kummler, J. W. Neidigh and N. H. Andersen, Protein Eng., Des. Sel., 2008, 21(3), 171 CrossRef CAS PubMed.
- S. H. Gellman and D. N. Woolfson, Nat. Struct. Biol., 2002, 9(6), 408 CrossRef CAS PubMed.
- H. Meuzelaar, K. A. Marino, A. Huerta-Viga, M. R. Panman, L. E. J. Smeenk, A. J. Kettelarij, J. H. van Maarseveen, P. Timmerman, P. G. Bolhuis and S. Woutersen, J. Phys. Chem. B, 2013, 117(39), 11490 CrossRef CAS PubMed.
- B. N. Markiewicz, H. Jo, R. M. Culik, W. F. DeGrado and F. Gai, J. Phys. Chem. B, 2013, 117(47), 14688 CrossRef CAS PubMed.
- L. Qiu, S. A. Pabit, A. E. Roitberg and S. J. Hagen, J. Am. Chem. Soc., 2002, 124(44), 12952 CrossRef CAS PubMed.
- A. Byrne, D. V. Williams, B. Barua, S. J. Hagen, B. L. Kier and N. H. Andersen, Biochemistry, 2014, 53(38), 6011 CrossRef CAS PubMed.
- M. Scian, J. C. Lin, I. Le Trong, G. I. Makhatadze, R. E. Stenkamp and N. H. Andersen, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(31), 12521 CrossRef CAS PubMed.
- W. W. Streicher and G. I. Makhatadze, Biochemistry, 2007, 46(10), 2876 CrossRef CAS PubMed.
- C. Simmerling, B. Strockbine and A. E. Roitberg, J. Am. Chem. Soc., 2002, 124(38), 11258 CrossRef CAS PubMed.
- S. Kannan and M. Zacharias, PLoS One, 2014, 9(2), e88383 CrossRef PubMed.
- J. Juraszek and P. G. Bolhuis, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(43), 15859 CrossRef CAS PubMed.
- R. Day, D. Paschek and A. E. Garcia, Proteins, 2010, 78(8), 1889 CAS.
- Matthias Lipfert, Design of a Stabilizing and Switching Module for α-Helical Peptides, Dissertation, Kiel, 2017 Search PubMed.
- E. J. Corey and N. Raju, Tetrahedron Lett., 1983, 24(50), 5571 CrossRef CAS.
- A. Bundi and K. Wüthrich, Biopolymers, 1979, 18(2), 285 CrossRef CAS.
- G. Merutka, H. J. Dyson and P. E. Wright, J. Biomol. NMR, 1995, 5(1), 14 CrossRef CAS PubMed.
- G. Cabré, A. Garrido-Charles, À. González-Lafont, W. Moormann, D. Langbehn, D. Egea, J. M. Lluch, R. Herges, R. Alibés, F. Busqué, P. Gorostiza and J. Hernando, Org. Lett., 2019, 21(10), 3780 CrossRef PubMed.
- E. R. Thapaliya, J. Zhao and G. C. R. Ellis-Davies, ACS Chem. Neurosci., 2019, 10(5), 2481 CrossRef CAS PubMed.
- J. B. Trads, K. Hüll, B. S. Matsuura, L. Laprell, T. Fehrentz, N. Görldt, K. A. Kozek, C. D. Weaver, N. Klöcker, D. M. Barber and D. Trauner, Angew. Chem., Int. Ed., 2019, 58(43), 15421 CrossRef CAS PubMed.
- L. Albert, A. Peñalver, N. Djokovic, L. Werel, M. Hoffarth, D. Ruzic, J. Xu, L.-O. Essen, K. Nikolic, Y. Dou and O. Vázquez, ChemBioChem, 2019, 20(11), 1417 CrossRef CAS PubMed.
- H. Sell, C. Näther and R. Herges, Beilstein J. Org. Chem., 2013, 9, 1 CrossRef CAS PubMed.
- S. S. Wong, Chemistry of protein conjugation and cross-linking, CRC Press, Boca Raton, FL, USA, 1991 Search PubMed.
- S. C. Gill and P. H. von Hippel, Anal. Biochem., 1989, 182(2), 319 CrossRef CAS PubMed.
- T.-L. Hwang and A. J. Shaka, J. Magn. Reson., Ser. A, 1995, 112(2), 275 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional experimental procedures and spectroscopic data. See DOI: 10.1039/c9ob02442e |
| ‡ IUPAC did not define any one-letter code for the artificial amino acid L-2,3-diaminopropionic acid. Therefore, in this work we assigned it the greek letter Γ. |
| § The recently developed N-bridged diazocines are a remarkable exception to that rule because of their nitrogen-atom incorporated into the bridge. This feature distinguishes them from ‘standard’ diazocines bearing an ethylene bridge. The NAc-CH2-bridged diazocine shows cis → trans conversion rates of 78% trans-isomer in D2O:MeOH-d4 (80:20) at 405 nm.24 |
| This journal is © The Royal Society of Chemistry 2020 |