
Head-to-head comparison of LNA, MPγPNA, INA and Invader probes targeting mixed-sequence double-stranded DNA†
Raymond G.
Emehiser
,
Eric
Hall
,
Dale C.
Guenther
,
Saswata
Karmakar
and
Patrick J.
Hrdlicka
 *
*
Department of Chemistry, University of Idaho, Moscow, ID-83844, USA. E-mail: hrdlicka@uidaho.edu
First published on 25th October 2019
Abstract
Four probe chemistries are characterized and compared with respect to thermal denaturation temperatures (Tms), thermodynamic parameters associated with duplex formation, and recognition of mixed-sequence double-stranded (ds) DNA targets: (i) oligodeoxyribonucleotides (ONs) modified with Locked Nucleic Acid (LNA) monomers, (ii) MPγPNAs, i.e., single-stranded peptide nucleic acid (PNA) probes that are functionalized at the γ-position with (R)-diethylene glycol (mini-PEG, MP) moieties, (iii) Invader probes, i.e., DNA duplexes modified with +1 interstrand zipper arrangements of 2′-O-(pyren-1-yl)methyl-RNA monomers, and (iv) intercalating nucleic acids (INAs), i.e., DNA duplexes with opposing insertions of 1-O-(1-pyrenylmethyl)glycerol bulges. Invader and INA probes, which are designed to violate the nearest-neighbor exclusion principle, denature readily, whereas the individual probe strands display exceptionally high affinity towards complementary DNA (cDNA) as indicated by increases in Tms of up to 8 °C per modification. Optimized Invader and INA probes enable efficient and highly specific recognition of mixed-sequence dsDNA targets with self-complementary regions (C50 = 30–50 nM), whereas recognition is less efficient with LNA-modified ONs and fully modified MPγPNAs due to lower cDNA affinity (LNA) and a proclivity for dimerization (LNA and MPγPNA). A Cy3-labeled Invader probe is shown to stain telomeric DNA of individual chromosomes in metaphasic spreads under non-denaturing conditions with excellent specificity.
Introduction
DNA-targeting probes are increasingly used to regulate gene expression, detect diagnostically relevant targets, and edit disease-causing mutations. However, most contemporary probe strategies face technological limitations, which has impeded progress in the life sciences and medical fields. For example, while the recent discovery of CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-associated (Cas) endonucleases1 has generated tremendous excitement, future efforts will need to focus on reducing off-target editing activities, increasing the targeting scope, and improving the efficiency of cellular delivery to realize the full potential of CRISPR/Cas.2 Prior to the advent of CRISPR/Cas, minor groove binding polyamides and triplex-forming oligonucleotides and peptide nucleic acids (PNAs) were the most extensively studied DNA-targeting probes.3–5 Their binding is generally restricted to short mixed-sequence and extended polypurine segments, respectively, thus limiting their targeting scope. Alternative strategies,6–10 including locked nucleic acids (LNAs),11 (R)-miniPEG-γPNAs (MPγPNA),12 Invader probes,13,14 and intercalating nucleic acids (INAs)15 have, accordingly, been developed to enable sequence-unrestricted recognition of double-stranded DNA (dsDNA) (Fig. 1). However, these probes have – for the most part – been restricted to a few laboratories and have not been compared. This is unfortunate, since the outcome from a head-to-head study could provide much-needed information about the relative dsDNA-targeting capabilities of these probes and inform future development efforts. The present study addresses this need by evaluating LNA, MPγPNA, INA, and Invader probes in a common model system.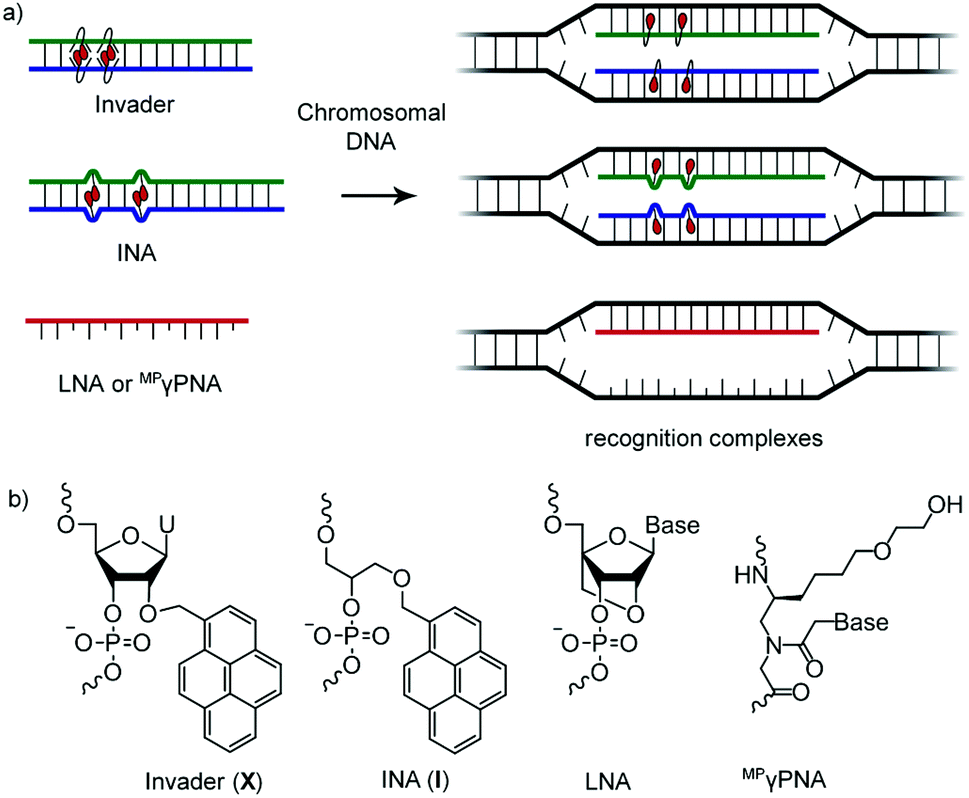 | ||
| Fig. 1 (a) Illustration of dsDNA-recognition mechanisms with Invader, INA, LNA, and MPγPNA probes. Droplets denote intercalators. (b) Structures of monomers used herein. | ||
LNAs, whilst primarily known for their RNA-targeting properties,16 have been shown to target mixed-sequence dsDNA at conditions in which the target regions appear to be unusually accessible.17–20 Along similar lines, MPγPNAs have been shown to recognize mixed-sequence genomic DNA in a β-globin/eGFP mouse model and induce targeted in vivo gene editing.21 The driving force for LNA- and MPγPNA-mediated dsDNA-recognition is linked to their high affinity for complementary DNA (cDNA). The C3′-endo restricted LNA monomers increase the North-type character of neighboring 2′-deoxyribonucleotides, whereby duplexes with cDNA adopt more compact and stable RNA-like helix geometries.22 MiniPEG-γPNAs owe their increased cDNA-affinity to favorable strand pre-organization, which is mediated by the chirality-inducing γ-substituent, which also increases aqueous solubility, thus reducing aggregation.12 In addition, the neutral backbone of MPγPNA reduces electrostatic repulsion in duplexes with cDNA, thus further facilitating DNA duplex invasion.
Invader probes, which were developed by us,13,14 are short DNA duplexes featuring one or more +1 interstrand zipper arrangements of intercalator-modified nucleotides like 2′-O-(pyren-1-yl)methyl-RNA (Fig. 1). The unique probe architecture forces two intercalating pyrene moieties to compete for the same inter-base-pair region, resulting in localized unwinding and probe destabilization23,24 as the nearest-neighbor exclusion principle25 – which asserts that intercalation is anti-cooperative at adjacent sites26 – is violated. In contrast, the two individual probe strands comprising an Invader probe, display very high affinity toward cDNA as duplex formation results in strongly stabilizing stacking interactions between the intercalators and flanking base-pairs.13,14,23,24 The greater stability of the duplexes between individual probe strands and cDNA vis-à-vis the Invader duplex and dsDNA target region, provides the driving force for recognition of complementary mixed-sequence dsDNA regions. Invader probes have been shown to recognize mixed-sequence target regions on Y-chromosomes in interphase and metaphase nuclei from male bovine kidney cells under non-denaturing conditions.14 INAs15 are based on a related principle with non-nucleotidic 1-O-(1-pyrenylmethyl)glycerol monomers placed opposite of each other, resulting in a partially unwound and destabilized probe duplex,27 in which the individual probe strands have high cDNA affinity.
Results and discussion
Design and synthesis of probes
To facilitate direct comparison and aid future DNA-targeting studies, we designed a series of Invader, INA, LNA, and MPγPNA probes against a 13-mer mixed-sequence dsDNA segment with an AT-rich central region that we have used routinely14 to evaluate Invader probes (Table 1). The self-complementary nature of the central region represents a challenging target for single-stranded high-affinity probes like LNA and MPγPNA probes as they are prone to intramolecular hairpin formation and/or dimerization which, in turn, may interfere with dsDNA-binding. Invader and INA probes, on the other hand, are engineered to form predictable and easily denaturing duplexes, irrespective of the sequence context of the target region, and are, therefore, expected to be less affected by partially self-complementary segments.| T m [ΔTm] (°C) | ||||
|---|---|---|---|---|
| Probe | Sequence | Probe duplex | Upper strand vs. cDNA | Lower strand vs. cDNA |
| a Invader, INA, and LNA strands are modified oligodeoxyribonucleotides (ONs). LNA monomers are denoted by lower case letters (“c” = 5-methylcytosine LNA monomer). ΔTm = change in Tm relative to the unmodified DNA duplex, Tm (5′-GGT ATA TAT AGG C:3′-CCA TAT ATA TCC G) = 37.5 °C. Buffer composition: [Na+] = 110 mM, [Cl−] = 100 mM, pH 7.0 (NaH2PO4/Na2HPO4) using 1.0 μM of each strand. | ||||
| INV3 | 5′-GG![[X with combining low line]](https://www.rsc.org/images/entities/b_char_0058_0332.gif) AA TA AGG C AA TA AGG C |
51.0 [+13.5] | 61.0 [+23.5] | 63.0 [+25.5] |
| 3′-CCA A ATA CC G |
||||
| INV4 | 5′-GG ATA A AGG C |
52.0 [+14.5] | 61.5 [+24.0] | 62.5 [+25.0] |
| 3′-CCA AT AA CC G |
||||
| INA3 | 5′-GGT ![[I with combining low line]](https://www.rsc.org/images/entities/b_char_0049_0332.gif) AT AT AT AGG C AT AT AT AGG C |
49.5 [+12.0] | 61.5 [+24.0] | 61.5 [+24.0] |
| 3′-CCA TA TA TA TCC G |
||||
| INA4 | 5′-GGT AT AT AT AGG C |
51.0 [+13.5] | 62.0 [+24.5] | 63.0 [+25.5] |
| 3′-CCA TA TA TA TCC G |
||||
| LNA1 | 5′-GgT AtA TaT AgG C | — | 54.0 [+16.5] | — |
| LNA2 | 3′-CCa TAt Ata TCc G | — | — | 55.0 [+17.5] |
| MP γPNA1 | H-Lys-GGT ATA TAT AGG C-Lys-NH2 | — | >85 [>47.5] | — |
| MP γPNA2 | NH2-Lys-CCA TAT ATA TCC G-Lys-H | — | — | 79.0 [+41.5] |
Invader, INA, LNA, and MPγPNA probes were synthesized and purified using established methods (see Experimental section). Preliminary studies – in which four Invader and four INA probes with varying levels and patterns of modification, were evaluated – revealed that probes with three energetic hotspots are particularly well-suited for dsDNA-recognition, striking a favorable balance between high binding affinity and excellent binding specificity (for full details of the preliminary study, see ESI†). These Invader and INA designs were, therefore, used for comparison with LNA and MPγPNA probes.
Thermal denaturation properties of probes
Thermal denaturation temperatures (Tms) were determined for probe duplexes and duplexes between individual probe strands and cDNA (Table 1). As expected,14,23,24 individual Invader probe strands display exceptionally high affinity for cDNA as evidenced by marked increases in Tms of the resulting duplexes vis-à-vis DNA reference duplexes (ΔTm = 23.5–25.5 °C; ΔTm/modification (mod) ∼8 °C). Conversely, double-stranded Invader probes denature at significantly lower temperatures (ΔTm = 13.5–14.5 °C; ΔTm/mod < 2.5 °C). INA probes display near-identical denaturation properties (e.g., compare Tms for duplexes involving INV3 and INA3). Also as expected,11,12 LNA-modified DNA (∼31% LNA content; mixmer design) and fully modified MPγPNA strands form very stable duplexes with cDNA (LNA: ΔTm ∼17 °C and ΔTm/mod ∼4.3 °C; MPγPNA: ΔTm ≥ 41.5 °C and ΔTm/mod ≥ 3.2 °C).Thermodynamic driving force for recognition of complementary dsDNA targets
The available free energy for recognition of complementary dsDNA targets can be parameterized as ΔG310rec = ΔG310 (upper probe vs. cDNA) + ΔG310 (lower probe vs. cDNA) – ΔG310 (probe) − ΔG310 (dsDNA) (see Experimental section for full details). Thermodynamic parameters associated with duplex formation were determined through baseline fitting of denaturation curves assuming bimolecular reactions, two-state melting behavior, and constant heat capacity (ΔG310 and ΔG310rec values are shown in Table 2; for additional thermodynamic parameters, see Tables S3–S5, S7 and S8†).28 Invader and INA probes are strongly activated for dsDNA-recognition as evidenced by highly negative ΔG310rec values (i.e., ΔG310rec between −73 kJ mol−1 and −56 kJ mol−1) which is a manifestation of relatively labile probe duplexes (i.e., ΔΔG310 between −12 and −7 kJ mol−1) and highly stable probe–target duplexes (i.e., ΔΔG310 between −49 and −29 and kJ mol−1). For single-stranded LNA and MPγPNA probes, the ΔG310rec term reduces to ΔG310rec = ΔG310 (probe vs. cDNA) − ΔG310 (dsDNA), which simply is the ΔΔG310 value for a given probe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cDNA duplex. Comparison of ΔG310rec values suggests that the available thermodynamic driving force for dsDNA-recognition is similar for MPγPNA, INA and Invader probes, but far less prominent for LNA-modified ONs (Table 2). It should be noted that the driving force for single-stranded MPγPNA and LNA probes is expected to be further decreased if secondary structures are adopted since melting thereof must be overcome as part of a dsDNA-recognition process. Indeed, these probes appear to be dimerizing (vide infra, ΔΔG310 = −11 and −12 kJ mol−1 for MPγPNA1 and MPγPNA2, respectively, Table S9†).
:cDNA duplex. Comparison of ΔG310rec values suggests that the available thermodynamic driving force for dsDNA-recognition is similar for MPγPNA, INA and Invader probes, but far less prominent for LNA-modified ONs (Table 2). It should be noted that the driving force for single-stranded MPγPNA and LNA probes is expected to be further decreased if secondary structures are adopted since melting thereof must be overcome as part of a dsDNA-recognition process. Indeed, these probes appear to be dimerizing (vide infra, ΔΔG310 = −11 and −12 kJ mol−1 for MPγPNA1 and MPγPNA2, respectively, Table S9†).
| ΔG310[ΔΔG310] (kJ mol−1) | |||||
|---|---|---|---|---|---|
| Probe | Sequence | Probe | Upper strand vs. cDNA | Lower strand vs. cDNA | ΔG310rec (kJ mol−1) |
| a ΔΔG310 is measured relative to the corresponding unmodified DNA duplex (ΔG = −42 kJ mol−1). For definition of ΔG310rec, see main text. nt = no transition; nd = not determined due to unclear upper baseline. | |||||
| INV3 | 5′-GG AA TA AGG C |
−53 [−11] | −78 [−36] | −78 [−37] | −62 |
| 3′-CCA A ATA CC G |
|||||
| INV4 | 5′-GG ATA A AGG C |
−53 [−12] | −80 [−38] | −76 [−34] | −60 |
| 3′-CCA AT AA CC G |
|||||
| INA3 | 5′-GGT AT AT AT AGG C |
−49 [−7] | −71 [−29] | −75 [−34] | −56 |
| 3′-CCA TA TA TA TCC G |
|||||
| INA4 | 5′-GGT AT AT AT AGG C |
−54 [−12] | −77 [−36] | −91 [−49] | −73 |
| 3′-CCA TA TA TA TCC G |
|||||
| LNA1 | 5′-GgT AtA TAt AgG C | — | −63 [−21] | — | −21 |
| LNA2 | 3′-CCa Tat Ata TCc G | — | — | −68 [−26] | −26 |
| MP γPNA1 | H-Lys-GGT ATA TAT AGG C-Lys-NH2 | — | nt | — | nd |
| MP γPNA2 | NH2-Lys-CCA TAT ATA TCC G-Lys-H | — | — | −109 [−67] | −67 |
The thermodynamic driving force for Invader- and INA-mediated dsDNA-recognition is strongly enthalpically favored (ΔHrec between −386 kJ mol−1 and −204 kJ mol−1, Table S4†), which is a manifestation of enthalpically destabilized probe duplexes (ΔΔH between +71 kJ mol−1 and +100 kJ mol−1, Table S4†) and enthalpically stabilized duplexes between individual probe strands and cDNA (ΔΔH between −212 kJ mol−1 to −35 kJ mol−1, Table S4†). These trends are in line with expectations inasmuch the high intercalator density of Invader and INA probe duplexes results in a violation of the nearest-neighbor exclusion principle25,26 and localized helix perturbation, whereas duplexes between individual probe strands and cDNA are stabilized due to strong stacking interactions between the intercalators and flanking base-pairs as the intercalator density is reduced and the nearest-neighbor exclusion principle no longer is violated. The favorable change in enthalpy upon Invader- or INA-mediated recognition of dsDNA is partly offset by a highly unfavourable decrease in entropy (i.e., −TΔS310rec > 148 kJ mol−1, Table S5†).
The stabilization of LNA:cDNA and MPγPNA:cDNA duplexes – and, thus, the driving force for LNA- and MPγPNA-mediated dsDNA-recognition – is also due to favorable enthalpic factors (Table S7†) that are partly offset by unfavorable decreases in entropy (Table S8†).
Recognition of model mixed-sequence dsDNA targets – design and initial screen
The ability of Invader, INA, LNA, and MPγPNA probes to recognize mixed-sequence dsDNA targets was evaluated using a previously developed electrophoretic mobility shift assay,14 which utilizes a digoxigenin (DIG)-labelled DNA hairpin (DH) as a model target (Fig. 2). DH1 is comprised of the 13-mer double-stranded target segment and a T10 loop that links the two stem strands, resulting in a high-melting hairpin (Tm = 58.5 °C, Table S10†). Successful recognition of DH1 is expected to result in the formation of binary and ternary complexes (with LNA/MPγPNA and Invader/INA probes, respectively) with reduced mobility in non-denaturing polyacrylamide gel electrophoresis (nd-PAGE) relative to DH1. The initial screen was performed using 5-fold molar probe excess and an incubation temperature of 37 °C (Fig. 3). Invader and INA probes display efficient recognition of DH1 (64–77%), whereas recognition appears to be slightly less efficient with MPγPNA1 (59%). Moderate levels of recognition were observed with LNA1 (27%), whereas MPγPNA2 and LNA2 only result in trace recognition (<15%). | ||
| Fig. 2 Illustration of assay used to evaluate dsDNA-recognition of various probes. | ||
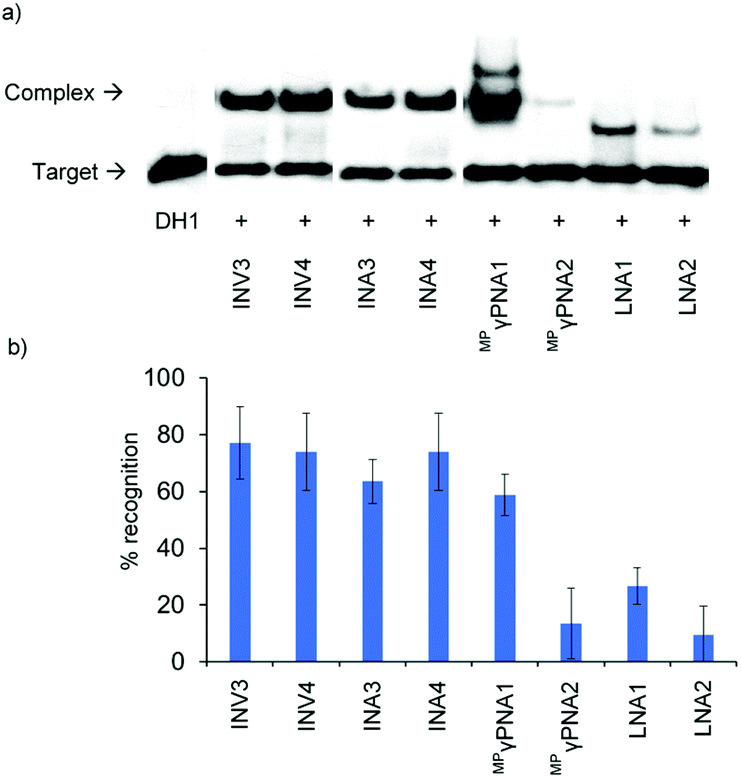 | ||
| Fig. 3 (a) Representative gel electrophoretograms from recognition experiments between DH1 and a 5-fold molar excess of Invader, INA, LNA or MPγPNA probes. (b) Histograms depict averaged results from at least three experiments with error bars representing standard deviation. DIG-labeled DH1 (34.4 nM, 5′-GGTATATATAGGC-T10-GCCTATATATACC-3′) was incubated with the specified probe in HEPES buffer (50 mM HEPES, 100 mM NaCl, 5 mM MgCl2, pH 7.2, 10% sucrose, 1.44 mM spermine tetrahyrdochloride) for 2.5 h at 37 °C. Lanes have been rearranged to facilitate presentation but are from the same electropherogram. Both recognition bands formed were considered in the quantification for MPγPNA1. | ||
Secondary structure formation of LNA and MPγPNA probes
The low levels of DH1-recognition observed with the LNA probes and the surprising discrepancy between MPγPNA1 and MPγPNA2, prompted us to conduct additional thermal denaturation experiments for these probes in absence of cDNA to establish if they undergo partial self-hybridization. Indeed, probe concentration-dependent thermal denaturation trends, consistent with dimerization, were observed for the LNA and MPγPNA probes (Fig. S6†), with Tms of 27–29 °C and 57.5–62.0 °C, respectively, at 1 μM probe concentration (Table S9†). The higher cDNA-affinity of MPγPNA1 – along with an additional binding mode (discussed below) – likely underlies the more effective recognition of DH1vis-à-visMPγPNA2.Formation of unexpected recognition complexes with MPγPNA1
Interestingly, two distinct recognition bands are consistently observed when using MPγPNA1, i.e., a band with similar electrophoretic mobility as ternary recognition complexes between DH1 and Invader or INA probes, and a band with lower relative mobility (Fig. 3). To explore if the slow-moving band corresponds to a ternary complex involving the non-target stem arm of DH1, MPγPNA1 or MPγPNA2 were annealed with 36-mer DIG-labeled single-stranded DNA targets. The targets were designed to have the same length as DH1, comprising three segments, i.e., a central T10 region, either a 13 nt complementary or isosequential arm relative to the MPγPNA probes, and a 13 nt scrambled arm (Fig. 4). As expected, duplex formation was observed between ON1 and MPγPNA2 and between ON2 and MPγPNA1 as a complementary duplex spanning the length of the MPγPNA probe is formed (lanes 1 and 7). Also as expected, there is no evidence of duplex formation between ON2 and MPγPNA2 (lane 6). Surprisingly, ON1 and MPγPNA1 form a double-stranded structure (lane 2), which might reflect formation of an antiparallel 8-mer duplex across the AT-rich region of the probe and isosequential DNA with 2 and 3 nt single-stranded overhangs (see Fig. 4 for illustration), or formation of a parallel 7-mer duplex across the AT-rich region with single-stranded overhangs that are 2 and 4 nt long. While conventional PNAs readily form duplexes with cDNA in parallel orientation, in addition to the preferred antiparallel orientation,29 γ-modified PNAs are far less likely to bind to DNA in parallel orientation.30 This indicates that the slower-moving band formed when MPγPNA1 is incubated with DH1, stems from a ternary complex in which two MPγPNA1 strands are bound to DH1, both binding with anti-parallel orientation, one to the fully complementary 13-mer region and the other to the partially complementary 8-mer region. The difference in binding behavior between MPγPNA1 and MPγPNA2 is not fully understood but may be a result of the lower cDNA-affinity of MPγPNA2 (Table 1). The unintended ternary complex formation seen with MPγPNA1 and the low dsDNA-recognition efficiency of MPγPNA2 serve as a reminder that target regions with high levels of self-complementarity are challenging for single-stranded high-affinity probes.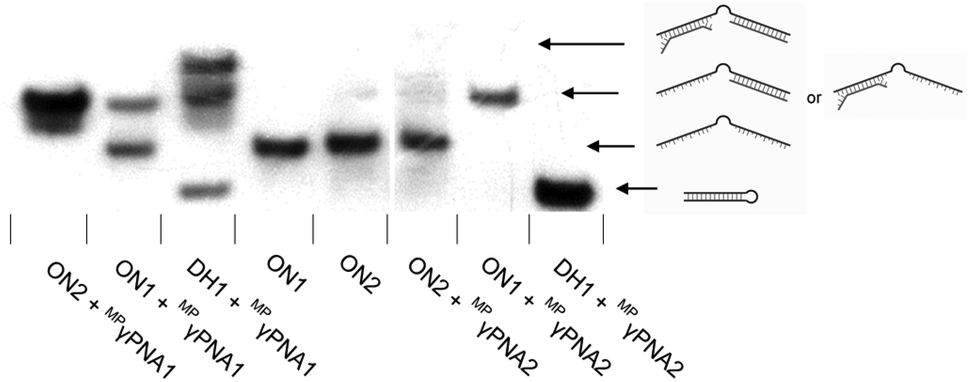 | ||
Fig. 4 Representative electrophoretograms for recognition of DIG-labelled DH1, ON1, or ON2 (34.4 mM) using a 5-fold excess of different probes under annealing conditions. Probes and DNA targets were heated to 80 °C for 2 min, followed by cooling to room temperature over 30 min, and non-denaturing PAGE as described in Fig. 3. ON1: 5′-![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) ![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) ![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) -T10-AACTGATCTACTC; ON2: 5′-GAGTAGATCAG-T10-. Regions complementary to MPγPNA1 (ON2) and MPγPNA2 (ON1) are underlined. -T10-AACTGATCTACTC; ON2: 5′-GAGTAGATCAG-T10-. Regions complementary to MPγPNA1 (ON2) and MPγPNA2 (ON1) are underlined. | ||
Recognition of model mixed-sequence dsDNA targets – dose–response, time-course and binding specificity studies
Dose–response experiments were performed for Invader, INA, and MPγPNA probes to determine C50 values, i.e., the probe concentrations resulting in 50% recognition of DH1 (Fig. 5 and Fig. S13 and S15†). Optimized Invader probes display C50 values in the 40–50 nM range, whereas the corresponding INA probes display minimally lower C50 values (30–40 nM). The C50 value for MPγPNA1 is nearly an order of magnitude higher (Table 3).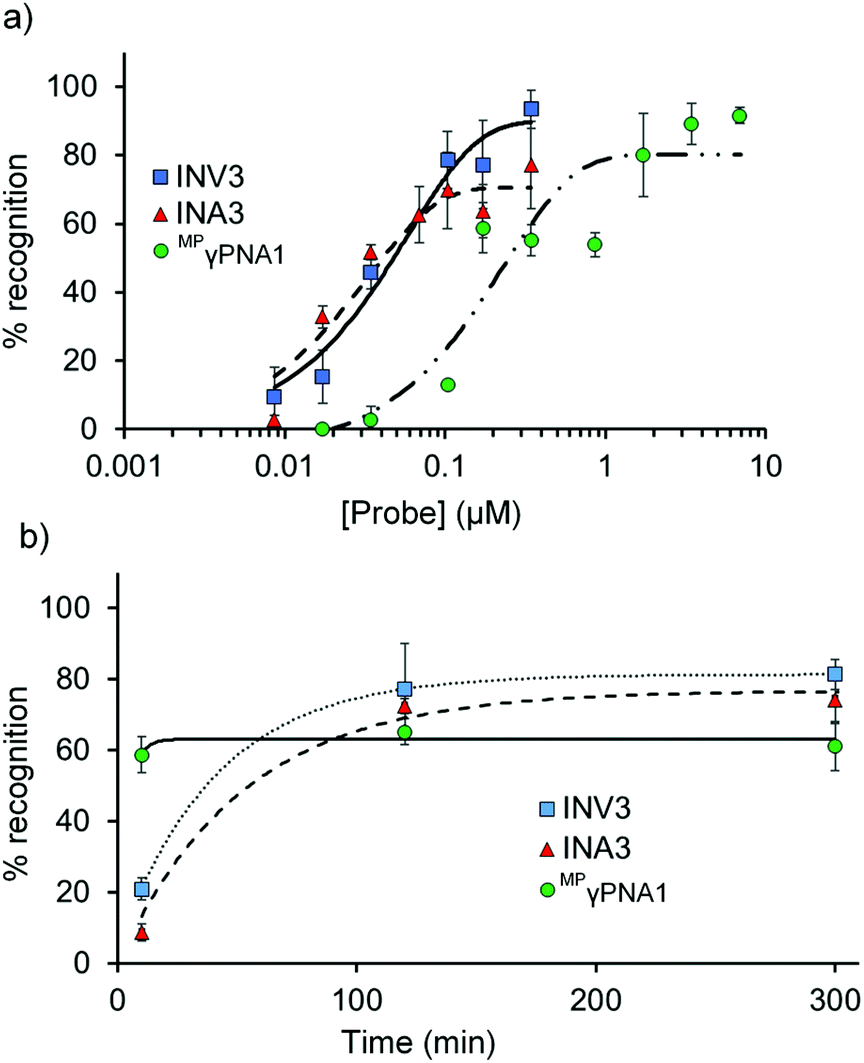 | ||
| Fig. 5 (a) Dose–response and (b) time-course relationships for DH1 recognition using Invader, INA, and MPγPNA probes. Except for variable (a) probe concentration or (b) incubation time, conditions are as described in Fig. 3. Corresponding curves for INV4 and INA4 are shown in Fig. S15.† Bars denote standard deviation. | ||
The dsDNA-recognition kinetics were studied using a 5-fold probe excess relative to DH1 (Fig. 5 and Fig. S14 and S15†). Interestingly, dsDNA-recognition by MPγPNA1 is rapid (50% recognition, t50 < 10 min), whereas recognition using Invader and INA probes is slower (37–68 min and 55–75 min, respectively; Fig. 5 and Table 3). This likely reflects the different recognition mechanisms utilized by MPγPNA and Invader/INA probes, i.e., single- vs. double-duplex invasion (Fig. 1), respectively, with the latter expected to require partial melting of the probes to reveal nucleation sites that initiate formation of recognition complexes with DH1. Along these lines, it is interesting to note that incubation of DH1 with a 100-fold molar excess of optimized Invader or INA probes at room temperature – i.e., conditions at which the probes (Tm = 49.5–52.0 °C, Table 1) and DH1 (Tm = 58.5 °C, Table S10†) display minimal denaturation – results in significant recognition of the target regions (72–92% and 66–75%, respectively, Fig. S9†).
Next, the binding specificity of the Invader, INA, and MPγPNA probes was evaluated. Probes were incubated with DIG-labelled DNA hairpins DH2–DH7, which have fully base-paired stems that differ in sequence at either the 6 or 9 position vis-à-vis the probes (Fig. 6). MPγPNA1 displays minimal discrimination of these singly mismatched DNA targets when used at 5-fold molar excess. This contrasts previous observations in which MPγPNA probes were shown to discriminate a singly mismatched mixed-sequence region located centrally in a 291 bp dsDNA target.12
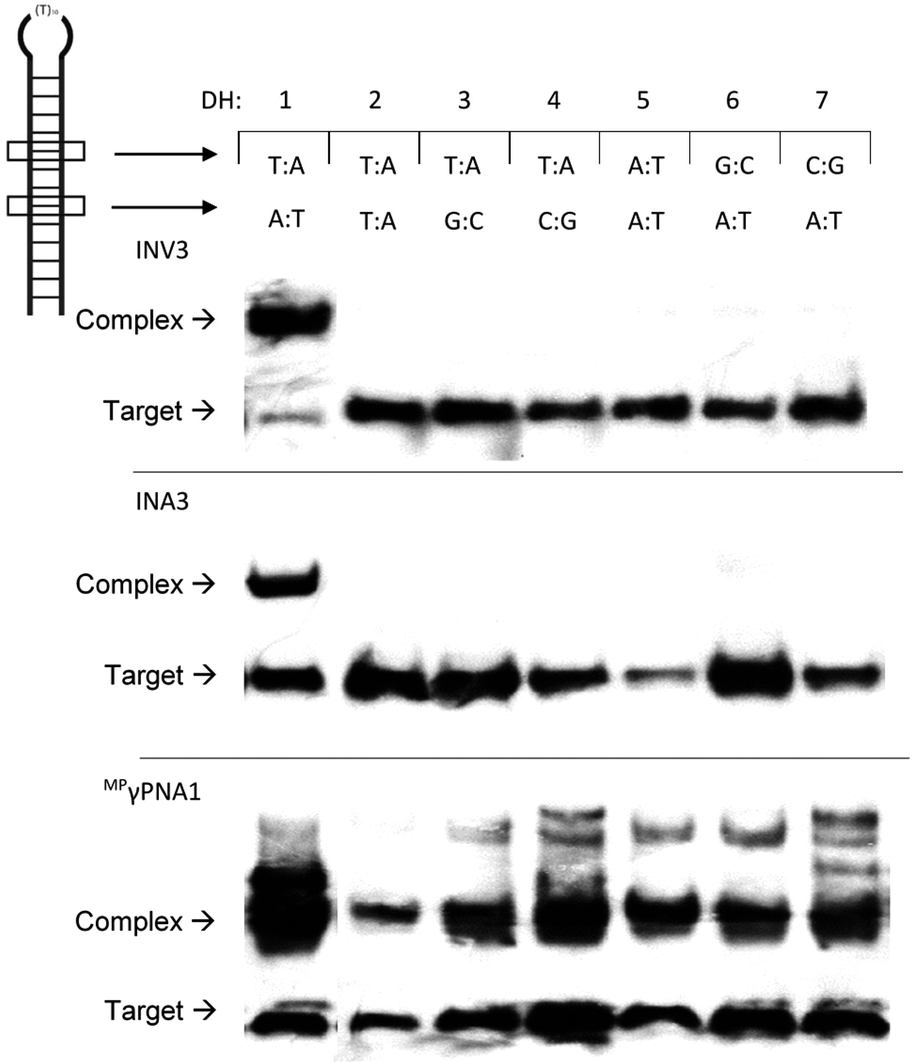 | ||
| Fig. 6 Binding specificity of Invader, INA and MPγPNA probes against non-complementary model targets DH2–DH7. Representative electrophoretograms from experiments in which a 5-fold molar excess of INV3, INA3, or MPγPNA1 was incubated with DH1–DH7 (34.4 nM) at 37 °C. Experimental conditions are as described in Fig. 3. For sequences and Tms of DH1–DH7, see Table S10.† Lanes have been rearranged to facilitate presentation but are from the same electropherogram. | ||
Different assays, incubation conditions, and target sequence lengths and contexts, likely account for these contrasting observations. Conversely, near-perfect discrimination of the singly mismatched targets is observed when a 5-fold excess of INV3 or INA3 is used (Fig. 6). In fact, INV3 and INA3 display reasonable discrimination of the singly mismatched targets even when used at a 75-fold molar excess (20–40% and 0–35% recognition, respectively, Fig. S16†). Presumably, double-stranded Invader and INA probes display improved discrimination of mismatched dsDNA targets due to stringency clamping effects,31 and because binding to DH2–DH7 would require the formation of recognition complexes with two energetically destabilized mismatched duplexes.32
Staining of telomeric DNA under non-denaturing conditions
Encouraged by the efficient and specific dsDNA-recognition displayed by optimized Invader probes, we set out to evaluate their ability to target telomeric DNA. Telomers are guanosine-rich tandem repeats (5′-TTAGGG-3′ in vertebrates) that cap the termini of chromosomes. Recently, there has been considerable interest in developing fluorescence-based diagnostic tools that enable interrogation of telomers33–37 due to their roles in aging, age-associated diseases, and cancer.38,39 Thus, shortening of telomers – a normal occurrence during cell replication – is linked to cellular senescence. In addition, telomerase, the enzyme that compensates for telomer attrition in stem cells through addition of TTAGGG repeats, is over-expressed in ∼85% of cancer cells. Modalities that stabilize telomers and/or prevent telomerase from binding and elongating telomers have been proposed as potential anticancer agents.40To this end, we designed a 16-mer Cy3-labeled Invader probe with three +1 interstrand zipper arrangements of X monomers (INV-T, Table 4). This probe displays the expected thermal denaturation properties, i.e., the probe duplex is less stable than the corresponding duplexes between individual probe strands and cDNA (Tm = 60.0 °C, 64.5 °C, and 66.0 °C), thus being activated for DNA-recognition as indicated by a TA value of 19.5 °C (Table 4).
| T m [ΔTm] (°C) | |||||
|---|---|---|---|---|---|
| Probe | Sequence | Probe | Upper strand vs. cDNA | Lower strand vs. cDNA | TA (°C) |
| a Each ON was used at 1.0 μM in Tm buffer. For experimental details, see Table 1. For definition of TA, see Experimentals. Tm for 5′-TTAGGGTTAGGGTTAG:3′-AATCCCAATCCCAATC = 51.0 °C. nt = no transition. nd = not determined. b Values are reported for the doubly mismatched duplexes with telomeric DNA target strands, i.e., 3′-AATCCCAATCCCAATC and 5′-TTAGGGTTAGGGTTAG for the upper and lower INV-T MM strands, respectively (mismatched nucleotides are italicized). | |||||
| INV-T | 5′-Cy3-TAGGGTAGGGTAG |
60.0 [+9.0] | 64.5 [+13.5] | 66.0 [+20.0] | +19.5 |
| 3′-AACCCAACCCAAC-Cy3 |
|||||
| INV-T MM | 5′-Cy3-TAGCGTAGCGTAG |
nt | 21.5 [−29.5]b | 48.5 [−2.5]b | nd |
| 3′-AACGCAACGCAAC-Cy3 |
|||||
Isolated nuclei from a male bovine kidney cell line were incubated with Invader probes under non-denaturing conditions (3 h, 37 °C, Tris-Cl/EDTA buffer at pH 8.0). Gratifyingly, incubation with the INV-T probe produces pairs of bright foci with limited background on most metaphasic chromosome termini consistent with binding to, and labelling of, telomeric DNA (Fig. 7a). Some termini are unlabeled, which might be due to loss of a telomere or telomeres that are too short to bind enough probes for detection. As expected, interphase nuclei are stained in a more erratic fashion, which is reflective of a less structured chromatin organization (Fig. 7b). Critically, incubation of isolated nuclei with INV-T MM, i.e., a 16-mer Invader probe that differs in sequence at one position relative to the telomeric repeat, does not result in labelling (Fig. 7c) since the individual probe strands display low affinity towards the telomeric DNA sequences (Table 4), which suggests that Invader-mediated binding to telomeric DNA is specific.
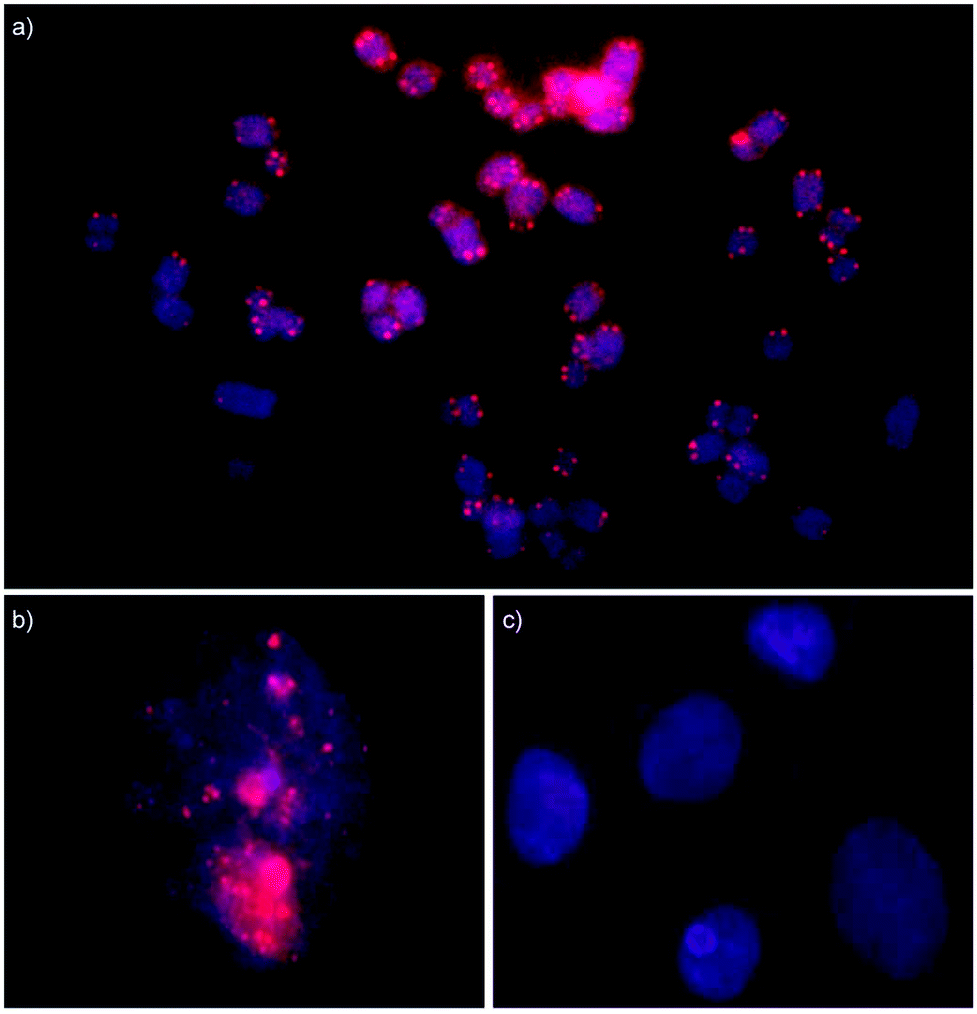 | ||
| Fig. 7 Fluorescence in situ hybridization experiments in which isolated nuclei from a bovine kidney cell line were incubated with Invader probes under non-denaturing conditions. Invader probe INV-T, which is fully complementary to the telomeric repeat, stains metaphase spreads (panel a) and interphase nuclei (panel b). Invader probe INV-T MM, which differs in sequence at one position relative to the telomeric repeat, does not stain interphase nuclei (panel c). For sequences of INV-T and INV-T MM, see Table 4. Images are overlays of images viewed through Cy3 (red) and DAPI channels (blue). Isolated nuclei were fixed using methanol and glacial acetic acid (3:1, v/v). Incubation conditions: 30 ng of Invader probes, 3 h at 37 °C in Tris-Cl pH 8.0 and 1 mM EDTA. | ||
The preliminary results seen with Invader-mediated staining of telomeric DNA are encouraging in view of existing approaches. Thus, the standard PNA-based fluorescence in situ hybridization (FISH) protocol used for staining of telomeric DNA requires denaturation of the chromosomal DNA prior to probe hybridization.37 Telomer-targeting MPγPNAs have been shown to effect efficient labelling, but still require denaturing conditions for optimal performance.33,34 Minor-groove binding polyamides have been used to stain telomers under non-denaturing conditions, although non-specific binding necessitated the use of tetrameric polyamides targeting 24 bp sequence segments that are comprised of four telomeric repeats for optimal results.35 The use of shorter Invader probes allows for localization of a higher density of fluorophores per length unit of telomere,33 which is expected to result in stronger signals. Thus, optimized Invader probes present themselves as promising agents for visualization of dsDNA in fixed and live cells.41
Conclusion
Optimized double-stranded Invader and INA probes allow for efficient and highly specific recognition of mixed-sequence dsDNA targets featuring self-complementary segments with minimal differences between the probe chemistries (C50 = 30–50 nM; t50 < 75 min; near-perfect discrimination of singly mismatched targets). In contrast, single-stranded LNA-modified ONs and fully modified MPγPNAs display less efficient and less specific recognition of these model targets, presumably due to their proclivity for dimerization. This serves as a reminder that targets with substantial self-complementarity should be avoided when using single-stranded high-affinity nucleic acid probes. In contrast, Invader and INA probes are designed to form predictable and easily denaturing double-stranded structures irrespective of sequence context and, therefore, do not exhibit similar limitations. The present findings call for additional comparative studies of Invader, INA, and MPγPNA probes across a wide range of sequence contexts, and for continued exploration of these mixed-sequence dsDNA-targeting probes for applications in molecular biology, biotechnology, and biomedicine, akin to staining of telomeric DNA as presented herein.Experimental section
Synthesis and purification of probe strands
Individual Invader and INA strands – i.e., ONs modified with 2′-O-(pyren-1-yl)methyl-uridine and (R)-1-O-(1-pyrenylmethyl)glycerol monomers, respectively – were synthesized on an automated DNA synthesizer (0.2 μmol scale) using solid support columns (long chain alkyl amine controlled pore glass (LCAA-CPG) with a pore size of 500 Å). Standard protocols were used for incorporation of DNA phosphoramidites. The corresponding Invader and INA phosphoramidites were prepared as previously described42,43 and incorporated into ONs via hand-couplings (∼45-fold molar excess, 0.02 M in anhydrous acetonitrile, using 0.01 M 4,5-dicyanoimidazole (15 min) as the activator) with extended oxidation (45 s) resulting in coupling yields of at least 85%. Cy3-labeling of Invader strands was accomplished by incorporating a commercially available Cy3 phosphoramidite (Glen Research) into ONs by hand-coupling (4,5-dicyanoimidazole, 3 min, anhydrous CH3CN). Treatment with 32% aq. ammonia (55 °C, 17 h) ensured deprotection and cleavage from solid support. DMT-protected ONs were purified via ion-pair reverse phase HPLC (XTerra MS C18 column: 0.05 M triethyl ammonium acetate and acetonitrile gradient) followed by detritylation (80% acetic acid, 20 min) and precipitation (NaOAc, NaClO4, acetone, −18 °C, 16 h). LNA-modified oligodeoxyribonucleotides and MPγPNA strands were synthesized and purified as previously described.44,45 The purity and identity of the synthesized ONs was verified using analytical HPLC (>85% purity) and MALDI-MS analysis recorded on a Quadrupole Time-of-Flight (Q-TOF) mass spectrometer using 3-hydoxypicolinic acid matrix (Table S1† and Fig. S1–S5†).Thermal denaturation experiments
Concentrations of Invader, INA and LNA strands were estimated using the following extinction coefficients (OD260/μmol): G (12.01), A (15.20), T (8.40), C (7.05), pyrene (22.4)46 and Cy3 (4.93).47 Extinction coefficients for MPγPNA were calculated as previously described.45 Thermal denaturation temperatures of duplexes (1.0 μM final concentration of each strand) were measured on a Cary 100 UV/VIS spectrophotometer equipped with a 12-cell Peltier temperature controller and determined as the maximum of the first derivative of the thermal denaturation curve (A260vs. T) recorded in medium salt buffer (Tm buffer: 100 mM NaCl, 0.2 mM EDTA, and pH 7.0 adjusted with 10 mM Na2HPO4 and 5 mM Na2HPO4) (for representative thermal denaturation curves, see Fig. S6 and S7†). Strands were mixed in quartz optical cells with a path length of 1.0 cm and annealed by heating to 85 °C (2 min), followed by cooling to the starting temperature of the experiment. The temperature of the denaturation experiments ranged from at least 15 °C below the Tm (though not below 15 °C) to 15 °C above the Tm (though not above 95 °C). A temperature ramp of 1.0 °C min−1 was used in all experiments, which resulted in minimal hysteresis (Tm values determined from heating and cooling cycles differed by less than 3 °C). Reported Tm values are averages of at least two experiments within ±1.0 °C.Determination of thermodynamic parameters
Thermodynamic parameters associated with duplex formation were determined through baseline fitting of denaturation curves (van't Hoff method) using software provided with the UV-Vis spectrophotometer. Bimolecular reactions, two-state melting behavior, and constant heat capacity are assumed.28 Two denaturation curves per duplex were analyzed at least three times to minimize errors arising from baseline choice.TA and ΔGrec terms
The driving force for recognition of complementary dsDNA targets using Invader and INA probes can be parameterized as ΔG310rec = ΔG310 (upper strand vs. cDNA) + ΔG310 (lower strand vs. cDNA) − ΔG310 (probe duplex) − ΔG310 (dsDNA) or, alternatively, by the term thermal advantage, which we define as TA = Tm (upper strand vs. cDNA) + Tm (lower strand vs. cDNA) − Tm (probe duplex) − Tm (dsDNA). It is important to note that the TA term expresses the difference in thermal denaturation temperatures of duplexes involved in Invader- or INA-mediated recognition of complementary isosequential dsDNA targets (e.g., 5′-GGTATATATAGGC:3′-CCATATATATCCG for the probes shown in Table 1). Equivalent considerations apply to ΔG310rec. Thus, neither TA nor ΔG310rec values are true measures of the thermodynamic driving force for recognition of model target DH1 or other complex targets harboring the target sequence (e.g., chromosomal DNA). However, we have found these terms to be useful in assessing the relative dsDNA-targeting potential of different probe designs.14 The calculations likely underestimate the stability of the dsDNA-component when complex targets are used (i.e., the Tms of such targets are higher than of the linear isosequential dsDNA target). The Tm (upper strand vs. cDNA) and Tm (lower strand vs. cDNA) values – as well as the equivalent ΔG310 values – are expected to remain approximately similar irrespective of the nature of the target, although potentially stabilizing capping effects from neighboring nucleotides are not accounted for. Consequently, the calculated TA and ΔG310rec values for Invader or INA probes, almost certainly, overestimate the driving force available for recognition of complex dsDNA targets.For single-stranded probes (e.g., LNA and MPγPNA), the TA term reduces to TA = Tm (probe strand vs. cDNA) – Tm (dsDNA), which is the ΔTm value for a probe:cDNA duplex. Similarly, the ΔG310rec term reduces to ΔG310rec = ΔG310 (probe vs. cDNA) − ΔG310 (dsDNA), which is the ΔΔG310 value for a given probe:cDNA duplex.
Electrophoretic mobility shift assay
The electrophoretic mobility shift assays were performed essentially as previously described.14 Thus, DNA hairpins (DH) were obtained from commercial sources and used without further purification. Hairpins were digoxigenin (DIG)-labeled using the 2nd generation DIG Gel Shift Kit (Roche Applied Bioscience) as recommended by the manufacturer. Briefly, 11-digoxigenin-ddUTP was incorporated at the 3′-end of the hairpin (100 pmol) using a recombinant terminal transferase. The reaction mixture was quenched through addition of EDTA (0.05 M), diluted to 68.8 nM, and used without further processing. Invader, INA, LNA or MPγPNA probes (concentration as specified) were heated (90 °C for 2 min) and cooled to room temperature, and subsequently incubated with the specified DIG-labeled DNA hairpin (final concentration 34.4 nM) in HEPES buffer (50 mM HEPES, 100 mM NaCl, 5 mM MgCl2, pH 7.2, 10% sucrose, 1.44 mM spermine tetrahydrochloride) at room temperature or 37 °C for the specified time.In experiments involving DIG-labelled ON1 and ON2, probes were incubated with DNA targets, and annealed by heating to 80 °C (2 min) and cooling to room temperature over 30 min.
In time-course experiments, aliquots were taken at specific time points, flash-frozen in liquid N2, and stored at −20 °C until subsequent analysis.
Following incubation, loading dye (6×) was added and the mixtures were loaded onto 12% non-denaturing TBE-PAGE slabs (45 mM tris-borate, 1 mM EDTA; acrylamide:bisacrylamide (19:1)). Electrophoresis was performed using constant voltage (∼70 V) at ∼4 °C for ∼2 h. Bands were subsequently blotted onto positively charged nylon membranes (∼100 V, 30 min, ∼4 °C) and cross-linked through exposure to UV (254 nm, 5 × 15 W bulbs, 5 min). Membranes were incubated with anti-digoxigenin-alkaline phosphatase Fab fragments as recommended by the manufacturer and transferred to a hybridization jacket. Membranes were incubated with the chemiluminescence substrate (CSPD) for 10 min at 37 °C, and chemiluminescence of the formed product was captured on X-ray films. Digital images of developed X-ray films were obtained using a BioRad ChemiDoc™ MP Imaging and used for densitometric quantification of the bands. The percentage of dsDNA recognition was calculated as the intensity ratio between the recognition complex band and unrecognized hairpin. Both recognition bands were considered when quantifying DH1-recognition for MPγPNA1. An average of three independent experiments is reported along with standard deviations (±). Electrophoretograms shown in the ESI† may be composite images of lanes from different runs. Non-linear regression was used to fit data points from dose–response experiments. A script written for the “Solver” module in Microsoft Office Excel,48 was used to fit data points from dose–response experiments to the following equation: y = C + A (1 − e−kt) where C, A and k are fitting constants. The resulting equation was used to calculate C50 values by setting y = 50 and solving for t.
Cell culture and nuclei preparation
Male bovine kidney cells (MDBK, ATCC: CCL-22, Bethesda, MD) were maintained in DMEM with GlutaMax (Gibco, 10569-010) and 10% fetal bovine serum (Invitrogen). The cells were cultured in separate 25 mL or 75 mL flasks at 38.5 °C in a 5% CO2 atmosphere for 24–72 h to achieve confluent growth. Following this, the medium was treated with colcemid for 2 h and then replaced with 0.25% trypsin-EDTA in DMEM. The cells were cultured for 20 min as stated above to detach adherent cells. The loosened cells were aspirated and transferred to 15 mL tubes and centrifuged for 4 min at 600g to pellet the cells. The supernatant was aspirated off, replaced with 75 mM KCl (hypotonic buffer), and the cells were incubated for 20 min at 37 °C. The cell suspension was centrifuged, and the supernatant aspirated off. The pellet – containing somatic nuclei – was resuspended in methanol and glacial acetic acid (3:1, v/v) and stored at −20 °C until use.
Fluorescence in situ hybridization
An aliquot (3–5 μL) was taken from the abovementioned somatic nuclei suspension and dropped onto a glass slide. The slide was placed in an environmental chamber at 28 °C and 39% humidity to fix nuclei and evaporate solvents. An aliquot (∼250 μL) of the labelling buffer [∼30 ng of a Cy3-labeled Invader probe in 500 μL TE buffer (10 mM Tris and 1 mM EDTA; pH 8.0)] was placed on top of the fixed nuclei. The slide was placed in a plastic culture dish, covered with a lid and moved to a 37 °C incubator for ∼3 h. Following incubation, the slide was rinsed in warm TE buffer (3 min, 37 °C) and left to dry at room temperature. An aliquot (∼3 μL) of Gold lowFade plus DAPI (Invitrogen) was placed directly on the slide and a round coverslip was mounted for fluorescence imaging. A Nikon Eclipse Ti-S/L100 fluorescent microscope, equipped with a SOLA SMII LED light source system and Cy3 and DAPI filter sets, was used to visualize the nuclei at 100× magnification. Images of fluorescently labelled nuclei were captured using a 14-bit CoolSNAP HQ2 cooled CCD camera and processed with NIS Elements software. Excellent slide-to-slide reproducibility was observed using this protocol within a given batch of nuclei preparations.Conflicts of interest
P. J. H. is an inventor on patents pertaining to Invader probes, which have been issued to the University Idaho.Acknowledgements
Preliminary contributions by Shannon Mecham and Jackson Davis (Univ. Idaho) are appreciated. We thank Prof. Jesper Wengel (Univ. Southern Denmark) and Dr Tumul Srivastava for providing us with LNAs and MPγPNAs, respectively. This study was supported by the Higher Education Research Council, Idaho State Board of Education [awards IF13-001, IF14-012].References
- P. D. Hsu, E. S. Lander and F. Zhang, Cell, 2014, 157, 1262–1278 CrossRef CAS.
- A. C. Komor, A. H. Badran and D. R. Liu, Cell, 2017, 168, 20–36 CrossRef CAS.
- M. Duca, P. Vekhoff, K. Oussedik, L. Halby and P. B. Arimondo, Nucleic Acids Res., 2008, 36, 5123–5138 CrossRef CAS.
- K. Kaihatsu, B. A. Janowski and D. R. Corey, Chem. Biol., 2004, 11, 749–758 CrossRef CAS.
- Y. Kawamoto, T. Bando and H. Sugiyama, Bioorg. Med. Chem., 2018, 26, 1393–1411 CrossRef CAS.
- J. Lohse, O. Dahl and P. E. Nielsen, Proc. Natl. Acad. Sci. U.S.A., 1999, 96, 11804–11808 CrossRef CAS.
- Y. Hari, S. Obika and T. Imanishi, Eur. J. Org. Chem., 2012, 2875–2887 CrossRef CAS.
- Y. Aiba, Y. Honda and M. Komiyama, Chem. – Eur. J., 2015, 21, 4021–4026 CrossRef CAS.
- P. R. Bohländer, T. Vilaivan and H.-A. Wagenknecht, Org. Biomol. Chem., 2015, 13, 9223–9230 RSC.
- M. Hibino, Y. Aiba, Y. Watanabe and O. Shoji, ChemBioChem, 2018, 19, 1601 CrossRef CAS.
- H. Kaur, B. R. Babu and S. Maiti, Chem. Rev., 2007, 107, 4672–4697 CrossRef CAS.
- R. Bahal, B. Sahu, S. Rapireddy, C.-M. Lee and D. H. Ly, ChemBioChem, 2012, 13, 56–60 CrossRef CAS.
- P. J. Hrdlicka, T. S. Kumar and J. Wengel, Chem. Commun., 2005, 4279–4281 RSC.
- D. C. Guenther, G. H. Anderson, S. Karmakar, B. A. Anderson, B. A. Didion, W. Guo, J. P. Verstegen and P. J. Hrdlicka, Chem. Sci., 2015, 6, 5006–5015 RSC.
- V. V. Filichev, B. Vester, L. H. Hansen and E. B. Pedersen, Nucleic Acids Res., 2005, 33, 7129–7137 CrossRef CAS.
- P. H. Hagedorn, R. Persson, E. D. Funder, N. Albæk, S. L. Diemer, D. J. Hansen, M. R. Møller, N. Papargyri, H. Christiansen, B. R. Hansen, H. F. Hansen, M. A. Jensen and T. Koch, Drug Discovery Today, 2018, 23, 101–114 CrossRef CAS.
- K. M. L. Hertoghs, J. H. Ellis and I. R. Catchpole, Nucleic Acids Res., 2003, 31, 5817–5830 CrossRef CAS.
- R. Beane, S. Gabillet, C. Montaillier, K. Arar and D. R. Corey, Biochemistry, 2008, 47, 13147–13149 CrossRef CAS.
- E. M. Zaghloul, A. S. Madsen, P. M. D. Moreno, I. I. Oprea, S. El-Andaloussi, B. Bestas, P. Gupta, E. B. Pedersen, K. E. Lundin, J. Wengel and C. I. E. Smith, Nucleic Acids Res., 2011, 39, 1142–1154 CrossRef CAS.
- E. M. Zaghloul, O. Gissberg, P. M. D. Moreno, L. Siggens, M. Hällbrink, A. S. Jørgensen, K. Ekwall, R. Zain, J. Wengel, K. E. Lundin and C. I. E. Smith, Nucleic Acids Res., 2017, 45, 5153–5169 CrossRef CAS.
- R. Bahal, E. Quijano, N. A. McNeer, Y. Liu, D. C. Bhunia, F. Lopez-Giraldez, R. J. Fields, W. M. Saltzman, D. H. Ly and P. M. Glazer, Curr. Gene Ther., 2014, 14, 331–342 CrossRef CAS.
- M. Petersen, C. B. Nielsen, K. E. Nielsen, G. A. Jensen, K. Bondensgaard, S. K. Singh, V. K. Rajwanshi, A. A. Koshkin, B. M. Dahl, J. Wengel and J. P. Jacobsen, J. Mol. Recognit., 2000, 13, 44–53 CrossRef CAS.
- S. P. Sau, A. S. Madsen, P. Podbevsek, N. K. Andersen, T. S. Kumar, S. Andersen, R. L. Rathje, B. A. Anderson, D. C. Guenther, S. Karmakar, P. Kumar, J. Plavec, J. Wengel and P. J. Hrdlicka, J. Org. Chem., 2013, 78, 9560–9570 CrossRef CAS.
- S. Karmakar, A. S. Madsen, D. C. Guenther, B. C. Gibbons and P. J. Hrdlicka, Org. Biomol. Chem., 2014, 12, 7758–7773 RSC.
- D. M. Crothers, Biopolymers, 1968, 6, 575–584 CrossRef CAS.
- S. C. Jain, C. Tsai and H. M. Sobell, J. Mol. Biol., 1977, 114, 317–331 CrossRef CAS.
- C. B. Nielsen, M. Petersen, E. B. Pedersen, P. E. Hansen and U. B. Christensen, Bioconjugate Chem., 2004, 15, 260–269 CrossRef CAS.
- J. L. Mergny and L. Lacroix, Oligonucleotides, 2003, 13, 515–537 CrossRef CAS PubMed.
- M. Egholm, O. Buchardt, L. Christensen, C. Behrens, S. M. Freier, D. A. Driver, R. H. Berg, S. K. Kim, B. Norden and P. E. Nielsen, Nature, 1993, 365, 3 CrossRef.
- R. Mitra and K. N. Ganesh, Chem. Commun., 2011, 47, 1198–1200 RSC.
- V. V. Demidov, E. Protozanova, K. I. Izvolsky, C. Price, P. E. Nielsen and M. D. Frank-Kamenetskii, Proc. Natl. Acad. Sci. U.S.A., 2002, 99, 5953–5958 CrossRef CAS.
- S. X. Chen, D. Y. Zhang and G. Seelig, Nat. Chem., 2013, 5, 782–789 CrossRef CAS.
- H. H. Pham, C. T. Murphy, G. Sureshkumar, D. H. Ly, P. L. Opresko and B. A. Armitage, Org. Biomol. Chem., 2014, 12, 7345–7354 RSC.
- A. Orenstein, S. A. Berlyoung, E. E. Rastede, H. H. Pham, E. Fouquerel, T. C. Murphy, J. B. Leibowitz, J. Yu, T. Srivastava, A. B. Armitage and L. P. Opresko, Molecules, 2017, 22, 2117 CrossRef.
- Y. Kawamoto, A. Sasaki, A. Chandran, K. Hashiya, S. Ide, T. Bando, K. Maeshima and H. Sugiyama, J. Am. Chem. Soc., 2016, 138, 14100–14107 CrossRef CAS.
- H. Ma, A. Naseri, P. Reyes-Gutierrez, S. A. Wolfe, S. Zhang and T. Pederson, Proc. Natl. Acad. Sci. U.S.A., 2015, 112, 3002–3007 CrossRef CAS.
- P. M. Lansdorp, J. Cell Biol., 1997, 139, 309–312 CrossRef CAS.
- J. W. Shay, Cancer Discovery, 2016, 6, 584–593 CrossRef CAS.
- P. Martínez and M. A. Blasco, J. Cell Biol., 2017, 216, 875–887 CrossRef.
- M. Ivancich, Z. Schrank, L. Wojdyla, B. Leviskas, A. Kuckovic, A. Sanjali and N. Puri, Antioxidants, 2017, 6, 15 CrossRef.
- A. S. Boutorine, D. S. Novopashina, O. A. Krasheninina, K. Nozeret and A. G. Venyaminova, Molecules, 2013, 18, 15357–15397 CrossRef CAS.
- S. Karmakar, B. A. Anderson, R. L. Rathje, S. Andersen, T. B. Jensen, P. Nielsen and P. J. Hrdlicka, J. Org. Chem., 2011, 76, 7119–7131 CrossRef CAS.
- U. B. Christensen and E. B. Pedersen, Nucleic Acids Res., 2002, 30, 4918–4925 CrossRef CAS.
- H. M. Pfundheller, A. M. Sørensen, C. Lomholt, A. M. Johansen, T. Koch and J. Wengel, in Oligonucleotide Synthesis, ed. P. Herdewijn, Humana Press, Totowa, NJ, 2005, pp. 127–145 Search PubMed.
- B. Sahu, I. Sacui, S. Rapireddy, K. J. Zanotti, R. Bahal, B. A. Armitage and D. H. Ly, J. Org. Chem., 2011, 76, 5614–5627 CrossRef CAS.
- N. N. Dioubankova, A. D. Malakhov, D. A. Stetsenko, M. J. Gait, P. E. Volynsky, R. G. Efremov and V. A. Korshun, ChemBioChem, 2003, 4, 841–847 CrossRef CAS.
- M. A. Morgan, K. Okamoto, J. D. Kahn and D. S. English, Biophys. J., 2005, 89, 2588–2596 CrossRef CAS.
- A. M. Brown, Comput. Methods Programs Biomed., 2001, 65, 191–200 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Protocols; HPLC and MS data for probes; representative thermal denaturation curves and electrophoretograms; preliminary biophysical characterization of Invader and INA probes; additional Tm, thermodynamic, dose–response, and time-course data; sequences and Tms for DNA hairpins; and binding specificity of INV3/INA3. See DOI: 10.1039/c9ob02111f |
| This journal is © The Royal Society of Chemistry 2020 |