
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-induced thiol–ene reactions for late-stage functionalization of unsaturated polyether macrocycles: regio and diastereoselective access to macrocyclic dithiol derivatives†‡
Elodie
Brun
 a,
Ke-Feng
Zhang
a,
Laure
Guénée
b and
Jérôme
Lacour
*a
a,
Ke-Feng
Zhang
a,
Laure
Guénée
b and
Jérôme
Lacour
*a
aDepartment of Organic Chemistry, University of Geneva, Quai Ernest Ansermet 30, 1211 Geneva 4, Switzerland. E-mail: jerome.lacour@unige.ch
bLaboratory of Crystallography, University of Geneva, Quai Ernest Ansermet 24, 1211 Geneva 4, Switzerland
First published on 6th December 2019
Abstract
Double hydrothiolation of bis enol ether macrocycles was achieved under photo-mediated conditions. The thiol–ene reactions afford a fully regioselective anti-Markovnikov post-functionalization. Thanks to the use of ethanedithiol as reagent, moderate to excellent diastereoselectivity was accomplished leading to macrocycles containing four defined stereocenters in only three steps from 1,4-dioxane, tetrahydrofuran (THF) or tetrahydropyran (THP).
The thiol–ene reaction, which is the formal coupling of olefins and thiols to form alkylsulfide derivatives, is widely used in polymer science, nanoengineering, chemical biology and medicinal chemistry.1 This process, also known as hydrothiolation, is considered to be a click reaction and much of its success is related to this reactivity.2 With terminal olefins, the hydrothiolation can lead to two types of products namely, branched and linear sulfides with respect to position of the carbon–sulfur (C–S) bond formation. For most applications, the anti-Markovnikov thiol–ene reactivity, that generates only the latter class of linear products, is most interesting and sought for. This regioselectivity is usually achieved following free radical or base-induced mechanisms, and this under metal-free, metal-catalyzed, and photo-induced reaction conditions.1a However, stereochemical control of newly generated stereocenter(s) is often problematic and low to excellent selectivity can be obtained for a same substrate depending on the thiol reagent, reaction conditions and additives.3 This situation is even more complicated when multiple thiol–ene reactions can occur on a substrate carrying several olefins.4 In this context, our group recently reported the stereoselective deconjugations of bis-α,β-unsaturated macrocycles 1 in presence of aromatic amines and t-BuOK.5 In a single step (Scheme 1, top), bis enol ether macrocycles are formed by double (tandem) amidations and olefin transpositions that yield products 2 as single stereoisomers (racemic, diastereomeric ratio dr >49
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Such bis(amide) derivatives 2 have been applied in fields ranging from chiroptical spectroscopy to molecular switches, nanosensors and heteroditopic receptors.6 These applications were achieved by the functionalization of the arylamide groups and not of the macrocyclic skeleton.
:1). Such bis(amide) derivatives 2 have been applied in fields ranging from chiroptical spectroscopy to molecular switches, nanosensors and heteroditopic receptors.6 These applications were achieved by the functionalization of the arylamide groups and not of the macrocyclic skeleton.
 | ||
| Scheme 1 Stereoselective synthesis of bis enol ether functionalized polyether macrocycles 2 (A, syn transposition, dr >49:1). Regio and diastereoselective thiol–ene additions (B). | ||
Herein, in an effort to achieve the late-stage functionalization of the unsaturated polyether fragments, double stereoselective hydrothiolations of compounds 2 are reported. Taking advantage of photo-initiated conditions, disulfides and dithiol derivatives 3 to 13 are generated in good to excellent yields and with moderate to excellent diastereocontrol (dr up to 7.3:1:0, Scheme 1 and Fig. 2). The reaction is general and can be applied to a variety of macrocyclic precursors (16 and 18-membered rings, aromatic and heteroaromatic amides). To achieve higher selectivity, the use of bis thiol reagents was developed; the diastereoselectivity depending on the chain length between the two S-atoms. Overall, two acid-sensitive exocyclic enol ethers have been transformed selectively into saturated chemically-robust functional groups that can behave as handle for future developments. The thiol–ene reactions can be considered orthogonal as the components react together in high yields and in the presence of the other functional groups and chromophores.7
Previously, it was shown that methyl α-diazo-β-ketoester reacts with THF, 1,4-dioxane or THP under dirhodium catalysis in formal [3 + Y + 3 + Y] multi-component condensations (Y = 5 or 6). The process is mild and affords the unsaturated macrocycles 1 on multi-gram scale (up to 20 grams) while using a low catalyst loading (0.01–0.001 mol%).8 As mentioned earlier, compounds 1 react with excesses of ArNH2 and t-BuOK (>3 equiv. each) to yield unsaturated bis(amide) derivatives 2. This reaction tolerates a large variety of aromatic amines.5 However, despite major efforts in the group, it was never possible to achieve the late stage functionalization of the exocyclic terminal olefins. Bis enol ethers 2 are particularly sensitive to Lewis and Brønsted acidic conditions (or workup). Care was thus taken to study the reactivity of compounds 2 under photo-induced thiol–ene conditions that are neutral essentially.
In a first attempt, the reaction was investigated in the presence of 2,2-dimethoxyphenylacetophenone (DMPA) as photo-initiator and under mercury lamp irradiation.9 Quite a few reactions were performed using macrocycle 2 (X![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) as model substrate, carrying two 3,5-bis(trifluoromethyl)phenyl carboxamide groups, and various thiols as reagents (14a–14h, Table 1). With thiolacetic acid 14a, the reaction worked smoothly giving only the corresponding anti-Markovnikov addition product 3a in 90% yield within 1 hour (entry 1). However, 3a was obtained as a mixture of three diastereoisomers as a consequence of the creation of two new stereogenic centers and the presence of a local symmetry. The three stereoisomers could be separated by preparative thin layer chromatography. By NMR spectroscopy, the two C2-symmetric derivatives were readily differentiated from the single C1 isomer. The assignment of their relative cis and trans configurations was achieved upon deprotection of the acetyl groups (K2CO3 4.0 equiv., MeOH/THF/H2O, 25 °C, 1 h). The resulting C2-cis-4 and C2-trans-4 bis thiols were found to be crystalline and structures were unambiguously determined by X-ray crystallography (Fig. 1). While the C2-symmetry of cis-4 was maintained in the solid state, a non-symmetric conformation was found for trans-4,10 unlike in solution for which duplicated functional groups are magnetically equivalent in the 1H and 13C NMR spectra. This signifies either fast positional interchanges on the NMR time scale or the adoption of a C2-symmetric conformation in solution.11
O) as model substrate, carrying two 3,5-bis(trifluoromethyl)phenyl carboxamide groups, and various thiols as reagents (14a–14h, Table 1). With thiolacetic acid 14a, the reaction worked smoothly giving only the corresponding anti-Markovnikov addition product 3a in 90% yield within 1 hour (entry 1). However, 3a was obtained as a mixture of three diastereoisomers as a consequence of the creation of two new stereogenic centers and the presence of a local symmetry. The three stereoisomers could be separated by preparative thin layer chromatography. By NMR spectroscopy, the two C2-symmetric derivatives were readily differentiated from the single C1 isomer. The assignment of their relative cis and trans configurations was achieved upon deprotection of the acetyl groups (K2CO3 4.0 equiv., MeOH/THF/H2O, 25 °C, 1 h). The resulting C2-cis-4 and C2-trans-4 bis thiols were found to be crystalline and structures were unambiguously determined by X-ray crystallography (Fig. 1). While the C2-symmetry of cis-4 was maintained in the solid state, a non-symmetric conformation was found for trans-4,10 unlike in solution for which duplicated functional groups are magnetically equivalent in the 1H and 13C NMR spectra. This signifies either fast positional interchanges on the NMR time scale or the adoption of a C2-symmetric conformation in solution.11
 | ||
| Fig. 1 Stick view of the crystal structures of C2-cis-4 and C2-trans-4. For clarity reasons, most hydrogen atoms are omitted. Disorder is observed for some CF3 groups. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | RSH, 14 | Time (h) | dr , | C 2-cisd | C 1 | C 2-transd |
|
a Reactions were performed with 0.1 mmol of 2.
b Diastereomeric ratio C2-cis:C1:C2-trans determined by 1H-NMR analysis of crude mixtures when possible.
c
dr based on isolated fraction yields.
d Isolated yields.
e Reaction conditions: 2 (0.1 mmol), 14a (4.0 equiv.), Et3B (1.0 M in hexane, 3.2 equiv.), TBC (2.4 equiv.), CH2Cl2 (0.15 mL), r.t., under air, 2.0 h. n.d. stands for not determined.
|
||||||
| 1 | 14a | 1 | 1.5:1.1:1c |
38% | 27% | 25% |
| 2 | 14b | 1 | 1.8:1:0c |
48% | 26% | — |
| 3 | 14c | 1.5 | 1.9:1:0c |
49% | 26% | — |
| 4 | 14d | 2 | 7.3:1:0b |
57% | n.d. | — |
| 5 | 14e | 2 | 4.0:1:0b |
60% | n.d. | — |
| 6 | 14f | 36 | 2.0:1:0b |
27% | 15% | — |
| 7 | 14g | 36 | 1.5:1:0b |
34% | 21% | — |
| 8 | 14h | 4 | 2.2:2.4:1c |
29% | 31% | 13% |
| 9e | 14a | 2 | 1:1.2:1.4c |
22% | 26% | 31% |
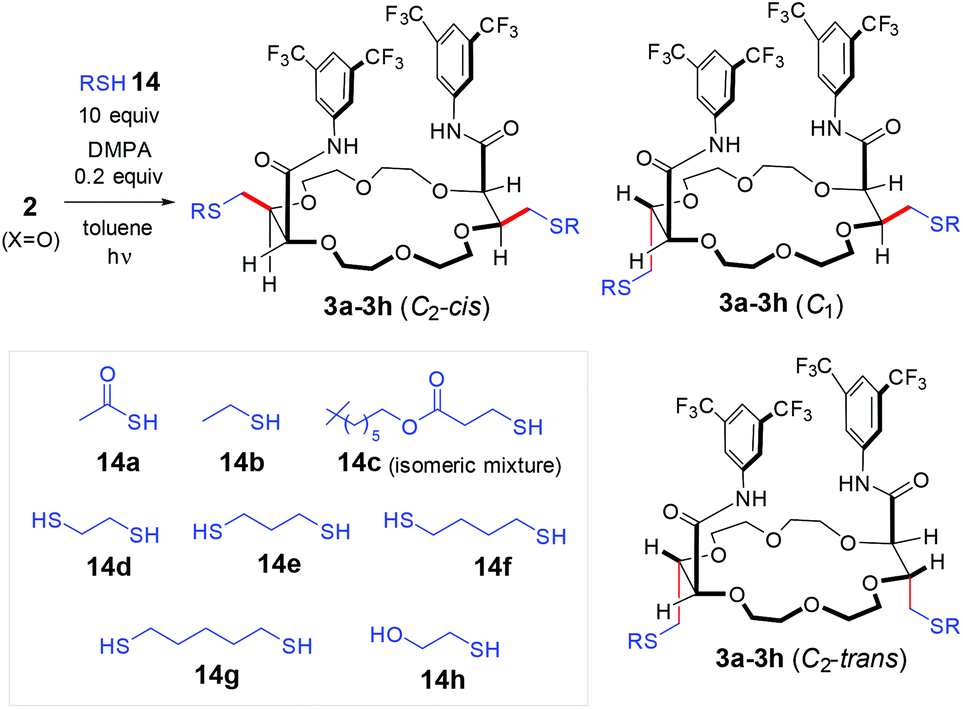
Care was then taken to increase the diastereoselectivity. As expected, classical thiol reagents participated to the reaction and, to our delight, an improvement was afforded with ethanethiol 14b and methylheptyl 3-mercaptopropanoate 14c. In these cases, only two instead of three products were formed in ca. 1.8:1:0 ratio. The major isomer of 3b presents a C2-cis configuration, as determined by the X-ray crystallography, and the minor one is the C1 product.12 Looking to improve the selectivity further and in view of the expected radical mechanism (vide infra), we turned our attention to dithiol derivatives as reagents.4,13 In thiol–ene reactions, it was our analysis that one of the two sulfur atoms would participate in the C–S bond forming reaction (propagation step) while the other could contribute as a H-donor atom. It was thus imagined that an intramolecular chain transfer step could favor an enhanced stereoselectivity; the discrimination depending then probably on the chain length between the two S-atoms. Several dithiols were selected, namely 14d–14g. With shorter ethanedithiol 14d, an excellent diastereoselectivity was obtained (7.3:1:0 dr, entry 4).14 Remarkably, step-by-step increase of the chain length between the thiols resulted in lower and lower diastereoselectivity ratios (Table 1, entries 4–7). With 2-mercaptoethan-1-ol 14h, poor selectivity was again obtained (2.2:2.4:1 dr, entry 8).15 All these results show the importance of the chain transfer step in the stereodetermining events. Finally, inspired by Renaud's studies on the Et3B-mediated radical chemistry,16 we wondered whether these mild and efficient conditions could improve or modify the selectivity. With thiolacetic acid 14a, a mixture of three stereoisomers was again obtained in essentially an equimolar ratio (entry 9).
With the optimal conditions for the thiol–ene reaction in hand (ethanedithiol 14b, Table 1, entry 4), the generality and limitations of the process were tested. Macrocycles 2 derived from THP (XCH2) and THF (X—) were reacted to afford the corresponding 18C4 and 16C4 dithiols 5 and 6 (Fig. 2). Only two stereoisomers albeit with lower diastereoselectivity (dr 4.0:1 and 2.6:1) and yields were obtained in favor of the major C2-cis derivatives (41% and 32% respectively). This shows, not too surprisingly, that the diastereoselectivity is influenced by the nature and size (conformation) of the starting macrocycles 2. Next, various substrates with heteroaromatic substituents were tested. In such instances, longer reaction times (15–36 h) were required to achieve full conversions. Reactions proceeded well with bis para and meta pyridines affording products 7 and 8 in good yields (64–73%) but moderate selectivity (dr ca. 3.2:1:0). When bis pyrimidine substituents were used, 9 was obtained in 70% yield and a 4.0:1:0 diastereomeric ratio. However, in the presence of ortho pyridines (product 10), three diastereomers were observed in 2.3:1.3:1 ratio among the C2-cis, C1 and C2-trans isomers. Currently, to explain this change in selectivity, only a proximity effect can be tentatively evoked with a participation of the Lewis basic pyridine nitrogen atoms that would interact with the pendant thiol groups.
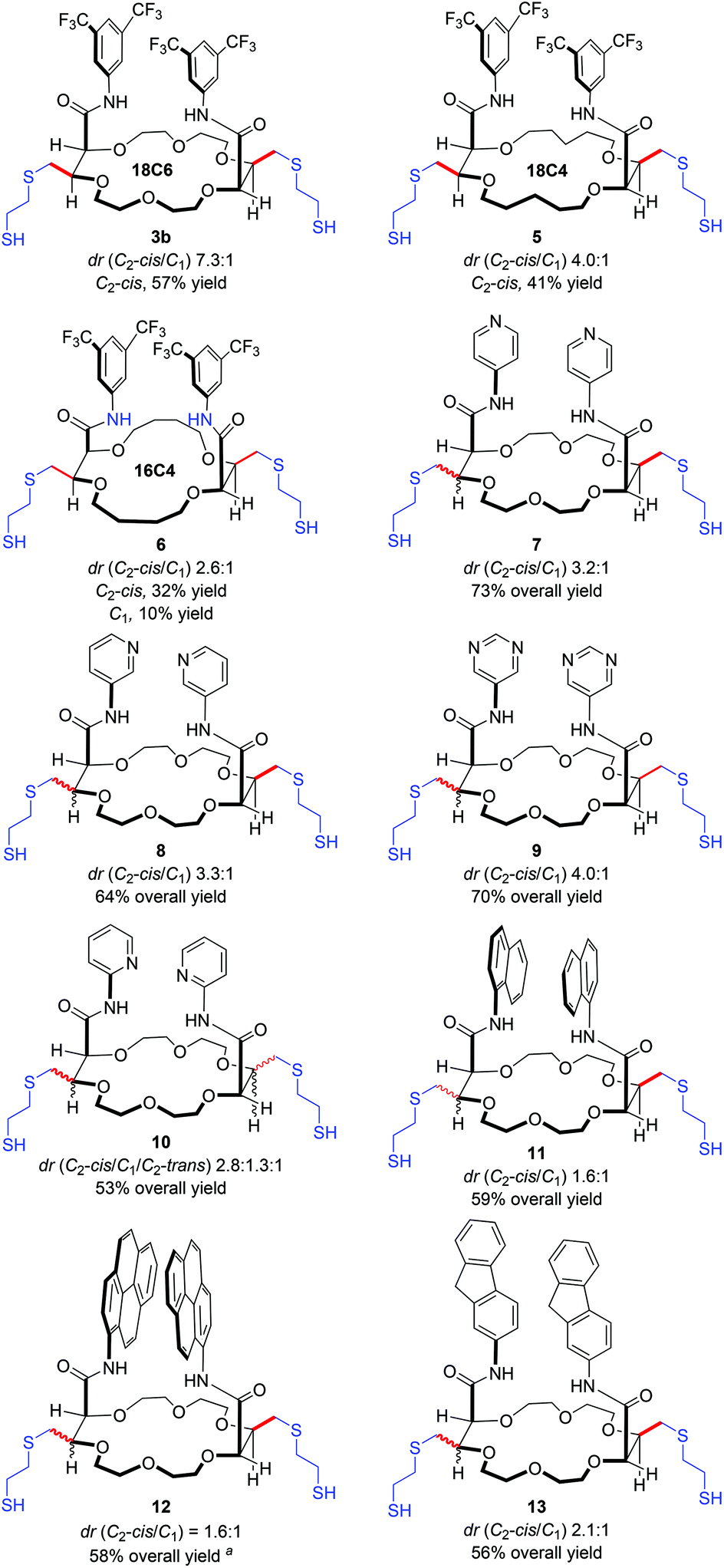 | ||
| Fig. 2 Substrate scope. aReaction performed in degassed THF rather than toluene. | ||
Then, we examined substrates carrying polyarenes, namely 1-naphthyl, 2-fluorenyl and 1-pyrenyl amide substituents. Under standard reaction conditions, only two diastereomers and good yields were obtained in all cases. With naphthalene and pyrene derivatives, dithiols 11 and 12 afforded the C2-cis isomer as the major component in a 1.6:1 ratio only; a slightly higher diastereoselectivity (2.1:1) was observed with 9H-fluorene 13. Finally, a macrocycle 2 bearing two perylene units was also tested as substrate. However, conversion could not be achieved using the current conditions; the perylene chromophores perturbing the photo-induced process.
Based on previous studies, a mechanistic rationale can be proposed for the thiol–ene reaction.1a,f In Scheme 2, it is exemplified with ethanedithiol 14d that gives the higher level of stereocontrol. Upon photo-initiation with cleavable DMPA, thiyl radicals A are readily generated. These moieties add to the less substituted ends of the enol ether CC bonds, forming carbon-centered radicals B reversibly.1f Free radicals B then abstract intramolecularly hydrogen atoms from the pendant thiol functional groups to deliver intermediates C. Then, reactions with 14d afford the targeted sulfide products along with the regenerated thiyl radical A that propagates the chain reaction. In fact, due to the presence of the second thiol group in reagents 14d–14g, intramolecular mechanisms are probably favored for the hydrogen atom abstraction. However, in such intramolecular steps, geometric factors are known to dominate.17 In our system, the shortest possible intramolecular process is a 1,7-hydrogen atom transfer with 14d that leads to the highest selectivity.18 With an increasing chain length between the two S-atoms, entropies of activation for the intramolecular process become less favorable and this may be the reason for the lower selectivity ratios.19 Reversible hydrogen atom abstractions adjacent to ethereal oxygen atoms have been reported with thiols and can lead to the epimerization of stereogenic centers but such reactions occur under more strenuous conditions (octane, 125 °C) than that employed.20 A thermodynamic control for step B to C and the issuing stereoselectivity are thus unlikely in the present situation.
 | ||
| Scheme 2 Mechanistic proposal. HT stands for hydrogen transfer. | ||
In conclusion, double hydrothiolation of bis enol ether macrocycles 2 was achieved under photo-induced radical-based conditions. The straightforward post-functionalization occurred with full anti-Markovnikov regioselectivity. Thanks to the use of ethanedithiol as reagent, C2-trans isomers were essentially removed from the reaction mixtures (with one exception). This greatly simplifies the isolation and purification of the remaining C2-cis and C1 isomers by chromatography. Overall, moderate to excellent diastereoselectivity could be accomplished leading to macrocycles containing four defined stereogenic centers in only three steps from 1,4-dioxane, THF or THP. The obtained products are currently considered for various applications in which the newly-introduced sulfur containing groups can bring reactivity and selectivity in intermolecular interactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Geneva and the Swiss National Science Foundation for financial support (SNF 200020-172497 and 200020-184843). We acknowledge the contributions of the Sciences Mass Spectrometry (SMS) platform at the Faculty of Sciences, University of Geneva. We thank Professor Philippe Renaud (Bern), Valentin Soulard and Dr Qi Huang for their assistance in the Et3B-mediated chemistry. We thank them and Dr Fabrice Dénès (Nantes) for their mechanistic insight.Notes and references
- (a) A. K. Sinha and D. Equbal, Asian J. Org.Chem., 2019, 8, 32–47 CrossRef CAS; (b) C. Ellingford, C. Bowen, T. McNally and C. Wan, Macromol. Rapid Commun., 2018, 39, 1800340 CrossRef PubMed; (c) R. Kowalczyk, P. W. R. Harris, G. M. Williams, S.-H. Yang and M. A. Brimble, in Peptides and Peptide-based Biomaterials and their Biomedical Applications, ed. A. Sunna, A. Care and P. L. Bergquist, Springer International Publishing, Cham, 2017, pp. 185–227 Search PubMed; (d) M. Firdaus, Asian J. Org. Chem., 2017, 6, 1702–1714 CrossRef CAS; (e) N. B. Cramer and C. N. Bowman, in Chemoselective and Bioorthogonal Ligation Reactions, ed. W. R. Algar, P. E. Dawson and I. L. Medintz, 2017, pp. 117–145 Search PubMed; (f) F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587–2693 CrossRef PubMed.
- (a) A. B. Lowe, Polym. Chem., 2014, 5, 4820–4870 RSC; (b) C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- (a) M. Bege, I. Bereczki, M. Herczeg, M. Kicsák, D. Eszenyi, P. Herczegh and A. Borbás, Org. Biomol. Chem., 2017, 15, 9226–9233 RSC; (b) D. Limnios and C. G. Kokotos, Adv. Synth. Catal., 2017, 359, 323–328 CrossRef CAS; (c) L. Lázár, M. Csávás, Á. Hadházi, M. Herczeg, M. Tóth, S. László, T. Barna, P. Herczegh and A. Borbás, Org. Biomol. Chem., 2013, 11, 5339–5350 RSC.
- H. Tagoshi and T. Endo, Chem. Lett., 1987, 16, 2363–2364 CrossRef.
- M. Vishe, R. Hrdina, A. I. Poblador-Bahamonde, C. Besnard, L. Guénée, T. Bürgi and J. Lacour, Chem. Sci., 2015, 6, 4923–4928 RSC.
- (a) Z. Jarolimova, M. Vishe, J. Lacour and E. Bakker, Chem. Sci., 2016, 7, 525–533 RSC; (b) S. Sinn, F. Biedermann, M. Vishe, A. Aliprandi, C. Besnard, J. Lacour and L. De Cola, ChemPhysChem, 2016, 17, 1829–1834 CrossRef CAS PubMed; (c) S. K. Ray, A. Homberg, M. Vishe, C. Besnard and J. Lacour, Chem. – Eur. J., 2018, 24, 2944–2951 CrossRef CAS PubMed; (d) M. Vishe, T. Lathion, S. Pascal, O. Yushchenko, A. Homberg, E. Brun, C. Piguet, E. Vauthey and J. Lacour, Helv. Chim. Acta, 2018, 101, e1700265 CrossRef; (e) A. Homberg, E. Brun, F. Zinna, S. Pascal, M. Górecki, L. Monnier, C. Besnard, G. Pescitelli, L. Di Bari and J. Lacour, Chem. Sci., 2018, 9, 7043–7052 RSC; (f) A. Aster, G. Licari, F. Zinna, E. Brun, T. Kumpulainen, E. Tajkhorshid, J. lacour and E. Vauthey, Chem. Sci., 2019, 10, 10629–10639 RSC; (g) A. Homberg, R. Hrdina, M. Vishe, L. Guénée and J. Lacour, Org. Biomol. Chem., 2019, 17, 6905–6910 RSC; (h) F. Zinna, S. Voci, L. Arrico, E. Brun, A. Homberg, L. Bouffier, T. Funaioli, J. Lacour, N. Sojic and L. Di Bari, Angew. Chem., Int. Ed., 2019, 58, 6952–6956 CrossRef CAS PubMed.
- C.-H. Wong and S. C. Zimmerman, Chem. Commun., 2013, 49, 1679–1695 RSC.
- (a) A. Homberg, D. Poggiali, M. Vishe, C. Besnard, L. Guénée and J. Lacour, Org. Lett., 2019, 687–691 CrossRef CAS PubMed; (b) D. Poggiali, A. Homberg, T. Lathion, C. Piguet and J. Lacour, ACS Catal., 2016, 6, 4877–4881 CrossRef CAS; (c) M. Vishe, R. Hrdina, L. Guénée, C. Besnard and J. Lacour, Adv. Synth. Catal., 2013, 355, 3161–3169 CrossRef CAS; (d) W. Zeghida, C. Besnard and J. Lacour, Angew. Chem., Int. Ed., 2010, 49, 7253–7256 CrossRef CAS PubMed.
- H. Fischer, R. Baer, R. Hany, I. Verhoolen and M. Walbiner, J. Chem. Soc., Perkin Trans. 2, 1990, 787–798 RSC.
- A non-symmetric conformation was also found for C2-trans-3a. See the ESI and CCDC 1952405.‡.
- Of the two hypotheses, the second is judged more likely but one hypothesis does not preclude the other.
- In comparison with unsaturated 2 (XO) which assumes an overall planar geometry for the macrocyclic platform, cis-3b and cis-4 present curved C2-symmetric basket-like structures for the heterocyclic macrocycle.
- For the use of the monosodium salt of 1,2-ethanedithiol as reducing agent, see: Y. Ando, S. Hori, T. Fukazawa, K. Ohmori and K. Suzuki, Angew. Chem., Int. Ed., 2015, 54, 9650–9653 CrossRef CAS PubMed.
- The high selectivity might be the result of the double addition and of the Horeau principle: A. M. Harned, Tetrahedron, 2018, 74, 3797–3841 CrossRef CAS; V. Rautenstrauch, Bull. Soc. Chim. Fr., 1994, 131, 515–524 Search PubMed; J. P. Vigneron, M. Dhaenens and A. Horeau, Tetrahedron, 1973, 29, 1055–1059 CrossRef . Unfortunately, in this instance, we cannot generate analogous substrates with a single enol ether functional group that would allow to test the selectivity for the mono-addition.
- Dissociation energies of O–H bonds (104.4 kcal mol−1, MeOH) are quite higher than that of thiols (88 kcal mol−1, MeSH). As such, and contrary to the later discussion in Scheme 2, an intermolecular mechanism is most likely in that case. Stereoselectivity differences between 14h and monothiols 14b–14c are then difficult to explain.
- (a) G. Povie, S. R. Suravarapu, M. P. Bircher, M. M. Mojzes, S. Rieder and P. Renaud, Sci. Adv., 2018, 4, eaat6031 CrossRef CAS PubMed; (b) G. Povie, A.-T. Tran, D. Bonnaffé, J. Habegger, Z. Hu, C. Le Narvor and P. Renaud, Angew. Chem., Int. Ed., 2014, 53, 3894–3898 CrossRef CAS PubMed.
- (a) D. Denenmark, P. Hoffmann, T. Winkler, A. Waldner and A. De Mesmaeker, Synlett, 1991, 621–624 CrossRef CAS; (b) B. Viskolcz, G. Lendvay, T. Körtvélyesi and L. Seres, J. Am. Chem. Soc., 1996, 118, 3006–3009 CrossRef CAS.
- D. Denenmark, T. Winkler, A. Waldner and A. De Mesmaeker, Tetrahedron Lett., 1992, 33, 3613–3616 CrossRef CAS.
- (a) A. E. Dorigo and K. N. Houk, J. Am. Chem. Soc., 1987, 109, 2195–2197 CrossRef CAS; (b) A. E. Dorigo, M. A. McCarrick, R. J. Loncharich and K. Houk, J. Am. Chem. Soc., 1990, 112, 7508–7514 CrossRef CAS.
- (a) H.-S. Dang, B. P. Roberts and D. A. Tocher, J. Chem. Soc., Perkin Trans. 1, 2001, 2452–2461 RSC; (b) H.-S. Dang and B. P. Roberts, Tetrahedron Lett., 2000, 41, 8595–8599 CrossRef CAS; (c) H.-S. Dang and B. P. Roberts, Tetrahedron Lett., 1999, 40, 4271–4274 CrossRef CAS.
Footnotes |
| † In addition, the dataset for this article can be found at the following DOI: 10.26037/yareta:omb24lj5dbat3c6sqqtsecu4lu. It will be preserved for 10 years |
| ‡ Electronic supplementary information (ESI) available: Experimental conditions, 1H NMR, 13C NMR, 19F NMR and IR spectra of all new compounds; Rf and HR-MS. CCDC 1952403–1952406. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ob02375e |
| This journal is © The Royal Society of Chemistry 2020 |