
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cationic iron porphyrins with sodium dodecyl sulphate for micellar catalysis of cyclopropanation reactions†
Ruben V.
Maaskant‡
,
Ehider A.
Polanco‡
,
Roos C. W.
van Lier
and
Gerard
Roelfes
 *
*
Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: j.g.roelfes@rug.nl
First published on 2nd January 2020
Abstract
Here, we report that the combination of cationic iron porphyrins with sodium dodecyl sulphate (SDS) gives rise to efficient micellar catalysis of cyclopropanation reactions of styrene derivatives, using diazoacetates as carbene precursors. This simple, yet effective approach for cyclopropanations illustrates the power of micellar catalysis.
Iron porphyrins have been proven excellent catalysts for carbene transfer reactions such as cyclopropanation reactions in aqueous environments.1–3 In recent years, this has been extended with much success to the creation of enzymes for cyclopropanations, by repurposing of heme enzymes and proteins such as cytochrome P450 and myoglobin,4–7 or by creation of artificial heme enzymes.8–10 In many cases this resulted in high enantioselectivities and tremendous rate accelerations compared to heme or related iron porphyrins.
Previously, we have reported on a DNA/cationic iron porphyrin hybrid that also showed highly accelerated catalysis of cyclopropanation reactions.11 It was proposed that the observed DNA-induced rate acceleration is due concentration of the reactants in hydrophobic spaces close to the DNA, resulting in a high effective molarity. This is reminiscent of micellar catalysis, where similar effects play a role. We now report that the combination of cationic iron porphyrins with anionic surfactants, such as SDS, indeed gives rise to efficient catalysis of cyclopropanation reactions of styrene derivatives.
Micellar catalysis enables and accelerates reactions of organic compounds in aqueous media, negating the need for organic solvents.12,13 By using surfactants in quantities above their critical micelle concentration (CMC), micelles with a polar exterior and apolar interior are obtained. Organic reagents and transition metal catalysts, which are normally insoluble in water, can be accommodated and concentrated in the hydrophobic interior of the micelle to obtain a high effective molarity, resulting in significantly increased reaction rates. C–C bond forming reactions have benefitted especially from the development of micellar catalysis.14–19
In the early 2000s, a number of studies reported that metalloporphyrin catalyzed epoxidations could be accelerated by the addition of surfactants.20–22 Since carbene transfer reactions, such as cyclopropanations, are mechanistically related to oxygen transfer reactions, it was hypothesized that this could also apply to iron porphyrin catalysed cyclopropanation reactions.
The cyclopropanation of p-methoxystyrene (1a), using ethyl diazoacetate (2a) as carbene precursor (Scheme 1), was investigated using neutral, cationic and anionic iron porphyrins in combination with nonionic, cationic or anionic surfactants. All surfactants were employed in concentrations above their critical micelle concentrations (CMC) in water without additives.
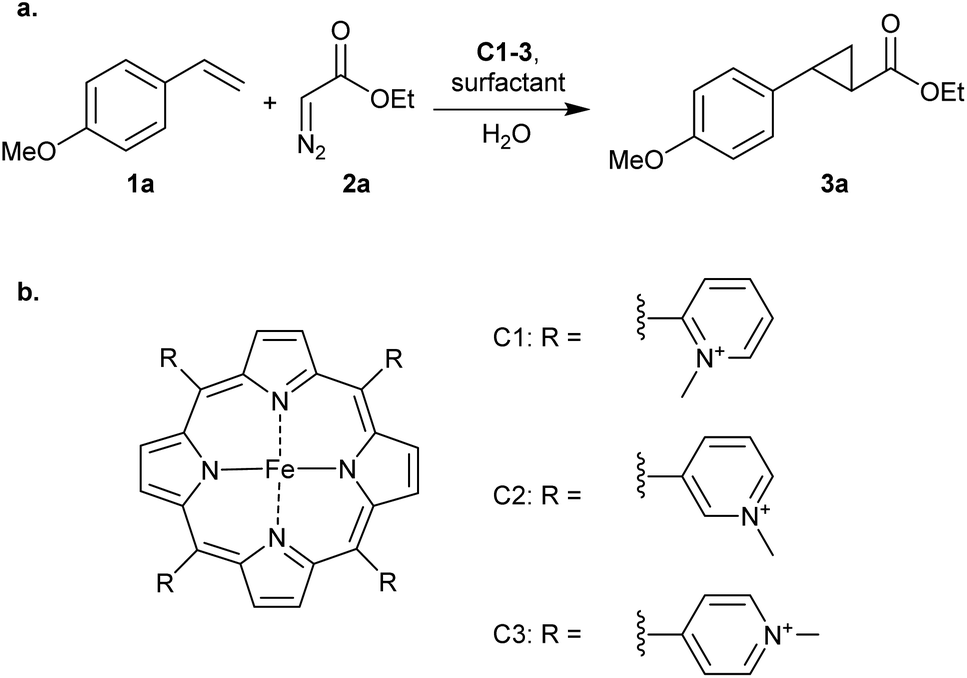 | ||
| Scheme 1 (a) Cyclopropanation of p-methoxystyrene (1a) catalysed by cationic iron porphyrins/surfactants. (b) Structures of cationic iron porphyrins C1–3 used in this study. | ||
Neutral and anionic iron porphyrin catalysts did not benefit from the addition of SDS or neutral surfactants such as TPGS-1000, whereas in some cases a modest increase in yield was found with DTAB, a cationic surfactant (Table SI1†).
Interestingly, the results were quite different when using cationic iron porphyrins C1–C3 as catalyst, which are the porhyrins we also used in our previous work on DNA-based catalytic cyclopropanation.11 These porhyrins carry four ortho-, meta- or para-N-methylpyridinium groups, respectively, on the meso positions. In the absence of surfactant, low yields of 3a were obtained. In the presence of 20 mM SDS the yield of 3a increased to 50% with C1 and an excellent yield of 98% was obtained with C2 and C3 (Table 1 entries 1–6). It is hypothesized that this acceleration is the result of ion pairing between the cationic catalyst and the anionic surfactant, effectively positioning the porphyrin on the micelle surface and in close proximity to the substrates that are in the micellar interior. The fact that the reaction with C1 is accelerated to a lesser extent than with C2 and C3 may be related to the higher steric hindrance from the o-N-methylpyridinium groups of C1.
| Entry | Catalyst | Surfactant | Time (h) | 3a (%) |
|---|---|---|---|---|
| a Reaction conditions: 75 μmol 1a, 2 eq. 2a, 1 mol% iron porphyrin, 10 ml H2O, room temperature, 1 hour, in duplo, unless stated otherwise. b 2 wt%/v is approximately 13 mM. c 2a added in 4 portions of 0.33 equivalents (total 1.33 eq.), 20 minutes between additions. d 2a added in 3 portions of 0.33 equivalents and a final portion of 0.1 equivalents (total 1.1 eq.), 20 minutes between additions. | ||||
| 1 | C1 | — | 1 | 12 ± 1 |
| 2 | C1 | 20 mM SDS | 1 | 50 ± 2 |
| 3 | C2 | — | 1 | 16 ± 8 |
| 4 | C2 | 20 mM SDS | 1 | 98 ± 2 |
| 5 | C3 | — | 1 | 13 ± 2 |
| 6 | C3 | 10 mM SDS | 1 | 60 ± 7 |
| 7 | C3 | 20 mM SDS | 1 | 98 ± 2 |
| 8 | C3 | 35 mM SDeS | 1 | 85 ± 0 |
| 9 | C3 | 3 mM STS | 1 | 28 ± 4 |
| 10 | C3 | 20 mM DTAB | 1 | 27 ± 8 |
| 11b | C3 | 2 wt%/v TPGS-1000 | 1 | 10 ± 0 |
| 12c | C3 | 20 mM SDS | 1.5 | >99 |
| 13d | C3 | 20 mM SDS | 1.5 | >99 |
Two surfactants similar to SDS but with varying alkyl chain length, sodium n-decyl sulphate (SDeS) and sodium tetradecylsulphate (STS), were employed in 20 mM concentration and in concentrations just above their CMC. Combining 1 mol% C3 with SDeS and STS in concentrations of 35 mM and 3 mM respectively, just above their CMCs (33 mM and 2 mM respectively),23–25 resulted in an acceleration to obtain 3a in respectively a good yield of 85% and a modest yield of 28% (Table 1 entries 8 and 9). When employing SDeS or STS in concentrations significantly higher or lower than the CMC, the reaction was not accelerated significantly (Table SI2 entries 2–4†). While all sodium alkyl sulphate surfactants accelerate the reaction above their CMCs, SDS proved to be the optimal surfactant for this reaction.
Surprisingly, the addition of cationic DTAB to C3, which are not expected to undergo ion pairing due to electrostatic repulsion, gave a slightly higher yield of 27% compared to 13% without surfactant (Table 1 entries 5 and 10). At present, it is unknown what causes the increase in yield. Addition of neutral TPGS-1000 does not result in a rate acceleration, probably as the water-soluble catalyst, which is not associated with the micelle, and the substrate are spatially separated (Table 1 entry 11).
Upon varying the concentration of SDS, the yield of 3a significantly increased once the concentration went above the CMC of SDS, going from 23% with 5 mM SDS to 98% with 20 mM SDS (Table SI2 entries 5–8†). Increasing the concentration of SDS even further decreased the yield (Table SI2, entry 9†). At these higher SDS concentrations the total volume of hydrophobic space is increased, thus decreasing the effective molarity of the substrates, which in turn leads to lower reaction rate and a reduction of the yield.
The reaction was optimized further using C3, which is readily accessible as it is commercially available. Increasing or decreasing the catalyst loading to respectively 20 mol% and 0.1 mol% resulted in a decrease of the yield of 3a or even a total loss of conversion to 3a (Table SI2 entries 10–14†). At high iron porphyrin loading there is most likely a relatively high concentration of metallocarbene species on the outside of the micelle which reacts with water or another molecule of 2a, resulting in the formation of diethyl malonate and/or fumarate.
Under the optimized conditions, 3a was obtained in an excellent yield of 98% using 2 equivalents of 2a. The remaining equivalent of 2a was not recovered. 18% product from a dimerization reaction of 2a (11% diethyl fumarate and 7% diethyl maleate, calculated from 1 remaining equivalent of 2a) was obtained after the reaction, the remainder of 2a most likely reacted with water to form ethyl glycolate. The efficiency of the use of 2a was improved by addition in multiple smaller portions rather than as a single portion; this resulted in the formation of 3a in a quantitative yield while the formation of side product was minimized (Table 1 entries 12 and 13). These results show that the side reactions are most likely also micelle-accelerated, but that these can be suppressed by judicious choice of concentrations of the catalyst and the carbene precursor.
To investigate the scope of reaction, a range of different alkene substrates was reacted with 2a in the presence of 1 mol% C3 and 0, 15 or 20 mM SDS. In the presence of SDS, increased yield of product was obtained with all styrene derivatives, albeit that yields were highly dependent on the substituent. While good to excellent yields were obtained with o- or p-methoxystyrene (Tables 1 and 2 entries 1 and 2), lower yields in the same time were found with styrene of p-chlorostyrene (Table 2, entries 3–5). Yet, these were still higher than the yields obtained without SDS. Using o-methylstyrene, p-methylstyrene or α-methylstyrene also a significant SDS acceleration was observed (Table 2 entries 8–13), suggesting the micellar effect is general for styrene derivatives.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Alkene | Diazo reagent | Product | [SDS] (mM) | Time (h) | Yield (%) |
| a Reaction conditions: 75 μmol olefin (final concentration 7.5 mM), 2 eq. diazo reagent, 1 mol% C3, H2O, SDS, room temperature unless stated otherwise. All reactions were performed in duplo. N.D. = not determined. b 1.1 eq. 2b–2c added. c Obtained as a mixture of cis and trans isomers. | ||||||
| 1 | 1b | 2a | 3b | — | 1.5 | <5 |
| 2 | 1b | 2a | 3b | 20 | 1.5 | 59 ± 0 |
| 3 | 1c | 2a | 3c | — | 1.5 | <5 |
| 4 | 1c | 2a | 3c | 15 | 1.5 | 30 ± 2 |
| 5 | 1c | 2a | 3c | 20 | 1.5 | 16 ± 1 |
| 6 | 1d | 2a | 3d | — | 1.5 | <5 |
| 7 | 1d | 2a | 3d | 20 | 1.5 | 8 ± 1 |
| 8 | 1e | 2a | 3e | — | 1 | 6 ± 0 |
| 9 | 1e | 2a | 3e | 20 | 1 | 23 ± 3 |
| 10 | 1f | 2a | 3f | — | 1 | <5 |
| 11 | 1f | 2a | 3f | 20 | 1 | 60 ± 13 |
| 12 | 1g | 2a | 3g | — | 1 | <5 |
| 13 | 1g | 2a | 3g | 20 | 1 | 25 ± 12c |
| 14 | 1h | 2a | 3h | — | 1.5 | <5 |
| 15 | 1h | 2a | 3h | 20 | 1.5 | <5 |
| 16 | 1i | 2a | 3i | — | 1.5 | <5 |
| 17 | 1i | 2a | 3i | 20 | 1.5 | <5 |
| 18 | 1j | 2a | 3j | — | 1.5 | 35 ± 0 |
| 19 | 1j | 2a | 3j | 20 | 1.5 | <5 |
| 20b | 1a | 2b | 3k | — | 1 | 15 ± 0 |
| 21b | 1a | 2b | 3k | 20 | 1 | 80 ± 8 |
| 22b | 1a | 2c | 3l | — | 1 | 21 ± 4 |
| 23b | 1a | 2c | 3l | 20 | 1 | 97 ± 2 |
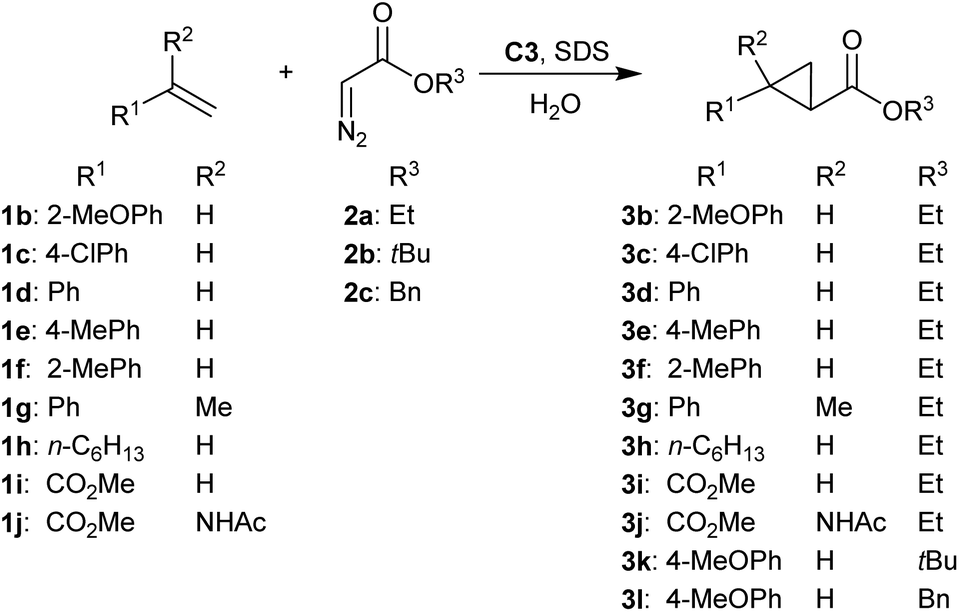
In contrast, other alkenes did not benefit from micellar catalysis upon addition from SDS. 1-Octene (1h) gave only trace amounts of product under these conditions, while no conversion was found when using electron poor acrylate substrates 1i and 1j (Table 2, entries 16–19). Notably, the reaction of 1j in the absence of SDS gave up to 35% 3j while to our surprise only trace amounts of 3j were obtained using 20 mM SDS (Table 2 entries 18 and 19). As the starting material was not recovered at the end of the reaction it is likely that 1j, being an activated α,β-unsaturated amino acid, polymerized under these reaction conditions.
Reactions of 1a with diazo acetates 2b and 2c to obtain cyclopropane products 3k and 3l showed significant acceleration by the addition of 20 mM SDS, obtaining 3k and 3l in 80% and 97% yield respectively (Table 2 entries 20–23).
Conclusions
We have reported the micelle accelerated cationic iron porphyrin catalyzed cyclopropanation. Using commercially available surfactants and catalysts this facile, yet effective approach has proven to be an efficient means to accelerate the cyclopropanation of styrene derivatives with a range of diazo acetates, while reducing the amount of side product formation. These results also suggest that effective molarity effects resulting from concentration of reagents in hydrophobic cavities is an important contributor to the rate accelerations observed in cyclopropanations catalysed by repurposed and artificial heme enzymes, as well as DNA-based catalysis. The simple, yet effective method is an attractive and cost-effective approach to the catalysis of cyclopropanation reactions, especially when enantioselectivity is not required, and further illustrates the power of micellar catalysis.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
Financial support from the Netherlands Ministry of Education, Culture and Science (Gravitation program no. 024.001.0035), the European Commission (Erasmus+ F PARIS005) and the Netherlands Organisation for Scientific Research (NWO, Vici grant 724.013.003) is gratefully acknowledged.Notes and references
- I. Nicolas, P. Le Maux and G. Simonneaux, Coord. Chem. Rev., 2008, 252, 727–735 CrossRef CAS.
- B. Morandi and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 938–941 CrossRef CAS.
- B. Morandi and E. M. Carreira, Science, 2012, 335, 1471–1474 CrossRef CAS.
- P. S. Coelho, Z. J. Wang, M. E. Ener, S. A. Baril, A. Kannan, F. H. Arnold and E. M. Brustad, Nat. Chem. Biol., 2013, 9, 485–487 CrossRef CAS.
- P. S. Coelho, E. M. Brustad, A. Kannan and F. H. Arnold, Science, 2013, 339, 307–310 CrossRef CAS.
- M. Bordeaux, V. Tyagi and R. Fasan, Angew. Chem., Int. Ed., 2015, 54, 1744–1748 CrossRef CAS PubMed.
- O. F. Brandenberg, R. Fasan and F. H. Arnold, Curr. Opin. Biotechnol., 2017, 47, 102–111 CrossRef CAS.
- L. Villarino, K. E. Splan, E. Reddem, L. Alonso-Cotchico, C. Gutiérrez de Souza, A. Lledós, J.-D. Maréchal, A. M. W. H. Thunnissen and G. Roelfes, Angew. Chem., Int. Ed., 2018, 57, 7785–7789 CrossRef PubMed.
- G. Sreenilayam, E. J. Moore, V. Steck and R. Fasan, ACS Catal., 2017, 7, 7629–7633 CrossRef CAS PubMed.
- K. Oohora, H. Meichin, L. Zhao, M. W. Wolf, A. Nakayama, J. Hasegawa, N. Lehnert and T. Hayashi, J. Am. Chem. Soc., 2017, 139, 17265–17268 CrossRef CAS PubMed.
- A. Rioz-Martínez, J. Oelerich, N. Ségaud and G. Roelfes, Angew. Chem., Int. Ed., 2016, 55, 14136–14140 CrossRef.
- T. Dwars, E. Paetzold and G. Oehme, Angew. Chem., Int. Ed., 2005, 44, 7174–7199 CrossRef CAS.
- G. L. Sorella, G. Strukul and A. Scarso, Green Chem., 2015, 17, 644–683 RSC.
- S. Kobayashi, T. Wakabayashi, S. Nagayama and H. Oyamada, Tetrahedron Lett., 1997, 38, 4559–4562 CrossRef CAS.
- S. Kobayashi and T. Wakabayashi, Tetrahedron Lett., 1998, 39, 5389–5392 CrossRef CAS.
- S. Otto, J. B. F. N. Engberts and J. C. T. Kwak, J. Am. Chem. Soc., 1998, 120, 9517–9525 CrossRef CAS.
- F. Rosati, J. Oelerich and G. Roelfes, Chem. Commun., 2010, 46, 7804–7806 RSC.
- B. H. Lipshutz and S. Ghorai, Aldrichimica Acta, 2012, 45, 3–16 CAS.
- N. A. Isley, Y. Wang, F. Gallou, S. Handa, D. H. Aue and B. H. Lipshutz, ACS Catal., 2017, 7, 8331–8337 CrossRef CAS.
- T. Omagari, A. Suzuki, M. Akita and M. Yoshizawa, J. Am. Chem. Soc., 2016, 138, 499–502 CrossRef CAS PubMed.
- D. Monti, P. Tagliatesta, G. Mancini and T. Boschi, Angew. Chem., Int. Ed., 1998, 37, 1131–1133 CrossRef CAS PubMed.
- L. J. P. van den Broeke, V. G. de Bruijn, J. H. M. Heijnen and J. T. F. Keurentjes, Ind. Eng. Chem. Res., 2001, 40, 5240–5245 CrossRef CAS.
- Z. Király and I. Dekány, J. Colloid Interface Sci., 2001, 242, 214–219 CrossRef.
- B. D. Flockhart, J. Colloid Sci., 1961, 16, 484–492 CrossRef CAS.
- L. Shedlovsky, C. W. Jakob and M. B. Epstein, J. Phys. Chem., 1963, 67, 2075–2078 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional catalysis data, full experimental details and characterisation of compounds. See DOI: 10.1039/c9ob02223f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |