
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A hybrid polymer to target blood group dependence of cholera toxin†
Diksha
Haksar
,
Linda
Quarles van Ufford
and
Roland J.
Pieters
 *
*
Department of Chemical Biology & Drug Discovery, Utrecht Institute for Pharmaceutical Sciences, Utrecht University, P.O.Box 80082, 3508 TB Utrecht, The Netherlands. E-mail: r.j.pieters@uu.nl
First published on 28th November 2019
Abstract
Cholera is a potentially fatal bacterial infection caused by the cholera toxin (CT), an AB5 toxin secreted by Vibrio cholera. GM1 has long been known as the receptor of the cholera toxin in the intestine. However, increasing evidence is pointing towards the role of fucosylated conjugates as additional attachment options of the toxin. In the present paper we have synthesized a polymeric hybrid which can inhibit both modes of attachment.
Cholera is an acute diarrhoeal infection that is caused by the ingestion of water or food contaminated with the Vibrio cholera bacterium.1 Cholera is endemic in countries with poor sanitation and inadequate drinking water facilities, with 3 to 5 million reported cases every year.1 The current cholera epidemic in Yemen that began in 2016 has so far resulted in more than 3500 fatalities.1 Cholera is caused by the cholera toxin (CT) which is an AB5 toxin secreted by the bacterium. The A subunit is the toxic portion whereas the B subunit attaches itself to GM1 gangliosides on the intestinal cell surface. This attachment is regarded as one of the strongest protein–carbohydrate interactions.2 Adhesion is followed by cellular endocytosis of the A subunit which catalyses the ADP ribosylation of G-proteins. The resulting stimulation of adenylate cyclase raises the intra-cellular cAMP levels followed by chloride outflow leading to water secretion and potentially fatal diarrhea.3 Many studies have focused on the inhibition of the toxin.4–8
CT has two major biotypes, classical (cCT) and El Tor (ET CT). Cholera has long been identified as a disease associated with a blood group-dependence. One of the first clinical findings of this dependence was noted in hospital settings in India and the Phillipines more than three decades ago, with an overrepresentation of blood group O patients.9,10 ABO blood groups are classified on the basis of the histo-blood group antigens (BGAs) present on red blood cells, with the H trisac-charide being the smallest determinant. Blood group O individuals carry the unmodified H antigen which has a terminal fucose residue while those with blood group A and B have terminal Gal and GalNAc residues, respectively. The BGAs are not only present in the blood but also in other body fluids such as mucus, saliva etc. in approximately 80% of the population termed as “secretors” while the rest are “non-secretors”.11,12
Recently, a second binding site on the cholera toxin has been identified. It was shown to recognize BGAs and was first detected for a chimeric toxin of CTB and the heat-labile enterotoxin of E. coli (LTB).13–16 Both cCT and ET CT were shown to bind BGAs with millimolar affinities at the second or secondary binding site on the lateral face of the toxin.16–18 Using surface plasmon resonance (SPR), it was observed that the H determinant binds more strongly than the A determinant, especially in the case of the ET CT variant.15 The enhanced binding of CT to the displayed H trisaccharide thus may lead to increased toxin uptake and more severe symptoms for blood group O individuals.19
GM1-deficient cell lines i.e. T84 and Colo205 have been used to demonstrate that GM1 is not the sole receptor for CT.13 Additionally, it has been shown that besides these immortal cell lines, human intestinal epithelia also contain relatively little GM1.13,20 Furthermore, CTB binding to primary human jejunal epithelial cells was shown to correlate with the amount of displayed Lewis X (Lex) glycan.21
A direct binding interaction between CTB and the LeY tetra-saccharide was studied by ITC and revealed a Kd of 1–2 mM.14 Crystal structures and SPR studies further showed that Lex and also L-fucose bind exclusively to the secondary site with millimolar Kd's.20 Clearly, fucose is the common component of all glycans with affinity for the secondary CT binding site. Although GM1 is the primary receptor in cell lines with both receptors, fucosylated glycoconjugates also contribute to CTB binding and internalization.22
So far only one fucose-based polymer has been reported with an IC50 of 1.5 μM derived from a cell-based assay.14 We set out to create a molecule that could block both GM1-based and fucose-based intoxication, by constructing a “hybrid” polymeric ligand. This was done in anticipation of multivalency enhancements as we have seen for other multivalent platforms.23–26 For this purpose, we used a dextran based polymer to which fucose and a galactoside were conjugated. Meta-nitrophenyl α-galactoside (MNPG) is an ideal candidate owing to its potency and we have demonstrated that when conjugated to polymers effective inhibition of cholera toxin is achieved in a GM1-based assay.27 In the present paper, we have synthesized a fucosylated and a hybrid polymer. The synthesized compounds were tested for their ability to inhibit the cholera toxin B-subunit by making use of the GM1-based ELISA assay along with the newly developed fucose-based version.
Results and discussion
Propargyl fucoside 1 and the MNPG derivative 2 were synthesized starting from L-fucose and galactose pentaacetate according to reported procedures (Fig. 1).27,28 Azido-functionalized dextran (Mw = 155 kDa) with 6% azide functionalization was used as the polymeric scaffold.29 Copper-catalysed alkyne–azide cycloaddition was used for the conjugation of the dextran polymer to the fucoside 1 in order to obtain the fucosylated polymer i.e.3. The hybrid polymer 4 was obtained by conjugating both MNPG propargyl and 1 in equimolar quantities to the dextran azide. Final polymers 3 and 4 were characterized by NMR and infrared spectroscopy, the latter of which was useful to see the disappearance of the azide signal at 2110 cm−1 (Scheme 1) (see ESI†).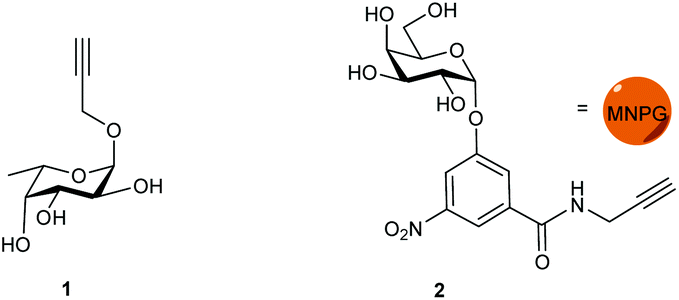 | ||
| Fig. 1 Monovalent ligands. | ||
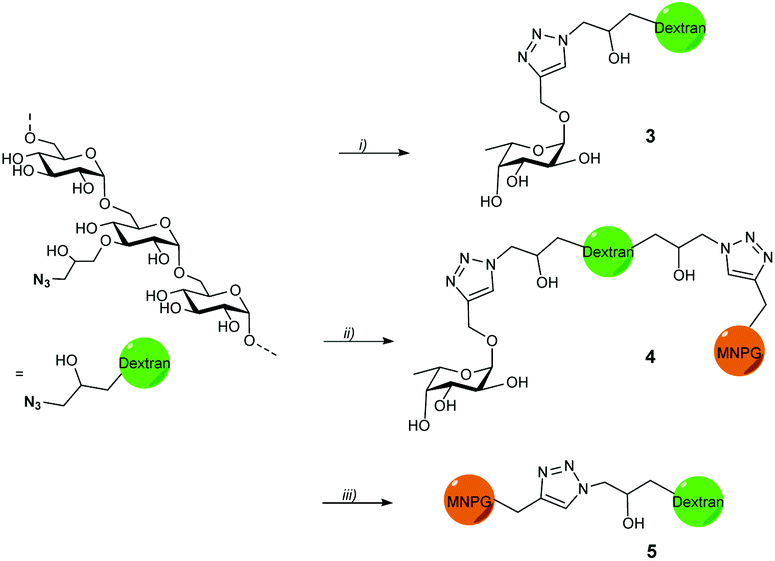 | ||
| Scheme 1 Synthesis of fucose- and hybrid polymers. (i) Dextran azide, 1, CuSO4, Na-CuAAC ascorbate, 100 °C, 75%, (ii) dextran azide, 1, 2, CuSO4, Na-ascorbate, 100 °C, 81%, (iii) dextran azide, 2, CuSO4, Na-ascorbate, 100 °C, 51–58%. | ||
Cholera toxin inhibition
The polymers were evaluated for CTB inhibition in an ELISA-type assay by immobilising the GM1 ganglioside and using a cholera toxin–biotin conjugate. Galactose was used as the monovalent reference compound and showed weak inhibition as before30,31 (IC50: 195 mM) whereas L-fucose was an extremely weak inhibitor with an IC50 of 1.6 M (Table 1). Polymer 3 did not inhibit CTB in this assay up to 200 μM, while hybrid 4 showed inhibition with an IC50 of 26 μM. This represents a large potency enhancement in comparison with the milimolar inhibitory potencies of galactose and MNPG derivatives.27 The dextran azide polymer was also tested and did not show any inhibition in the assay.| Entry | Construct | Ligand | Valency (% functionalization of polymer) | IC50 (μM) | Rel. pot.b | Rel. pot. per sugarc |
|---|---|---|---|---|---|---|
| a Determined in an ELISA-like assay with CTB5-biotin (40 ng mL−1) and wells coated with GM1. b Relative to the potency of galactose. c Relative potency divided by the MNPG valency. | ||||||
| 1 | Galactose | D-Gal | 1 | 195![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ± 21000 000 ± 21000 |
1 | 1 |
| 2 | L-Fucose | L-Fuc | 1 | 1581000 ± 171000 |
||
| 3 | 3 | L-Fuc | 52 (5.6%) | No inhibition | — | — |
| 4 | 4 |
L-Fuc + MNPG (1:1) |
52 (5.6%) | 26 ± 10 | 7500 | 288 |
| 5 | 5 | MNPG | 55 (6%) | 3.2 ± 0.9 | 61000 |
1108 |
Previously a fucosylated polymer has been synthesized and tested for aggregation based inhibition in an ELISA with T84 cells, Colo 205 cells and primary human jejunal epithelial cells.14 These cells, notably T84, are not easy to culture, so as an alternative assay unambiguously focused on fucose-CT interactions, we utilized immobilized polyacrylamide-conjugated L-fucose (PAA-fucose) and the same biotinylated toxin. PAA-fucose has been previously used to test fucosylated glycodendrimers.32 We first compared the PAA-fucose assay with the T84 cell assay to evaluate the assay sensitivity and concluded that 15.3 μg mL−1 was an appropriate concentration for the toxin to be used for further inhibition assays (Fig. 2). This is high in comparison to that required in the GM1 ELISA (40 ng mL−1). L-Fucose was used as a reference in the PAA-fucose ELISA and showed an IC50 of 12 mM (Table 2). Both polymers 3 and 4 inhibited in the low micromolar range (0.6 μM and 1 μM respectively) whereas polymer 5 did not inhibit the toxin.
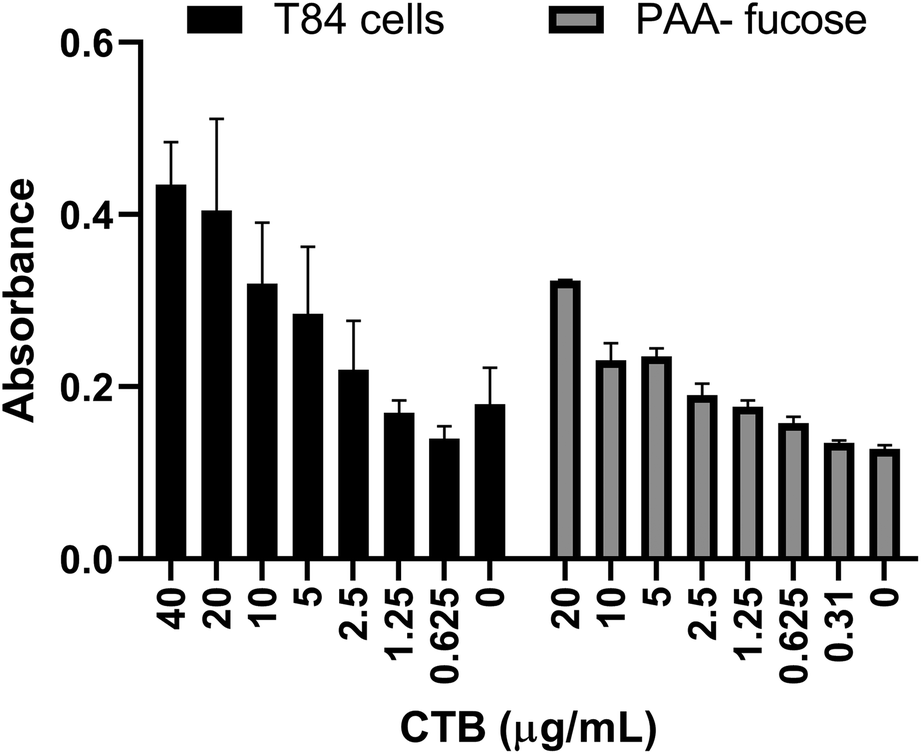 | ||
| Fig. 2 Comparison of T84 cell ELISA and PAA-fucose ELISA. Observed HRP-based signal as a function of toxin concentration. | ||
| Entry | Construct | Ligand | Valency (% functionalization of polymer) | IC50 (μM) | Rel. pot.b | Rel. pot. per sugarc |
|---|---|---|---|---|---|---|
| a Determined in an ELISA-like assay with CTB5-biotin (15.3 μg mL−1 final concentration) and wells coated with PAA-fucose. b Relative to the potency of L-fucose. c Relative potency divided by the L-fucose valency. | ||||||
| 1 | L-Fucose | L-Fuc | 1 | 11730 ± 9000 |
1 | 1 |
| 2 | 3 | L-Fuc | 52 (5.6%) | 0.63 ± 0.2 | 18619 |
358 |
| 3 | 4 |
L-Fuc + MNPG (1:1) |
52 (5.6%) | 1.1 ± 0.6 | 10663 |
205 |
| 4 | 5 | MNPG | 55 (6%) | No inhibition | — | — |
For compounds 3 and 4 these inhibitory potencies represent close to 11000 and 19000-fold potency enhancements or 358 and 205-fold per sugar ligand.
Conclusions
We have synthesized a hybrid inhibitor for the cholera toxin that can inhibit both the GM1-based adhesion of the primary binding site and the fucose-based adhesion of the secondary binding site. Additionally, an ELISA using PAA-fucose made it possible to test the fucose-based adhesion in a short span of time. Although the hybrid is not quite as active as the homopolymers in either assay, the inhibition was still strong in the low micromolar range with large multivalency enhancements in either case. The fact that the hybrids were less active that the homopolymer was expected as the ‘wrong’ ligand may obstruct multivalent binding at times, but the reductions were only minor, especially for the fucose-based assay. Furthermore, the fucose polymer 3 did not show any inhibition in the GM1-based assay, and the MNPG based polymer was not active in the fucose-based assay. The hybrid glycopolymer is a practical approach to cover both intoxication scenario's with a single agent. The agent is easy to synthesize, in a likely scalable synthesis at relatively low costs and contains a pharmaceutically benign33,34 dextran backbone.Conflicts of interest
There are no conflicts to declare.Notes and references
- World Health Organization. https://www.who.int/en/news-room/fact-sheets/detail/cholera (accessed Aug. 27, 2019).
- V. Kumar and W. B. Turnbull, Beilstein J. Org. Chem., 2018, 14, 484–498 CrossRef CAS PubMed.
- K. J. Baldauf, J. M. Royal, K. T. Hamorsky and N. Matoba, Toxins, 2015, 7, 974–996 CrossRef CAS PubMed.
- E. K. Fan, E. A. Merritt, C. Verlinde and W. G. J. Hol, Curr. Opin. Struct. Biol., 2000, 10, 680–686 CrossRef CAS PubMed.
- H. Zuilhof, Acc. Chem. Res., 2016, 49, 274–285 CrossRef CAS PubMed.
- T. R. Branson and W. B. Turnbull, Chem. Soc. Rev., 2013, 42, 4613–4622 RSC.
- A. Bernardi and P. Cheshev, Chem. – Eur. J., 2008, 14, 7434–7441 CrossRef CAS PubMed.
- V. Wittmann and R. J. Pieters, Chem. Soc. Rev., 2013, 42, 4492–4503 RSC.
- A. Chaudhuri and S. De, Lancet, 1977, 310, 404 CrossRef.
- D. Barua and A. S. Paguio, Ann. Hum. Biol., 1977, 4, 489–492 CrossRef CAS PubMed.
- Å. Holmner, A. Mackenzie and U. Krengel, FEBS Lett., 2010, 584, 2548–2555 CrossRef PubMed.
- J. E. Heggelund, A. Varrot, A. Imberty and U. Krengel, Curr. Opin. Struct. Biol., 2017, 44, 190–200 CrossRef CAS PubMed.
- A. M. Wands, A. Fujita, J. E. McCombs, J. Cervin, B. Dedic, A. C. Rodriguez, N. Nischan, M. R. Bond, M. Mettlen, D. C. Trudgian, A. Lemoff, M. Quiding-Järbrink, B. Gustavsson, C. Steentoft, H. Clausen, H. Mirzaei, S. Teneberg, U. Yrlid and J. J. Kohler, eLife, 2015, 4, 1–38 CrossRef PubMed.
- A. M. Wands, J. Cervin, H. Huang, Y. Zhang, G. Youn, C. A. Brautigam, M. Matson Dzebo, P. Björklund, V. Wallenius, D. K. Bright, C. S. Bennett, P. Wittung-Stafshede, N. S. Sampson, U. Yrlid and J. J. Kohler, ACS Infect. Dis., 2018, 4, 758–770 CrossRef CAS PubMed.
- J. E. Heggelund, D. Burschowsky, V. A. Bjornestad, V. Hodnik, G. Anderluh and U. Krengel, PLoS Pathog., 2016, 12, 1–19 Search PubMed.
- Å. Holmner, M. Lebens, S. Teneberg, J. Ångström, M. Ökvist and U. Krengel, Structure, 2004, 12, 1655–1667 CrossRef PubMed.
- J. E. Heggelund, E. Haugen, B. Lygren, A. Mackenzie, Å. Holmner, F. Vasile, J. J. Reina, A. Bernardi and U. Krengel, Biochem. Biophys. Res. Commun., 2012, 418, 731–735 CrossRef CAS PubMed.
- F. Vasile, J. J. Reina, D. Potenza, J. E. Heggelund, A. Mackenzie, U. Krengel and A. Bernardi, Glycobiology, 2014, 24, 766–778 CrossRef CAS PubMed.
- J. B. Heim, V. Hodnik, J. E. Heggelund, G. Anderluh and U. Krengel, Sci. Rep., 2019, 9, 1–14 CrossRef CAS PubMed.
- M. E. Breimer, G. C. Hansson, K.-A. Karlsson, G. Larson and H. Leffler, Glycobiology, 2012, 22, 1721–1730 CrossRef CAS PubMed.
- J. Cervin, A. M. Wands, A. Casselbrant, H. Wu, S. Krishnamurthy, A. Cvjetkovic, J. Estelius, B. Dedic, A. Sethi, K. L. Wallom, R. Riise, M. Bäckström, V. Wallenius, F. M. Platt, M. Lebens, S. Teneberg, L. Fändriks, J. J. Kohler and U. Yrlid, PLoS Pathog., 2018, 14, e1006862 CrossRef PubMed.
- A. Sethi, A. M. Wands, M. Mettlen, S. Krishnamurthy, H. Wu and J. J. Kohler, Interface Focus, 2019, 9, 20180076 CrossRef PubMed.
- H. Zhang, D. Laaf, L. Elling and R. J. Pieters, Bioconjugate Chem., 2018, 29, 1266–1275 CrossRef CAS PubMed.
- N. Parera Pera, A. Kouki, S. Haataja, H. M. Branderhorst, R. M. J. Liskamp, G. M. Visser, J. Finne and R. J. Pieters, Org. Biomol. Chem., 2010, 8, 2425–2429 RSC.
- G. Yu, A. C. Vicini and R. J. Pieters, J. Org. Chem., 2019, 84, 2470–2488 CrossRef CAS PubMed.
- L. Ballell, M. Van Scherpenzeel, K. Buchalova, R. M. J. Liskamp, R. J. Pieters and M. van Scherpenzeel, Org. Biomol. Chem., 2006, 4, 4387–4394 RSC.
- D. Haksar, E. de Poel, L. Q. van Ufford, S. Bhatia, R. Haag, J. Beekman and R. J. Pieters, Bioconjugate Chem., 2019, 30, 785–792 CrossRef CAS PubMed.
- E. Fernandez-Megia, J. Correa, I. Rodriguez-Meizoso and R. Riguera, Macromolecules, 2006, 39, 2113–2120 CrossRef CAS.
- N. Pahimanolis, A.-H. Vesterinen, J. Rich and J. Seppala, Carbohydr. Polym., 2010, 82, 78–82 CrossRef CAS.
- H. M. Branderhorst, R. M. J. Liskamp, G. M. Visser and R. J. Pieters, Chem. Commun., 2007, 5043–5045 RSC.
- D. D. Zomer-van Ommen, A. V. Pukin, O. Fu, L. H. C. Quarles van Ufford, H. M. Janssens, J. M. Beekman and R. J. Pieters, J. Med. Chem., 2016, 59, 6968–6972 CrossRef CAS PubMed.
- B. Thomas, N. Berthet, J. Garcia, P. Dumy and O. Renaudet, ChemComm, 2013, 49, 10796–10798 RSC.
- J. Varshosaz, Expert Opin. Drug Delivery, 2012, 9, 509–523 CrossRef CAS PubMed.
- W. Kim, Y. Yang, D. Kim, S. Jeong, J. W. Yoo, J. H. Yoon and Y. Jung, Drug Des., Dev. Ther., 2017, 11, 419–429 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data, NMR, inhibition curves for ELISA assay, inhibition curves for PAA-fucose assay and IR spectra. See DOI: 10.1039/c9ob02369k |
| This journal is © The Royal Society of Chemistry 2020 |