
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
How did correlative atomic force microscopy and super-resolution microscopy evolve in the quest for unravelling enigmas in biology?
Adelaide
Miranda†
 a,
Ana I.
Gómez-Varela†
*ab,
Andreas
Stylianou
cd,
Liisa M.
Hirvonen
e,
Humberto
Sánchez
f and
Pieter A. A.
De Beule
*a
a,
Ana I.
Gómez-Varela†
*ab,
Andreas
Stylianou
cd,
Liisa M.
Hirvonen
e,
Humberto
Sánchez
f and
Pieter A. A.
De Beule
*a
aInternational Iberian Nanotechnology Laboratory, Avenida Mestre José Veiga s/n, Braga, Portugal. E-mail: pieter.de-beule@inl.int
bDepartment of Applied Physics, University of Santiago de Compostela, E-15782, Santiago de Compostela, Spain. E-mail: anaisabel.gomez@usc.es
cCancer Biophysics Laboratory, University of Cyprus, Nicosia, Cyprus
dSchool of Sciences, European University Cyprus, Nicosia, Cyprus
eCentre for Microscopy, Characterisation and Analysis (CMCA), The University of Western Australia, 35 Stirling Highway, Perth, WA 6009, Australia
fFaculty of Applied Sciences, Department of Bionanoscience, Kavli Institute of Nanoscience, Delft University of Technology, 2629 HZ, Delft, The Netherlands
First published on 18th December 2020
Abstract
With the invention of the Atomic Force Microscope (AFM) in 1986 and the subsequent developments in liquid imaging and cellular imaging it became possible to study the topography of cellular specimens under nearly physiological conditions with nanometric resolution. The application of AFM to biological research was further expanded with the technological advances in imaging modes where topographical data can be combined with nanomechanical measurements, offering the possibility to retrieve the biophysical properties of tissues, cells, fibrous components and biomolecules. Meanwhile, the quest for breaking the Abbe diffraction limit restricting microscopic resolution led to the development of super-resolution fluorescence microscopy techniques that brought the resolution of the light microscope comparable to the resolution obtained by AFM. The instrumental combination of AFM and optical microscopy techniques has evolved over the last decades from integration of AFM with bright-field and phase-contrast imaging techniques at first to correlative AFM and wide-field fluorescence systems and then further to the combination of AFM and fluorescence based super-resolution microscopy modalities. Motivated by the many developments made over the last decade, we provide here a review on AFM combined with super-resolution fluorescence microscopy techniques and how they can be applied for expanding our understanding of biological processes.
1. Introduction
The invention of the Atomic Force Microscope (AFM) in 1986![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1 and subsequent developments in liquid imaging2 of soft samples,3,4e.g., molecules,5,6 proteins,7–9 cells,11–13 tissues,10,11 and viruses12,13 enabled the study of biological samples with nanometric resolution under almost physiological conditions. The AFM is a unique microscope for imaging biological samples, as specimens require minimum or no preparation, and AFM can be performed under environmental control (i.e., temperature and CO2). Furthermore, AFM can not only image biological samples at the nanoscale, but also quantitatively characterise nanomechanical properties, such as elasticity, viscosity and adhesion.14–16 The first imaging mode used in AFM was the so-called contact mode, but nowadays a myriad of modes17 are available to image the diverse and complex biological specimens: dynamic, force–distance curve-based, multiparametric, molecular recognition and multifrequency. Technological developments18 have paved the way to high-speed AFM (HS-AFM) where the image acquisition speed can be increased by a factor of approximately 1000 compared to conventional imaging.19 This enables the measurement of dynamic behaviour in many biological processes, e.g. biomolecules,19–24 live bacteria25,26 and eukaryotic cells,27 which cannot be achieved with conventional AFM. Both AFM and HS-AFM have found extensive applications in the biological field.15,28–38
1 and subsequent developments in liquid imaging2 of soft samples,3,4e.g., molecules,5,6 proteins,7–9 cells,11–13 tissues,10,11 and viruses12,13 enabled the study of biological samples with nanometric resolution under almost physiological conditions. The AFM is a unique microscope for imaging biological samples, as specimens require minimum or no preparation, and AFM can be performed under environmental control (i.e., temperature and CO2). Furthermore, AFM can not only image biological samples at the nanoscale, but also quantitatively characterise nanomechanical properties, such as elasticity, viscosity and adhesion.14–16 The first imaging mode used in AFM was the so-called contact mode, but nowadays a myriad of modes17 are available to image the diverse and complex biological specimens: dynamic, force–distance curve-based, multiparametric, molecular recognition and multifrequency. Technological developments18 have paved the way to high-speed AFM (HS-AFM) where the image acquisition speed can be increased by a factor of approximately 1000 compared to conventional imaging.19 This enables the measurement of dynamic behaviour in many biological processes, e.g. biomolecules,19–24 live bacteria25,26 and eukaryotic cells,27 which cannot be achieved with conventional AFM. Both AFM and HS-AFM have found extensive applications in the biological field.15,28–38
The main drawback of AFM is that imaging is limited to the sample surface. Hence, the advantages of combining microscopy approaches were soon realised.39,40 The possibility of retrieving physical, chemical and biological information in complex systems using correlative microscopy methods set the stage for the emergence of hybrid systems.41,42 Over the past two decades AFM has been combined with electron microscopy,43,44 confocal Raman microspectroscopy45 and optical46–48 and fluorescence microscopy techniques. Fluorescence microscopy is a popular imaging technique especially in the biological sciences field, where it allows the tagging of intracellular molecules and cellular components with high specificity, and their observation inside cells in a minimally invasive manner using non-destructive wavelengths of light in the visible spectrum.49 AFM has been combined with confocal laser scanning microscopy,50–57 fast58 and differential spinning disks,59,60 and volumetric light sheet microscopy,61–63 as well as some specialised fluorescence characterisation techniques, e.g. Förster Resonance Energy Transfer (FRET),64 Fluorescence Lifetime Imaging (FLIM),65 and Fluorescence Correlation Spectroscopy (FCS).66 Although many biologically oriented AFM systems, the so-called Bio-AFMs, include an optical microscope coupled to the AFM, the correlation of these techniques is complicated by the diffraction limit of light restricting the resolution in optical microscopy to two orders of magnitude more than AFM.
The development of super-resolution microscopy techniques has brought the resolution of light microscopy down by approximately an order of magnitude, similar to the typical lateral resolution achieved with AFM when imaging soft biological samples. In the 1990s, at the same time as the first AFM results emerged in the biological field, the optical microscopy field was in a quest of going beyond the Abbe diffraction limit,67 which would allow an optical microscope to resolve structures separated by less than approximately 200 nm in the lateral dimension. This would lead to the development of Super-Resolution (SR) fluorescence microscopy techniques,68–70 first as theoretical concepts and then as experimental techniques in both far-field and near-field. This concept of super-resolution was introduced by Giuliano Toraldo di Francia.71,72 In his 1955 paper he defined super-resolution as the discrimination of details below the Abbe resolution limit.72 Toraldo di Francia proposed an original approach to overcome the diffraction barrier, showing in a theoretical study that with finely tuned pupil filters a precisely tailored sub-diffracted spot can be produced. In 1994, Stefan Hell and Jan Wichmann's theoretical work showed, for the first time since Abbe formulated the diffraction limit 180 years ago, that it was possible to image beyond the diffraction limit in the optical far-field.73 With this new concept, referred to as STimulated Emission Depletion (STED), Stefan Hell opened the far-field microscopy field to the nanometric spatial resolution74,75 and was one of the three winners of the Nobel Prize in Chemistry in 2014 that was awarded for the development of super-resolution microscopy techniques.
Soon afterwards, other techniques were developed that could also reach resolution beyond Abbe's limit. Gustafsson et al. (2000) were the first to experimentally verify lateral resolution enhancement beyond the diffraction limit using Super-Resolution Structured Illumination Microscopy (SR-SIM),76 and in 2006, yet another class of SR techniques were demonstrated, based on single-molecule localisation microscopy (SMLM). SMLM was first described theoretically by Eric Betzig in 1995,77 a study that contributed to the award of the 2014 Nobel prize in Chemistry jointly with Stefan Hell and W. E. Moerner, and 10 years later Xiaowei Zhuang's group,78 Eric Betzig and Harald Hess,79 and Michael Mason's group80 presented similar practical solutions on how to resolve single fluorescent molecules in a highly populated sample: Stochastic Optical Reconstruction Microscopy (STORM), Photo-Activated Light Microscopy (PALM), and Fluorescence PALM (FPALM), respectively.
In the past decade, these far-field SR methods have become popular imaging techniques in biological research, and commercial instruments are now widely available. All of these SR microscopy techniques have been combined with AFM:81,82 the combination of STED and AFM was first reported by Chacko et al. in 2012,83 STORM and PALM were combined with AFM in 201384,85 and 2015,86 respectively, and recently, in 2020, Gómez-Varela et al. presented a hybrid system combining SR-SIM and AFM for simultaneous co-localized operation of both systems.87
Subwavelength resolution and imaging can also be achieved by placing the sample in the near-field of the illumination or by probing the near-field radiation emitted from a specimen; these techniques are termed Total Internal Reflection Fluorescence Microscopy (TIRFM) and Scanning Near-Field Optical Microscopy (SNOM/NSOM), respectively. Imaging platforms based on combined TIRFM and AFM have been demonstrated over the years for a variety of biological applications, e.g., lipid bilayer dynamics and cellular dynamics and structures, among others.88 SNOM also takes advantage of the confinement of radiation fields for resolution enhancement and has been integrated with both AFM and HS-AFM.89,90
These impressive advances in SR optical microscopy have helped bridge the gap between the resolution achievable in optical microscopy and AFM, and significantly enrich the tool-box available for biological system characterisation. The variety of hybrid systems and key accomplishments that have been made in this field over the years are summarized in Fig. 1.
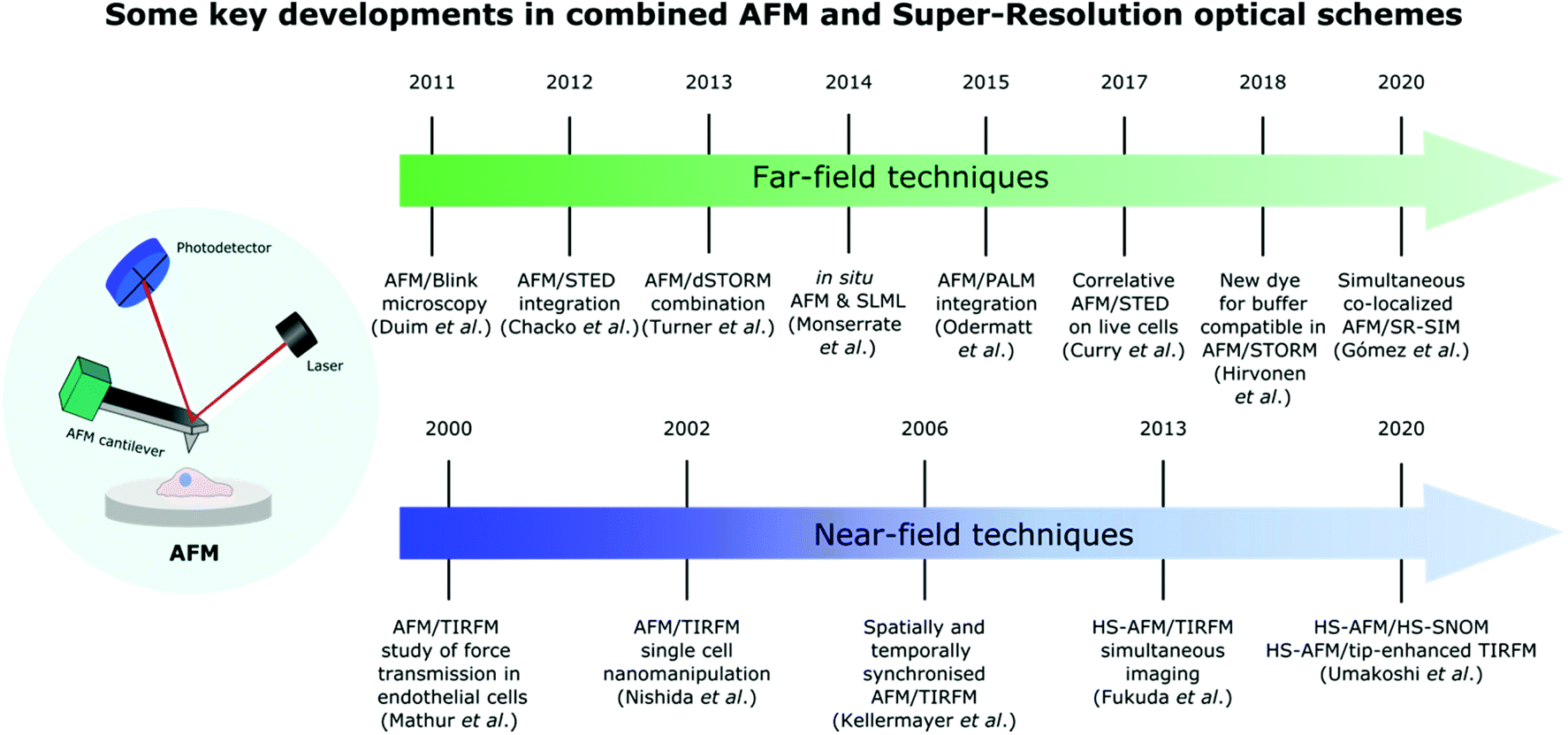 | ||
| Fig. 1 Timeline showing some major advances in the development of different far-field and near-field SR techniques combined with AFM for life science applications. | ||
In this review, we provide an overview of how correlative AFM and SR fluorescence microscopy techniques in both far-field and near-field have developed in the context of biological applications. In the far-field we review the combination of AFM with SMLM, STED, and SR-SIM microscopy techniques, and in the near-field we describe the combination of AFM with TIRFM and SNOM. After a general introduction to AFM, a brief overview of the main principles of each SR technique is given in the sections dedicated to these techniques, followed by examples of biological applications using the combined SR and AFM system. Different combined AFM/SR techniques are compared in a dedicated section, and finally, we discuss the future perspectives of combining AFM with SR fluorescence microscopy techniques in the quest for answers in the field of biology.
2. Atomic force microscopy background
2.1. Physical principles
AFM operation is based on the interactions of an AFM probe with the specimen surface. The AFM probe is usually a nanometer-sized tip mounted on the free end of a cantilever, which is a flat spring. Depending on the application, different tips can be used ranging from sharp nanoscale tips to ball-shaped tips with a diameter of several micrometers. Cantilevers also come in many shapes and sizes; the more popular ones are beam-like and V-shaped cantilevers. Cantilevers are characterized by their resonance frequency and spring constants, which range from few kilohertz (kHz) up to MHz and from 0.01 N m−1 to several hundred N m−1, respectively. During AFM operation, the AFM probe scans the specimen surface line by line in a grid pattern. Accurate movement of either the AFM probe or the sample stage, depending on the design, is achieved by piezoelectric scanners. Due to the forces acting between the tip and the sample surface, the cantilever bends. The cantilever movement and bending are measured with an optical system, where a laser beam is reflected from the back of the cantilever and directed to a four-quadrant position-sensitive photodetector. The photodetector records the position of the laser, which is then converted into lateral and vertical deflection signals. Considering the cantilever spring constant and the photodetector sensitivity, these signals are then converted into a force for each pixel, and a digital pseudo-colored image of the specimen surface can be built. The development of the optical detection system and the fluid cell that enables AFM to operate in aqueous solution led to the first Bio-AFM and was a significant milestone in the history of AFM technology.3,4,172.2. Typical set-up and modes
AFM systems can operate in a number of different modes.17 The most widely used modes for topography imaging are the contact, intermittent and non-contact modes. In the non-contact mode, the tip does not encounter the sample surface, while in the contact mode the tip is always in contact with the sample surface. In the dynamic or intermittent mode, also known as tapping, AC or oscillation mode, the tip taps the sample surface, touching the surface only for a short time. In this mode the cantilever oscillates at or near its resonant frequency, thus minimizing the energy in the tip–sample system, and the friction. A number of other dynamic modes have also been developed, which take into account different signals as feedback parameters, or excite the cantilever at different frequencies (multifrequency imaging).17,91–93 In biological applications silicon nitride cantilevers are often used, with an aluminium or gold coating on the back surface to improve the reflection of the laser beam under liquid conditions. For imaging biological samples the tapping mode is widely used as it is less destructive than the contact mode, and issues with lateral and frictional forces, which are observed in the non-contact mode, are minimized.94,95Besides surface imaging, AFM can operate in the force spectroscopy mode.96 In this mode, force curves representing the force versus tip–sample distance are recorded at a specific location.97 A calibration procedure including deflection sensitivity calibration and spring constant calibration is typically required to obtain quantitative data.98,99 Although initially it was possible to record only Force–Distance (FD) curves at a single spatial location, nowadays AFMs can collect force curves for a given grid whereby the cantilever scans a specific area of the sample's surface; this mode is called the force–volume mode or FD curve-based imaging mode.35 Recent advances, including optimization of hardware control and cantilevers, have enabled the development of fast force spectroscopy and parallel characterization of topographical, mechanical, and chemical characteristics of the sample. Force curves are recorded pixel-by-pixel on well-defined grids, which can achieve the same resolution as conventional imaging modes. Force measurements (force–spectroscopy, force–volume mode, etc.) can provide a vast amount of information on the nanomechanical and biophysical properties of the specimen, such as mechanical properties of living cells including the Young's modulus and interaction forces in various biological systems, e.g., cell–cell adhesion and protein unfolding. AFM-based techniques are not only appropriate for imaging and characterizing complex biological systems, but they are also highly effective for the design of novel biointerfaces.98,100
AFM is emerging as a unique tool and nanoscopic platform in the mechanobiology field. However, the measurement of the mechanical properties of complex biological systems at the nanoscale is not trivial, and a number of parameters and issues have to be taken into account, including the appropriate cantilever and probe, the mechanical model (e.g. Hertz/Sneddon), the environmental conditions and the AFM parameters such as the loading rate.16,101,102
Besides the characterization of a sample, AFM can also be used as a nano-manipulation tool either for the mechanical manipulation of the sample by using the tip–sample interactions, or by using a chemically functionalized AFM tip to manipulate targeted specimen regions.17,103 The AFM probe can be applied for cutting, picking up and pasting biomolecules.104
3. AFM and far-field super-resolution microscopy techniques
This section describes the combination of AFM and SR far-field techniques and is divided into three main parts: single-molecule localisation microscopy techniques, STED and SR-SIM. Each section describes the principles of the SR technique, and the applications of the combined AFM-SR system.3.1. Single-molecule localisation microscopy (SMLM)
The first result on single molecule localisation microscopy was achieved by Yildiz et al. in 2003 to obtain super-resolution images of myosin.105 This technique was named Fluorescence Imaging with One Nanometer Accuracy (FIONA), and the super-resolved images were achieved by distributing the fluorescently labelled myosin molecules sparsely over the sample such that the diffraction-limited image of each molecule did not overlap with the images of the other molecules. It was then possible to calculate the centre position of each probe from the diffraction-limited image with great accuracy.
FIONA only works with a sample where the fluorescent labels are very sparsely distributed, and therefore has limited use in imaging biological specimens. In 2006 Xiaowei Zhuang's group78 presented a new strategy based on the photoswitching properties of fluorescent cyanine dyes. These dyes can switch between a fluorescent state and a dark state by light activation. The researchers used a low activation light intensity to switch on a small subset of the probes in the sample, took an image of these molecules, and then calculated their centre position. By illuminating the sample with another wavelength of light, this subset of probes was turned off and another subset was turned on. This procedure was iterated many times, until enough positions of molecules were collected to form a high-resolution image of the underlying structure. This method, termed STORM, can achieve a typical lateral resolution of 20 to 30 nm. PALM, published by Betzig and Hess also in 2006,79 is based on the same principle, but uses photoswitchable fluorescent proteins instead of dyes.
Many other variants of SMLM have been published over the years. In 2008, Heilemann et al. introduced direct STORM (dSTORM),106 which simplified the experimental method by using conventional cyanine dyes (Cy5 and Alexa647) and making them blink by the addition of a switching buffer containing a thiol and an oxygen scavenging system (often glucose oxidase (GLOX)) instead of relying on the proximity of two fluorophores attached to an antibody in a specific ratio and at a specific distance. Furthermore, blink microscopy was presented in 2011107–109 widening the pool of fluorescent labels available for SMLM. The point accumulation for imaging in nanoscale topography (PAINT) method110,111 on the other hand achieves the on/off switching behaviour by using freely diffusing dyes that only become fluorescent when they are transiently bound to the target structure.
In an early technical note, Hermsdörfer et al. (2013) discussed the integration of AFM/STORM to study fluorescently labelled fixed human cervical cancer cells (HeLa cells).84 The microtubules in HeLa cells were labelled with Alexa647 via immuno-fluorescence staining. The combination of the two microscopy techniques provided information of the cell surface appearance with respect to the microtubules and their distribution: 3D STORM gave information about the distribution and structure of individual microtubules in a z-range of around 800 nm with an axial resolution of around 50–75 nm, while AFM yielded topographical and biophysical information.
In the same year Diaspro's laboratory employed AFM/STORM to image cytoskeletal structures (e.g. microtubule filaments) in HeLa and fibroblast cells.85 For STORM experiments, α-tubulin in the cells was immunostained with Alexa647, showing a drastic resolution increase when compared to the wide-field image, and allowing precise localization of microtubules in all three dimensions. Tubulin was chosen to give a comparison with correlative AFM/STED images presented in the same manuscript.
In 2014, the laboratory of Cristina Flors presented a new correlative microscopy tool that combined in situ AFM and SMLM microscopy. This novel hybrid system was able to reveal artefacts in SR imaging related to labelling and image reconstruction.115 The authors used stretched λ-DNA labelled with the intercalating cyanine dye YOYO-1, which blinks in the presence of a GLOX buffer containing a reducing thiol compound,116 and found out that although most of the λ-DNA seen in the AFM image appears also in the SR image, there are patchy sections. The authors suggested that this could be related to incomplete labelling, rapid photo-bleaching of YOYO-1 in that region, or poor signal-to-noise ratio leading to exclusion during image analysis.115 In 2017, the same group pioneered a hybrid system combining AFM and two-colour SR fluorescence imaging of β-lactoglobulin amyloid-like fibrils functionalized with organic fluorophores and quantum dots.117 The AFM topography image provided information on the number of filaments that composed the amyloid-like fibrils, while the SR image provided the identification of emissive and non-emissive quantum dots, which allowed differentiating between real localizations in the SR image and spurious ones. Recently a similar method has been used with a dual-colour probe for correlative AFM and SR imaging of polymer fibroid micelles.118
In 2015, Georg Fantner's laboratory showed the capabilities of combining AFM/STORM for quantifying the density of localization of F-actin cytoskeletal filaments along cells’ 3D topography.86 The authors correlated the height and location of actin filaments obtained with an AFM and fluorescence data obtained with STORM. The results revealed that: (1) in areas with one filament (topographical data derived from AFM) labelling fluctuations are attributed to insufficient binding of tagged phalloidin and (2) in areas where AFM revealed two or three filaments the labelling fluctuations originate partially from their presence and not necessarily from insufficient labelling. The authors also suggested that the localization intensity can eventually be used for quantifying the number of actin filaments in a bundle, where AFM information cannot be obtained. In the same work86 the authors pioneered the combination of AFM with PALM by expressing the fusion protein RNP-mEos2 in chemically fixed E. coli and live CHO-K1 cells. The use of photoswitchable proteins, i.e. the PALM technique, instead of the toxic dyes and high intensity illumination required for STORM enabled the first demonstration of combined AFM and SR imaging in living cells.
When combining AFM and STORM, AFM imaging is usually performed first before the addition of the STORM buffer,86,115–117 because a STORM buffer containing an enzymatic oxygen scavenger is not compatible with AFM imaging. To overcome this limitation Hirvonen et al. replaced the Alexa647 dye by a structurally similar cyanine dye called iFluor647,119 which exhibits excellent fluorescence and blinking properties in a buffer that contains a thiol, but no enzymatic oxygen scavenger.120 The authors demonstrated that the results obtained with AFM/STORM are independent of which technique is acquired first: the AFM laser with a wavelength of 850 nm did not have a significant impact in bleaching iFluor647, and there was no evidence of sample damage caused by the STORM laser. They also demonstrated that imaging could be performed for more than 5 h after the addition of the thiol-only buffer, unlike a traditional STORM buffer where the oxygen scavenger induces a pH change that degrades the sample and limits the imaging time to a couple of hours. This work was recently extended to demonstrate 2-colour localization microscopy and AFM imaging of podosomes in THP-1 cells.121 Two-colour SR images were acquired by combining the STORM imaging of iFluor647-labelled actin with PALM imaging of mEOS3.2-labelled talin, and the authors were able to correlate the podosomes seen in the SR images with higher stiffness areas in AFM images; see Fig. 2.
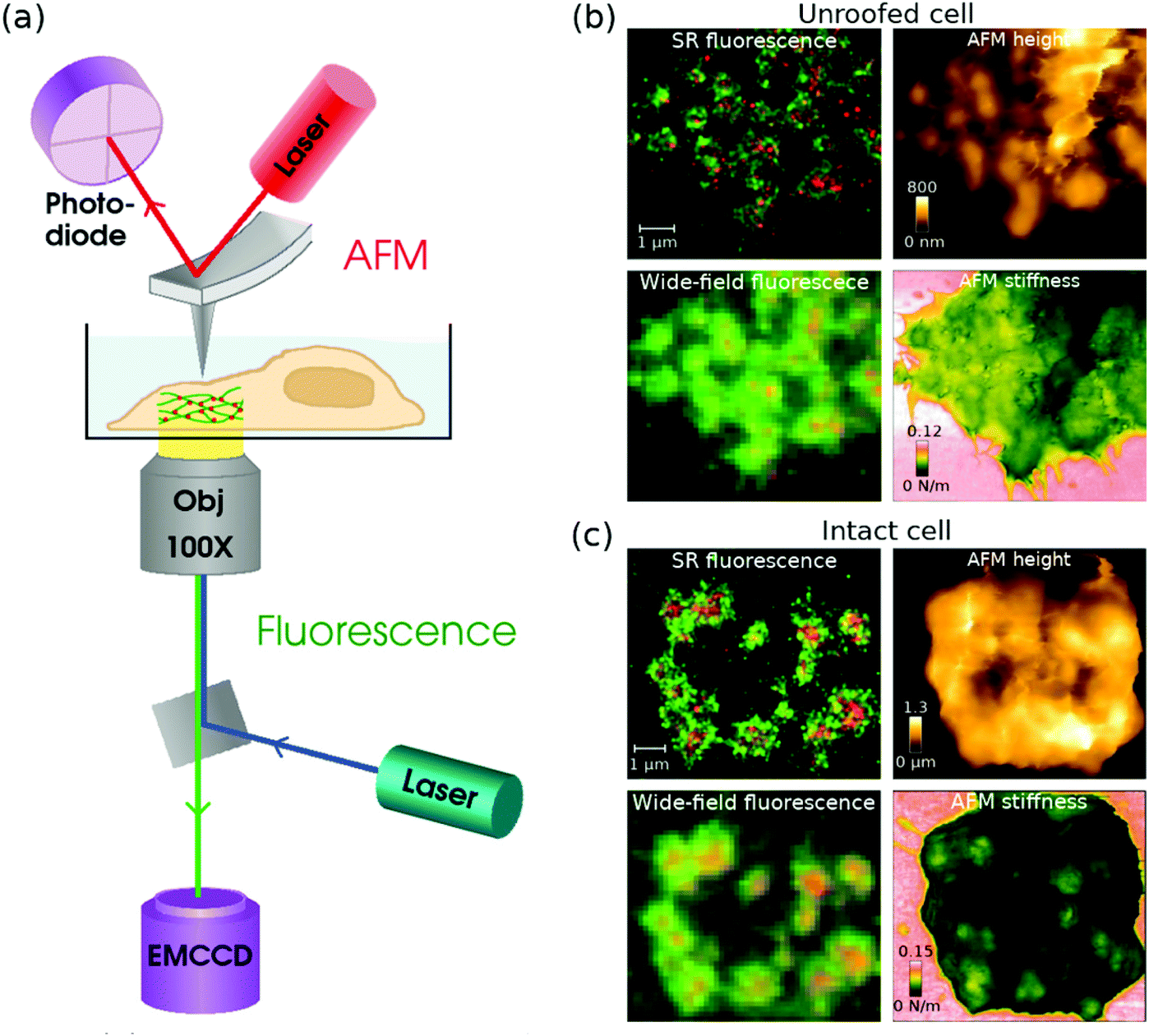 | ||
| Fig. 2 (a) A schematic diagram of a combined AFM + STORM microscope set-up, where an AFM collects an image of the sample from top, and STORM imaging is performed from underneath with an inverted fluorescence set-up. (b and c) Combined fluorescence and AFM images of podosomes in an (b) unroofed and (c) intact THP-1 cell. In the (b) unroofed cell, podosome cores can be seen as higher areas in the AFM height and stiffness images. In the (c) intact cell, the podosomes are difficult to see in the AFM height image due to a fluffy membrane, but the stiffness image clearly shows podosome cores as stiffer areas under the membrane. Modified from ref. 121 with permission. | ||
In addition to the systems described above, the combination of AFM and dSTORM has been recently extended to other biological topics such as the study of virus–host interactions122 and membrane proteins.123
3.2. AFM and stimulated emission depletion (STED)
 | ||
| Fig. 3 (a–f) Correlative AFM + STED imaging of Cos7 cells labelled with Atto 647 N. (a) Confocal raw image, (b) STED raw image, (c) 3D rendered view of AFM measured height extracted from AFM force curves, deconvolved (d) confocal and (e) STED images, and (f) an elasticity map calculated from AFM force curves. (g) Schematic diagram of a combined AFM/STED imaging set-up. The AFM cantilever is aligned such that STED and AFM have a common scan area. Fluorescence excitation pulses are combined with depletion pulses using a dichroic mirror (DM2), and fluorescent emission is separated using a dichroic mirror (DM1). AFM images are acquired by translating the sample. For each pixel a force curve is measured by approaching the tip toward the sample and recording the tip–sample interaction force as a function of the cantilever z-position (see inset), and the Young's modulus is estimated from the gradient. Scale bars in (a and b): 2 μm. (a–f) Reproduced from ref. 83 with permission, (g) reproduced from ref. 127 with permission. | ||
Yu et al. reported the integration of AFM and STED using a super-continuum fibre laser and a lateral resolution of 42 nm.125 The super-continuum laser source was used to provide both excitation and STED beams with an energy level higher than 1 nJ nm−1. The authors achieved temporal synchronization between the two techniques and tested their system with nanobeads and human cervical carcinoma cells (CaSki cells) where the actin filaments were labelled with phalloidin-ATTO655.
In 2014, Diaspro's laboratory pioneered the manipulation of cells using AFM/STED.126 In this proof of principle work, the authors used the AFM tip to manipulate microtubules labelled with Abberior Star 635P inside fixed cells. The results show that in areas where extremely high force was applied with the AFM tip there is a resolution enhancement of the microtubule in the STED image, corresponding to the stretching of a single microtubule in the bundle. In another proof of principle experiment, the authors used the AFM tip to cut a single microtubule filament in a bundle with the help of the STED image.
The combination of AFM and STED was first applied to live cell imaging in 2017.127 The authors used correlative microscopy to simultaneously investigate the contribution of fine cytoskeletal elements (actin and tubulin) in astrocyte subcellular compartments, and assessed their involvement in cell topography and mechanical properties under nearly physiological conditions or during polarized migration in vitro. For STED imaging, the authors used a previously described set-up,128 see Fig. 3(g), and labelled the cells with SiR-actin and SiR-tubulin.129 The authors chose actin and tubulin because these cytoskeletal elements are among the very few proteins that can be visualized with STED imaging with a resolution down to 50 nm. In addition, these can be manipulated by pharmacological agents, e.g. nocodazole, that disrupt their assembly. The authors found that the actin networks are highly organized in astrocytes, in those featuring polarization, and that these networks are well reflected in cell topography and are key determinants of membrane stiffness. They also found that tubulin patterns differ from actin patterns in astrocytes, tubulin displays a non-polarized structure in control conditions, and tubulin does not contribute significantly to astrocyte stiffness.
In 2019, Diaspro's laboratory used correlative AFM/STED microscopy to assess the effect of fluorescence labelling on molecular activity.130 They studied the in vitro aggregation of insulin from bovine pancreas covalently labelled with N-hydroxysuccinimide dye with two alloforms of Aβ amyloid peptides (Aβ1-42 and Aβ1-40) at different dye-to-protein ratios, and concluded that dye-labelled proteins do not form aggregates as readily as non-labelled proteins, and that STED alone is not capable of characterising all products derived from the in vitro aggregation of misfolded proteins. They also highlighted that AFM can identify fibrillary aggregates that were not shown in the fluorescence microscopy images.
In the same year, AFM/STED was employed to characterise the density and function of actin clusters in neurofibromatosis type 1, opening the path to suggest a novel actomyosin-dependent mechanism during osteoclast migration and resorption.131 Based on the data obtained with correlative microscopy, the authors also demonstrated the timescale of the substrate-specific signalling through which osteoclasts adapt to and polarize towards bone. They also showed differences in actin structures of neurofibromatosis type 1 osteoclast that follow the hyperphosphorylation of cofilin.
3.3. AFM and super-resolved structured illumination microscopy (SR-SIM)
 | ||
| Fig. 4 AFM measurements on fixed U2Os cells in medium/buffer with (a) and without N-SIM illumination (b). For enhanced feature/noise contrast, both AFM topography images in the AFM/SR-SIM overlays are displayed with an edge detection algorithm using a pixel difference operator in X. The topography images from a Petri dish surface on three positions (labelled in the figures) were plane fitted (first order polynomial function) to compensate for tilts in the sample surface, and subjected to surface roughness analysis (c). For comparison reasons, the average roughness (Ra), RMS roughness (Rq) and peak-to-valley roughness (Rt) values are given below the corresponding height profiles. XY-scales of the AFM images in (a and b) are 13.59 μm and 12.16 μm respectively, recorded at a resolution of 256 × 229 pixels. The insets used for analysis in (c) have a resolution of 37 × 37 pixels. (d) Simplified schematic diagram of AFM/SR-SIM imaging conditions within the sample region. Modified from ref. 87 with permission. | ||
The type of cantilever used in this work, qp-bioAC-CI, has only a limited partial Au coating in order to avoid uncontrolled and disruptive cantilever deflection due to fluorescence excitation light absorption. A schematic drawing of the sample area of this set-up is shown in Fig. 4d.
4. AFM and near-field super-resolution microscopy techniques
In this section we focus on the near-field techniques combined with AFM: Total Internal Reflection Fluorescence Microscopy (TIRFM) and Scanning Near-Field Optical Microscopy (SNOM).4.1. AFM and total internal reflection fluorescence microscopy
The versatility of AFM/TIRFM systems for recognition and simultaneous localization of different fluorescent-tagged proteins interacting with DNA has been shown in the last few years.112,154–156 A common challenge in hybrid microscopes, is the registration of different types of data. When imaging single molecules the challenge is amplified by the requirement of nanometer precision for localization. The use of fluorescent fiducials for image registration has been shown invaluable,112,157,158 and after the development and validation of software routines for consistent, accurate and convenient image registration, AFM/TIRFM systems can be used in a routine manner.155,156,159–161 These tools have helped researchers to understand mechanistic aspect of the cellular DNA repair systems, like Homologous Recombination (HR), that efficiently restore genome integrity in healthy cells.162 Mutations that affect the function of DNA repair proteins induce cancer and by capturing pictures of these proteins with DNA, how they work can be investigated. HR is a DNA rearrangement where protein recombinases, like RAD51, make filaments with the damaged DNA assisted by BRCA2, a tumour suppressor protein, and RAD54, a motor protein.163 To determine the mechanisms of how these mediators influence RAD51 activity, the arrangement of the proteins in complex with DNA has been resolved, demonstrating that different localization of RAD54 on the filaments (terminal or interspersed) promotes different functions,155 and how BRCA2 facilitates loading of the repair factor RAD51 by dynamic structural transitions of the complexes.156
Correlative AFM/TIRFM schemes often take advantage of AFM force measurement capability to examine sub-cellular variations when mechanical forces are applied.148,149 AFM micro- and nano-manipulation capabilities have also been exploited with such hybrid microscopy systems.164,165 Mathur et al. described a combination of AFM and TIRFM designed to provide simultaneous mechanical force transmission measurements and focal contact dynamics in human umbilical vein endothelial cells (HUVECs).166 TIRFM excites fluorescence in the basal membrane of cells attached to the waveguide surface, while an AFM tip introduced from above the coverslip probes the apical membrane of the same cells. The authors observed that the precise localized mechanical perturbations induced by the AFM tip resulted in a global rearrangement of focal contacts at the basal membrane. In 2002, Nishida et al. reported the combination of AFM with a TIRFM set-up for nanomanipulation of single cells,164 describing the delivery of small macromolecules into live cells while simultaneously monitoring with TIRFM the dynamics of these molecules inside the cells. Kellermayer et al. demonstrated in 2006 an AFM/TIRFM system capable of simultaneous spatial and temporal operation.165 With this synchronized AFM/TIRFM it is possible to correlate topography and fluorescence features of the examined specimen to mechanically manipulate soft biomolecular samples in a targeted fashion. Furthermore, it allows the monitoring of changes in the fluorescence properties of mechanically stretched biomolecules with high temporal resolution. Because of the spatial synchrony provided by the combined system, the authors were capable of imaging the specimen with scanning TIRFM, subsequently manipulating the same at specific positions with AFM via nanolithography, and finally reimaging the sample with TIRFM to detect the changes. Furthermore, they applied this method to show the ablation of parts of cells and individual actin filaments. A microscopy platform integrating AFM, TIRFM and fast-spinning disk (FSD) confocal microscopy for real-time mechanotransduction studies in live cells was later presented by Trache and Lim.167 With this system they later studied the response of vascular smooth muscle cells (VSMCs) to mechanical stimulation using a functionalised AFM tip with extracellular matrix (ECM) proteins laminin or fibronectin, finding that the VSMC-adaptive response is adapted to the applied tensile stress and modulated by the pre-existing cytoskeletal tension.168 An interesting study by Christenson et al.169 described a novel method based on the combination of single cell manipulation by AFM, TIRFM and microcontact printing170 to study the stability of multilayer fibrinogen matrices. In order to better understand how the DNA genome is released from the adenovirus during mechanical disassembly, Ortega-Esteban et al. triggered the disruption of single human adenovirus capsids with AFM and monitored the genome exposure with a DNA-specific intercalating fluorescent dye (YOYO-1) that could only access the DNA after the capsid had been opened up.171 The fluorescent signals from the released virus genomes during AFM manipulation were observed using a TIRFM set-up. Sarkar et al. demonstrated that TIRFM can measure distances in the axial direction perpendicular to the sample substrate with a length and time resolution comparable to that of an AFM.172 They made use of the depth-dependent intensity profile of the TIRF-generated evanescent wave to track sub-nanometer changes in the vertical displacement of fluorescent beads and quantum dots attached to single ubiquitin proteins tethered between a glass surface and an AFM cantilever during forced unfolding experiments.
In 2017, Harris et al. performed experimental measurements of nanomechanical properties of secretory vesicle-plasma membrane tethers using a combined AFM force clamp and TIRF microscopy on membrane sheets from PC12 cells expressing the vesicle marker ANF-eGFP.173 The length and frequency of tether-unfolding events were measured using the AFM cantilever while TIRFM was used to locate vesicles and track the movement of vesicles attached to the AFM cantilever tip within the TIRF evanescent wave. The experiments revealed a distribution of tether extension steps around ∼5 nm consistent with sequential unfolding of helical domains. More recently, mechanisms of intercellular tension involving cytoskeletal proteins, like catenin and vinculin, have been observed in real time by combining AFM and TIRFM.174 The authors visualized α-catenin stretched using AFM and the simultaneous recruitment of fluorescently labeled vinculins. The work highlights the potential of the hybrid instrument in the field of mechanobiology (Fig. 5).
 | ||
| Fig. 5 Example of one of the different AFM/TIFRM combined set-ups reported in the literature. (a) α-Catenin molecules (residues 276–634; the mechano-sensitive M1–M3 domain), modified on coverslips, were extended using AFM and simultaneously observed their recruitment of Alexa-labelled full-length vinculin molecules, dissolved in solution, using TIRFM; (b) α-catenin molecules attached to coverslips were tethered by AFM in programmed piezo-height control phases shown in the figure. Reproduced from ref. 174 with permission. | ||
4.2. AFM and scanning near-field optical microscopy
 | ||
| Fig. 6 Various configurations of cantilever based SNOM. (a) Aperture SNOM relying on a specially designed cantilever chip. The tip has a small optical opening at the apex, where a localized evanescent light is produced. (b) Aperture-less SNOM based on a metal-coated cantilever tip that acts as a local electromagnetic antenna (plasmon resonator). (c) SNOM combining a small aperture with an electromagnetic nano-antenna. The nano-antenna is a metal particle with the shape of a bowtie, rod, sphere or dumb-bell aperture-less. Reproduced from ref. 41 with permission. | ||
5. Comparison between correlative microscopy techniques
Table 1 shows a summary of the hybrid AFM/SR systems described in this manuscript, along with their strengths, limitations, and biological applications. The SR methods explained in this manuscript have significant differences, but all have become popular methods in biological sciences and have their advantages for specific applications. There is usually a trade-off between spatial and temporal resolution, and especially with living samples illumination intensity and labelling methods are critical issues. For hybrid AFM/SR systems additional trade-offs need to be considered, such as synchronization of data acquisition, image registration, and spatial overlap of the field of view.| Biological samples | Strengths | Limitations | Ref. | |
|---|---|---|---|---|
| AFM/STORM and AFM/BLINK and AFM/in situ SMLM | Fixed Gram-negative bacteria; fixed HeLa, THP-1 and fibroblast cells; virus-host interactions; membrane proteins of human bronchial epithelium; fixed amyloid disease protein aggregates – huntingtin proteins; λ-DNA; protein fibrils | Good fluorescence resolution, ∼20–50 nm typical; molecular tracking; AFM can reveal labelling and image reconstruction artefacts in SR images | Long image acquisition time; need to use specific dyes; staining protocols not compatible with live cell imaging; high illumination intensity required for SMLM; AFM and GLOX buffer non-compatibility; not simultaneous | 84–86, 112, 114, 115, 117 and 121–123 |
| AFM/PALM | E. coli. CHO-K1 cells | Good fluorescence resolution, ∼20–50 nm typical; lower power laser than STORM; live cell imaging possible; molecular tracking | Long image acquisition time; need to transfect; not simultaneous | 86 |
| AFM/STED | Fixed Cos7 cells; fixed CaSki cells; live astrocytes; bovine insulin from bovine pancreas; alloforms of Aβ amyloid peptides; neurofibromatosis type 1 osteoclasts | Image reconstruction not required; possibility to set common scan area; data shows how fluorophores influence molecular activity | Not simultaneous; bleaching, specific dyes for STED; need to use fiduciary marks; high illumination intensity | 83, 124–127, 130 and 131 |
| AFM/SR-SIM | Gene-edited U2OS cells to expressing MCT1 | Simultaneous co-localized operation; simple sample preparation protocols; multicolour imaging; live cell-imaging: fast image acquisition; low illumination intensity | Moderate resolution improvement compared to other SR techniques | 87 |
| AFM/TIRFM | Myosin filaments; fibronectin fibrillogenesis in rat embryonic living fibroblasts (REF52); DOPC/DSPC/cholesterol model membranes; supported phospholipid bilayers comprised of POPE/TOCL; RNA polymerases (RNAP); BRCA2-RAD51 protein complexes; RAD54 on RAD51-ssDNA complexes; living BALB/3T3 cells; HeLa cells and Human pancreatic cancer cells (Panc-1) expressing K8 and K18 keratins; HUVECs; VSMCs; HEK 293 (HEK WT) and HEK 293 cells expressing leukocyte integrin Mac-1ubiquitin protein; PC12 cells expressing the vesicle marker ANF-eGFP; α-catenin and vinculin molecules; human fibrinogen; wildtype HAdV-C2 and mutant HAdV-C2_TS1 grown in A549 (human lung carcinoma) and KB (HeLa-subclone) cells; GroEL, GroES and BSA; chitin crystalline fiber and myosin V walking on an actin filament; EcoRV–DNA nucleoprotein ensemble | Fast acquisition time; amenable for simultaneous operation with AFM; selective excitation at interface; low illumination intensity | Limited to the glass-sample interface | 150–156, 163–169, 171–174, 190 and 191 |
| AFM/SNOM | Carotene crystal in a model carrot cell system; DNA | Simultaneous collection of morphological characteristics of the sample from AFM imaging combined with optical and chemical properties observed in the SNOM image | Slow scanning speed; low near-field intensity in solution | 187, 189 and 190 |
With both SMLM and STED, typical resolution is in the order of few tens of nanometres, the same order of magnitude as the resolution of an AFM when imaging soft biological samples. However, both techniques also usually require high intensity illumination, which interferes with the AFM operation and limits the possibility of simultaneous data acquisition with AFM. As a scanning technique, STED–AFM has the advantage that the STED scan area can be matched with the AFM scan area. This allows the investigation of structures with the same magnification and can help with image correlation.127,129
A major limitation in combining AFM and STORM has been the need to change imaging buffers between techniques. Although the frequently used Alexa647 dye has excellent brightness and switching properties in a GLOX buffer, the buffer components crystallise on the AFM cantilever, preventing AFM imaging. As a result, the buffer has to be changed between imaging modes, although recently it has been suggested that this problem can be overcome by replacing Alexa647 by a similar dye iFluor647.120 With PALM, the use of photoswitchable proteins instead of dyes solves this problem, but fluorescence labelling requires transfection, which complicates sample preparation. The long image acquisition time of SMLM is also a drawback for imaging living samples.
SR-SIM, on the other hand, uses low illumination intensity, and is therefore well suited for simultaneous data capture with AFM;87 the main charm of SR-SIM for its integration with AFM relies on a fluorescent excitation light pattern favourable to avoid AFM cantilever disruption during simultaneous operation, and the possibility of live cell imaging. The simultaneous operation of AFM and SR fluorescence techniques has attracted great interest for observations at the nanoscale, and simultaneous operation of AFM and SR-SIM has been recently demonstrated.87 SIM data acquisition is also fast compared to other far-field SR methods, which is a significant advantage especially when imaging live samples. However, resolution of the SR-SIM image is limited to ∼120 nm, which is substantially above the resolution achievable with AFM.
TIRFM is especially amenable for integration with AFM as the illumination does not interfere with cantilever operation, allowing for imaging biological phenomena at the glass–sample interface with high axial resolution.88 TIRFM is therefore well suited for applications for surface structures and dynamics located near to the cover glass–sample interface because of the finite penetration depth of the evanescent excitation field (e.g., plasma membrane, cytoskeleton, and ligand–receptor interactions145). Consequently, it is important to note that its applications are restricted to the bottom cell membrane and it is not suitable for the study of the interior of a cell.
One major challenge in combined AFM/SR is image correlation between the two imaging modes. Different approaches have been used for matching the two images: the use of fiducial markers,113 mapping of the AFM tip position with the optical system,115,116 and using features that are present in both images.120,129 However, there is no standard solution to this problem, and especially with living samples, sample movement between imaging modes or during acquisition adds to the complications. Different fields of view, independent pixel sizes, rotation of the images with respect to each other and sample drift during acquisition all pose significant challenges to image correlation.
As with any hybrid system, a significant challenge for simultaneous AFM/SR data acquisition will be the synchronisation of the different imaging modes. Currently in combined AFM/SR systems the instruments are operated independently, making it difficult to integrate data acquisition sequences and match the field of view from the two independent instruments. Development of instruments that were designed for hybrid operation would allow seamless synchronisation, for example, between SR illumination and AFM operation, and would also make image correlation easier.
6. Conclusions and future directions
Over the past decade, AFM has been combined with a myriad of SR techniques available for unravelling biological enigmas. The SR method of choice critically depends on the biological system under observation, as there are trade-offs to be considered regarding fluorescent labelling, optical resolution in three dimensions, frame recording speed and imaging timeframe. Correlative multimodal systems with AFM and SR techniques offer the possibility to retrieve optical, chemical, and biophysical data from many types of biological samples, such as DNA, proteins or cells. One challenge of hybrid AFM and SR optical systems will be the development of single instruments to simplify data acquisition and control software. Currently, in combined AFM–SR systems the imaging modes are operated independently, and image correlation is performed afterwards.Hybrid AFM/SR fluorescence systems have been used for the identification of artefacts in the SR images attributed to poor labelling, photobleaching, or image reconstruction issues. AFM data have been used to validate data obtained with SR, for example, to assess the quality of the labelling. AFM has evolved and the data acquisition rates have vastly increased allowing the monitoring of cytoskeletal and cell membrane dynamics on the millisecond to second timescale in living cells.192,193 Simultaneous AFM and SR experiments on living cells remain a challenge, mainly due to a variety of technological difficulties such as the use of high-power laser sources for SR that interrupt the AFM cantilever operation, the different scan rates of the systems, and the sometimes incompatible imaging buffers. However, SR–SIM is especially amenable to simultaneous data capture with AFM and it has been shown that these two instruments can be operated simultaneously.87 This hybrid system is a promising development for the recording of biophysical data and cellular dynamics visualization at the same time. Further resolution improvement can be achieved by Saturated Structured Illumination Microscopy (SSIM), which is based on the nonlinear relationship between the excitation intensity and the emission rate of fluorophores to produce higher order harmonics, further boosting SIM's potential for resolution improvement. Resolution below 50 nm was demonstrated by Gustafsson et al. in 2005.194 One of the drawbacks of SSIM is the high laser power that is required to create the saturation conditions, which – as with SMLM and STED techniques – is likely to disrupt the AFM cantilever operation, and cause photobleaching and damage to living samples.
Recent technological developments are opening new opportunities for correlative SR and AFM imaging adding high temporal resolution by means of high-speed AFM scanners.190,191,195 The development of HS-AFM has permitted the simultaneous assessment of structure and dynamics of single proteins during their functional activity.195–198 In order to further extend the HS-AFM capabilities, Fukuda et al. presented a combined HS-AFM/TIRFM system and demonstrated simultaneous imaging of linear motions of proteins chitinase A and myosin V.191 Later on, simultaneous, correlative tip-enhanced TIRFM and HS-AFM imaging of fluorescently labelled protein molecules in solution at relatively high concentration was demonstrated.190 Fluorescence enhancement achieved by attaching a single Au particle to the AFM tip is limited to a factor of approximately two due to the distance of the attached Au particle of about 50 nm to the tip. This is expected to be improved by modifying the fabrication of the tip so that enough area can be provided for the tip–Au particle contact near the tip end. Even in this new scenario with localized intense light to be achieved after repositioning the Au particle attachment, photobleaching effects would be reduced compared to conventional TIRFM, as brighter images can be obtained even with lower excitation laser power. In this work the authors go further and show how to achieve higher spatiotemporal resolution with simultaneous high-speed near-field fluorescence microscopy and AFM (HS-SNOM/HS-AFM) by imaging DNA labelled with YOYO-1 in solution.190 The authors coated AFM tips with magnetron-sputtered Ag, and achieved a ca. 39 nm resolution with HS-SNOM and imaging rate of ca. 3 to 8 s per frame for SNOM imaging, which is 100 times higher than the typical SNOM imaging rate. This proof of concept is the first step towards further development of the use HS-SNOM on biological studies in combination with HS-AFM.
HS-AFM has significantly decreased AFM data acquisition time, and further improvement to harmonize AFM and the fluorescence data acquisition rate could be achieved by combining HS-AFM with an SSIM system.194 Another potential pathway consists of combining HS-AFM and video-rate SIM134 for living cell studies. The combination of HS-AFM with other far-field SR fluorescence schemes, e.g., STED, is expected to be readily accomplished, although the high illumination intensities required for most SR techniques remain a challenge for simultaneous data collection. In the next decade, we also envisage the consolidation of hybrid systems like HS-AFM/SNOM as well as the appearance of multimodal hybrid systems integrating AFM with two to three microscopy techniques.
Abbreviations
| AFM | Atomic force microscope |
| CaSki | Human cervical carcinoma cells |
| CHO | Chinese hamster ovarian |
| COS7 | CV-1 in origin with SV40 genes African green monkey kidney fibroblast |
| dSTORM | Direct stochastic optical reconstruction microscopy |
| E. coli | Escherichia coli |
| ECM | Extracellular matrix |
| FIONA | Fluorescence imaging with one nanometer accuracy |
| FN | Fibronectin |
| FWHM | Full width at half maximum |
| GLOX | Glucose oxidase |
| HeLa | Human cervical cancer cells |
| HR | Homologous recombination |
| HUVECs | Human umbilical vein endothelial cells |
| HS-AFM | High-speed atomic force microscopy |
| iSIM | Instant structured illumination microscopy |
| PALM | Photo-activated localization microscopy |
| pTIRFM | Polarized TIRFM |
| ROI | Region of interest |
| SIM | Structured illumination microscopy |
| SMLM | Single-molecule localization microscopy |
| SNOM | Scanning near-field optical microscopy |
| SR | Super-resolution |
| SSIM | Saturated structured illumination microscopy |
| STED | Stimulated emission depletion |
| STORM | Stochastic optical reconstruction microscopy |
| TIRFM | Total internal reflection fluorescence microscopy |
| VSMC | Vascular smooth muscle cells |
| YOYO-1 | Tetracationic homodimer of oxazole yellow |
Author contributions
All authors listed have made substantial, direct, and intellectual contribution to the work, and approved it for publication.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Ana I. Gómez Varela acknowledges support from the Xunta de Galicia, Consellería de Cultura, Educación e Ordenación Universitaria e da Consellería de Economía, and Emprego e Industria (Programa de axudas de apoio á etapa de formación posdoutoral 2017). Adelaide Miranda and Pieter De Beule acknowledge financial support from Norte's Regional Operational Programme 2014-2020-Norte2020 (NORTE-1-145-FEDER-19). This work was supported by the Fundação para a Ciência e a Tecnologia project SAM – [PTDC/NAN-OPT/31596/2017]. Andreas Stylianou acknowledges financial support from the European Research Council and from the University of Cyprus under the projects “CancerFingerPrints” (ERC-2018-PoC-838414) and “PacaFingerPrints” (Advanced Post-doctoral Research Fellowship), respectively.References
- G. Binnig, C. F. Quate and C. Gerber, Phys. Rev. Lett., 1986, 56, 930–933 CrossRef PubMed.
- A. L. Weisenhorn, P. Maivald, H.-J. Butt and P. K. Hansma, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 11226–11232 CrossRef PubMed.
- B. Drake, C. B. Prater, A. L. Weisenhorn, S. A. C. Gould, T. R. Albrecht, C. F. Quate, D. S. Cannell, H. G. Hansma and P. K. Hansma, Science, 1989, 243, 1586–1589 CrossRef CAS PubMed.
- M. Radmacher, R. Tillamnn, M. Fritz and H. Gaub, Science, 1992, 257, 1900–1905 CrossRef CAS PubMed.
- S. M. Lindsay, L. A. Nagahara, T. Thundat, U. Knipping, R. L. Rill, B. Drake, C. B. Prater, A. L. Weisenhorn, S. A. C. Gould and P. K. Hansma, J. Biomol. Struct. Dyn., 1989, 7, 279–287 CrossRef CAS PubMed.
- H. G. Hansma, J. Vesenka, C. Siegerist, G. Kelderman, H. Morrett, R. L. Sinsheimer, V. Elings, C. Bustamante and P. K. Hansma, Science, 1992, 256, 1180–1184 CrossRef CAS PubMed.
- R. D. Edstrom, X. Yang, G. Lee and D. F. Evans, FASEB J., 1990, 4, 3144–3151 CrossRef CAS PubMed.
- A. L. Weisenhorn, B. Drake, C. B. Prater, S. A. Gould, P. K. Hansma, F. Ohnesorge, M. Egger, S. P. Heyn and H. E. Gaub, Biophys. J., 1990, 58, 1251–1258 CrossRef CAS PubMed.
- C. A. Bippes and D. J. Muller, Rep. Prog. Phys., 2011, 74, 086601 CrossRef.
- N. J. Tao, S. M. Lindsay and S. Lees, Biophys. J., 1992, 63, 1165–1169 CrossRef CAS PubMed.
- M. Kammoun, R. Ternifi, V. Dupres, P. Pouletaut, S. Même, W. Même, F. Szeremeta, J. Landoulsi, J.-M. Constans, F. Lafont, M. Subramaniam, J. R. Hawse and S. F. Bensamoun, Sci. Rep., 2019, 9, 7733 CrossRef PubMed.
- A. J. Malkin, T. A. Land, Y. G. Kuznetsov, A. McPherson and J. J. DeYoreo, Phys. Rev. Lett., 1995, 75, 2778–2781 CrossRef CAS PubMed.
- Y. G. Kuznetsov and A. McPherson, Microbiol. Mol. Biol. Rev., 2011, 75, 268–285 CrossRef CAS PubMed.
- H. Schillers, C. Rianna, J. Schäpe, T. Luque, H. Doschke, M. Wälte, J. J. Uriarte, N. Campillo, G. P. A. Michanetzis, J. Bobrowska, A. Dumitru, E. T. Herruzo, S. Bovio, P. Parot, M. Galluzzi, A. Podestà, L. Puricelli, S. Scheuring, Y. Missirlis, R. Garcia, M. Odorico, J. M. Teulon, F. Lafont, M. Lekka, F. Rico, A. Rigato, J. L. Pellequer, H. Oberleithner, D. Navajas and M. Radmacher, Sci. Rep., 2017, 7, 5117 CrossRef PubMed.
- A. Stylianou, M. Lekka and T. Stylianopoulos, Nanoscale, 2018, 10, 20930–20945 RSC.
- M. Krieg, G. Fläschner, D. Alsteens, B. M. Gaub, W. H. Roos, G. J. L. Wuite, H. E. Gaub, C. Gerber, Y. F. Dufrêne and D. J. Müller, Nat. Rev. Phys., 2019, 1, 41–57 CrossRef.
- Y. F. Dufrêne, T. Ando, R. Garcia, D. Alsteens, D. Martinez-Martin, A. Engel, C. Gerber and D. J. Müller, Nat. Nanotechnol., 2017, 12, 295–307 CrossRef PubMed.
- M. B. Viani, T. E. Schäffer, A. Chand, M. Rief, H. E. Gaub and P. K. Hansma, J. Appl. Phys., 1999, 86, 2258–2262 CrossRef CAS.
- T. Ando, N. Kodera, E. Takai, D. Maruyama, K. Saito and A. Toda, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12468–12472 CrossRef CAS PubMed.
- T. Ando, N. Kodera, Y. Naito, T. Kinoshita, K. Furuta and Y. Y. Toyoshima, ChemPhysChem, 2003, 4, 1196–1202 CrossRef CAS PubMed.
- M. Shibata, H. Yamashita, T. Uchihashi, H. Kandori and T. Ando, Nat. Nanotechnol., 2010, 5, 208–212 CrossRef CAS PubMed.
- N. Kodera, D. Yamamoto, R. Ishikawa and T. Ando, Nature, 2010, 468, 72–76 CrossRef CAS PubMed.
- T. Uchihashi, R. Iino, T. Ando and H. Noji, Science, 2011, 333, 755–758 CrossRef CAS PubMed.
- Y. Sakiyama, A. Mazur, L. E. Kapinos and R. Y. H. Lim, Nat. Nanotechnol., 2016, 11, 719–723 CrossRef CAS PubMed.
- G. E. Fantner, R. J. Barbero, D. S. Gray and A. M. Belcher, Nat. Nanotechnol., 2010, 5, 280–285 CrossRef CAS PubMed.
- H. Yamashita, A. Taoka, T. Uchihashi, T. Asano, T. Ando and Y. Fukumori, J. Mol. Biol., 2012, 422, 300–309 CrossRef CAS PubMed.
- M. Shibata, T. Uchihashi, T. Ando and R. Yasuda, Sci. Rep., 2015, 5, 8724 CrossRef CAS PubMed.
- P. Hinterdorfer and Y. F. Dufrêne, Nat. Methods, 2006, 3, 347–355 CrossRef CAS PubMed.
- D. Ristic, H. Sanchez and C. Wyman, in Methods in Molecular Biology, 2011, vol. 783, pp. 213–231 Search PubMed.
- Y. L. Lyubchenko and L. S. Shlyakhtenko, Crit. Rev. Eukaryotic Gene Expression, 2016, 26, 63–96 CrossRef PubMed.
- D. J. Müller, M. Krieg, D. Alsteens and Y. F. Dufrêne, Curr. Opin. Biotechnol., 2009, 20, 4–13 CrossRef PubMed.
- S.-H. Jung, D. Park, J. H. Park, Y.-M. Kim and K.-S. Ha, Exp. Mol. Med., 2010, 42, 597 CrossRef CAS PubMed.
- D. J. Müller and Y. F. Dufrêne, Trends Cell Biol., 2011, 21, 461–469 CrossRef PubMed.
- T. Ando, T. Uchihashi and N. Kodera, Annu. Rev. Biophys., 2013, 42, 393–414 CrossRef CAS PubMed.
- Y. F. Dufrêne, D. Martínez-Martín, I. Medalsy, D. Alsteens and D. J. Müller, Nat. Methods, 2013, 10, 847–854 CrossRef PubMed.
- K. Haase and A. E. Pelling, J. R. Soc., Interface, 2015, 12, 20140970 CrossRef PubMed.
- F. A. Carvalho and N. C. Santos, IUBMB Life, 2012, 64, 465–472 CrossRef CAS PubMed.
- C. Bustamante, D. Keller and G. Yang, Curr. Opin. Struct. Biol., 1993, 3, 363–372 CrossRef CAS.
- S. Moreno Flores and J. L. Toca-Herrera, Nanoscale, 2009, 1, 40 RSC.
- J. Caplan, M. Niethammer, R. M. Taylor and K. J. Czymmek, Curr. Opin. Struct. Biol., 2011, 21, 686–693 CrossRef CAS PubMed.
- T. Ando, S. P. Bhamidimarri, N. Brending, H. Colin-York, L. Collinson, N. De Jonge, P. J. de Pablo, E. Debroye, C. Eggeling, C. Franck, M. Fritzsche, H. Gerritsen, B. N. G. Giepmans, K. Grunewald, J. Hofkens, J. P. Hoogenboom, K. P. F. Janssen, R. Kaufmann, J. Klumperman, N. Kurniawan, J. Kusch, N. Liv, V. Parekh, D. B. Peckys, F. Rehfeldt, D. C. Reutens, M. B. J. Roeffaers, T. Salditt, I. A. T. Schaap, U. S. Schwarz, P. Verkade, M. W. Vogel, R. Wagner, M. Winterhalter, H. Yuan and G. Zifarelli, J. Phys. D: Appl. Phys., 2018, 51, 443001 CrossRef PubMed.
- A. Walter, P. Paul-Gilloteaux, B. Plochberger, L. Sefc, P. Verkade, J. G. Mannheim, P. Slezak, A. Unterhuber, M. Marchetti-Deschmann, M. Ogris, K. Bühler, D. Fixler, S. H. Geyer, W. J. Weninger, M. Glösmann, S. Handschuh and T. Wanek, Front. Phys., 2020, 8, 47 CrossRef.
- J. Ma, X. Lv, S. Yang, G. Tian and X. Liu, Microsc. Microanal., 2015, 21, 1304–1313 CrossRef CAS PubMed.
- M. Shemesh, S. Addadi, Y. Milstein, B. Geiger and L. Addadi, ACS Appl. Mater. Interfaces, 2016, 8, 14932–14943 CrossRef CAS PubMed.
- G. D. McEwen, Y. Wu, M. Tang, X. Qi, Z. Xiao, S. M. Baker, T. Yu, T. A. Gilbertson, D. B. DeWald and A. Zhou, Analyst, 2013, 138, 787–797 RSC.
- O. Chaudhuri, S. H. Parekh, W. A. Lam and D. A. Fletcher, Nat. Methods, 2009, 6, 383–387 CrossRef CAS PubMed.
- R. Ghosh Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef CAS PubMed.
- P. Bondia, J. Torra, C. M. Tone, T. Sawazaki, A. del Valle, B. Sot, S. Nonell, M. Kanai, Y. Sohma and C. Flors, J. Am. Chem. Soc., 2020, 142, 922–930 CrossRef CAS PubMed.
- N. A. Geisse, Mater. Today, 2009, 12, 40–45 CrossRef CAS.
- A. R. Burns, Langmuir, 2003, 19, 8358–8363 CrossRef CAS.
- G. Pfister, C. M. Stroh, H. Perschinka, M. Kind, M. Knoflach, P. Hinterdorfer and G. Wick, J. Cell Sci., 2005, 118, 1587–1594 CrossRef CAS.
- R. Kassies, K. O. van der Werf, A. Lenferink, C. N. Hunter, J. D. Olsen, V. Subramaniam and C. Otto, J. Microsc., 2005, 217, 109–116 CrossRef CAS PubMed.
- Z. Deng, T. Zink, H. Chen, D. Walters, F. Liu and G. Liu, Biophys. J., 2009, 96, 1629–1639 CrossRef CAS PubMed.
- M. Krause, J. te Riet and K. Wolf, Phys. Biol., 2013, 10, 065002 CrossRef CAS PubMed.
- M. S. Kuyukina, I. B. Ivshina, I. O. Korshunova and E. V. Rubtsova, J. Microbiol. Methods, 2014, 107, 23–29 CrossRef CAS PubMed.
- J. R. Staunton, B. L. Doss, S. Lindsay and R. Ros, Sci. Rep., 2016, 6, 19686 CrossRef CAS PubMed.
- S. V. Bhat, T. Sultana, A. Körnig, S. McGrath, Z. Shahina and T. E. S. Dahms, Sci. Rep., 2018, 8, 1–10 CAS.
- A. Trache and S.-M. Lim, J. Visualized Exp., 2010, 4–7 Search PubMed.
- A. Miranda, M. Martins and P. A. A. De Beule, Rev. Sci. Instrum., 2015, 86, 093705 CrossRef PubMed.
- C. C. Moura, A. Miranda, R. O. C. Oreffo and P. A. A. De Beule, Biochem. Biophys. Res. Commun., 2020, 529, 392–397 CrossRef CAS PubMed.
- K. Beicker, E. T. O'Brien, M. R. Falvo and R. Superfine, Sci. Rep., 2018, 8, 1504 CrossRef PubMed.
- E. Nelsen, C. M. Hobson, M. E. Kern, J. P. Hsiao, E. T. O'Brien III, T. Watanabe, B. M. Condon, M. Boyce, S. Grinstein, K. M. Hahn, M. R. Falvo and R. Superfine, Sci. Rep., 2020, 10, 8133 CrossRef CAS PubMed.
- C. M. Hobson, M. Kern, E. T. O’Brien, A. D. Stephens, M. R. Falvo and R. Superfine, Mol. Biol. Cell, 2020, 31(16), 1788–1801 CrossRef CAS PubMed.
- S. A. Vickery and R. C. Dunn, J. Microsc., 2001, 202, 408–412 CrossRef CAS PubMed.
- M. Micic, D. Hu, Y. D. Suh, G. Newton, M. Romine and H. P. Lu, Colloids Surf., B, 2004, 34, 205–212 CrossRef CAS PubMed.
- S. Chiantia, N. Kahya, J. Ries and P. Schwille, Biophys. J., 2006, 90, 4500–4508 CrossRef CAS PubMed.
- E. Abbe, Arch. Mikrosk. Anat., 1873, 9, 413–468 CrossRef.
- B. Huang, M. Bates and X. Zhuang, Annu. Rev. Biochem., 2009, 78, 993–1016 CrossRef CAS PubMed.
- L. Schermelleh, R. Heintzmann and H. Leonhardt, J. Cell Biol., 2010, 190, 165–175 CrossRef CAS PubMed.
- A. M. Sydor, K. J. Czymmek, E. M. Puchner and V. Mennella, Trends Cell Biol., 2015, 25, 730–748 CrossRef CAS.
- G. T. Di Francia, Nuovo Cimento, 1952, 9, 426–438 CrossRef.
- G. Toraldo di Francia, J. Opt. Soc. Am., 1955, 45, 497 CrossRef.
- S. W. Hell and J. Wichmann, Opt. Lett., 1994, 19, 780–782 CrossRef CAS PubMed.
- M. Dyba and S. W. Hell, Phys. Rev. Lett., 2002, 88, 163901 CrossRef PubMed.
- V. Westphal, L. Kastrup and S. W. Hell, Appl. Phys. B, 2003, 77, 377–380 CrossRef CAS.
- M. G. L. Gustafsson, J. Microsc., 2000, 198, 82–87 CrossRef CAS PubMed.
- E. Betzig, Opt. Lett., 1995, 20, 237 CrossRef CAS PubMed.
- M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–795 CrossRef CAS PubMed.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS PubMed.
- S. T. Hess, T. P. K. Girirajan and M. D. Mason, Biophys. J., 2006, 91, 4258–4272 CrossRef CAS PubMed.
- H. Miller, Z. Zhou, J. Shepherd, A. J. M. Wollman and M. C. Leake, Rep. Prog. Phys., 2018, 81, 024601 CrossRef.
- M. Hauser, M. Wojcik, D. Kim, M. Mahmoudi, W. Li and K. Xu, Chem. Rev., 2017, 117, 7428–7456 CrossRef CAS.
- B. Harke, J. V. Chacko, H. Haschke, C. Canale and A. Diaspro, Opt. Nanosc., 2012, 1, 3 CrossRef.
- A. Hermsdörfer, J. Madl and W. Römer, Tech. note JPK Instruments AG, Ger., 2013, 1–9 Search PubMed.
- J. V. Chacko, F. C. Zanacchi and A. Diaspro, Cytoskeleton, 2013, 70, 729–740 CrossRef CAS.
- P. D. Odermatt, A. Shivanandan, H. Deschout, R. Jankele, A. P. Nievergelt, L. Feletti, M. W. Davidson, A. Radenovic and G. E. Fantner, Nano Lett., 2015, 15, 4896–4904 CrossRef CAS.
- A. I. Gómez-Varela, D. R. Stamov, A. Miranda, R. Alves, C. Barata-Antunes, D. Dambournet, D. G. Drubin, S. Paiva and P. A. A. De Beule, Sci. Rep., 2020, 10, 1122 CrossRef PubMed.
- J. E. Shaw, J. Oreopoulos, D. Wong, J. C. Y. Hsu and C. M. Yip, Surf. Interface Anal., 2006, 38, 1459–1471 CrossRef CAS.
- A. Rygula, T. Oleszkiewicz, E. Grzebelus, M. Z. Pacia, M. Baranska and R. Baranski, Spectrochim. Acta, Part A, 2018, 197, 47–55 CrossRef CAS PubMed.
- E. Bailo and V. Deckert, Angew. Chem., Int. Ed., 2008, 47, 1658–1661 CrossRef CAS PubMed.
- R. Garcia and E. T. Herruzo, Nat. Nanotechnol., 2012, 7, 217–226 CrossRef CAS PubMed.
- Z. Al-Rekabi and S. Contera, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2658–2663 CrossRef CAS PubMed.
- P. Eaton and P. West, Atomic Force Microscopy, Oxford University Press, 2019 Search PubMed.
- A. Stylianou and D. Yova, Mater. Sci. Eng., C, 2013, 33, 2947–2957 CrossRef CAS.
- N. Gadegaard, Biotech. Histochem., 2006, 81, 87–97 CrossRef CAS.
- D. J. Müller, A. C. Dumitru, C. Lo Giudice, H. E. Gaub, P. Hinterdorfer, G. Hummer, J. J. De Yoreo, Y. F. Dufrêne and D. Alsteens, Chem. Rev., 2020 DOI:10.1021/acs.chemrev.0c00617.
- H. J. Butt, B. Cappella and M. Kappl, Surf. Sci. Rep., 2005, 59, 1–152 CrossRef CAS.
- A. Stylianou, V. Gkretsi and T. Stylianopoulos, MethodsX, 2018, 5, 503–513 CrossRef.
- A. Stylianou, V. Gkretsi, C. S. Patrickios and T. Stylianopoulos, Exploring the nano-surface of collagenous and other fibrotic tissue with AFM, in Fibrosis, ed. S. N. Y. L. Rittié, New York, 2017, pp. 453–489 Search PubMed.
- D. Alsteens, H. E. Gaub, R. Newton, M. Pfreundschuh, C. Gerber and D. J. Müller, Nat. Rev. Mater., 2017, 2, 1–16 Search PubMed.
- S. V. Kontomaris, A. Stylianou, K. S. Nikita, A. Malamou and T. Stylianopoulos, Phys. Biol., 2019, 16, 056003 CrossRef CAS PubMed.
- S. V. Kontomaris, A. Stylianou, K. S. Nikita and A. Malamou, Mater. Res. Express, 2019, 6, 115410 CrossRef.
- D. J. Müller and Y. F. Dufrêne, Nat. Nanotechnol., 2008, 3, 261–269 CrossRef.
- S. K. Kufer, E. M. Puchner, H. Gumpp, T. Liedl and H. E. Gaub, Science, 2008, 319, 594–596 CrossRef CAS.
- A. Yildiz, J. N. Forkey, S. A. McKinney, T. Ha, Y. E. Goldman and P. R. Selvin, Science, 2003, 300, 2061–2065 CrossRef CAS.
- M. Heilemann, S. Van De Linde, M. Schüttpelz, R. Kasper, B. Seefeldt, A. Mukherjee, P. Tinnefeld and M. Sauer, Angew. Chem., Int. Ed., 2008, 47, 6172–6176 CrossRef CAS.
- J. Vogelsang, T. Cordes, C. Forthmann, C. Steinhauer and P. Tinnefeld, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8107–8112 CrossRef CAS PubMed.
- T. Cordes, M. Strackharn, S. W. Stahl, W. Summerer, C. Steinhauer, C. Forthmann, E. M. Puchner, J. Vogelsang, H. E. Gaub and P. Tinnefeld, Nano Lett., 2010, 10, 645–651 CrossRef CAS PubMed.
- S. van de Linde, U. Endesfelder, A. Mukherjee, M. Schüttpelz, G. Wiebusch, S. Wolter, M. Heilemann and M. Sauer, Photochem. Photobiol. Sci., 2009, 8, 465 CrossRef CAS PubMed.
- A. Sharonov and R. M. Hochstrasser, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18911–18916 CrossRef CAS PubMed.
- R. Jungmann, M. S. Avendaño, J. B. Woehrstein, M. Dai, W. M. Shih and P. Yin, Nat. Methods, 2014, 11, 313–318 CrossRef CAS PubMed.
- D. N. Fronczek, C. Quammen, H. Wang, C. Kisker, R. Superfine, R. Taylor, D. A. Erie and I. Tessmer, Ultramicroscopy, 2011, 111, 350–355 CrossRef CAS.
- W. C. Duim, B. Chen, J. Frydman and W. E. Moerner, ChemPhysChem, 2011, 12, 2387–2390 CrossRef CAS.
- R. D. Turner, A. F. Hurd, A. Cadby, J. K. Hobbs and S. J. Foster, Nat. Commun., 2013, 4, 1496 CrossRef.
- A. Monserrate, S. Casado and C. Flors, ChemPhysChem, 2014, 15, 647–650 CrossRef CAS.
- P. Bondia, S. Casado and C. Flors, in Methods in Molecular Biology, 2017, vol. 1663, pp. 105–113 Search PubMed.
- P. Bondia, R. Jurado, S. Casado, J. M. Domínguez-Vera, N. Gálvez and C. Flors, Small, 2017, 13, 1603784 CrossRef.
- J. Wang, Z. Wang, Y. Xu, X. Wang, Z. Yang, H. Wang and Z. Tian, Nanoscale, 2020, 12, 17203–17212 RSC.
- L. M. Hirvonen, G. E. Jones and S. Cox, Microsc. Anal., 2018, 32, 17–19 Search PubMed.
- L. M. Hirvonen and S. Cox, Methods Appl. Fluoresc., 2018, 6, 045002 CrossRef CAS.
- L. M. Hirvonen, R. J. Marsh, G. E. Jones and S. Cox, Eur. J. Cell Biol., 2020, 99, 151106 CrossRef CAS.
- S. Dahmane, C. Doucet, A. Le Gall, C. Chamontin, P. Dosset, F. Murcy, L. Fernandez, D. Salas, E. Rubinstein, M. Mougel, M. Nollmann and P.-E. Milhiet, Nanoscale, 2019, 11, 6036–6044 RSC.
- L. Zhou, J. Gao, H. Wang, Y. Shi, H. Xu, Q. Yan, Y. Jing, J. Jiang, M. Cai and H. Wang, Nanoscale, 2020, 12, 9950–9957 RSC.
- J. V. Chacko, C. Canale, B. Harke and A. Diaspro, PLoS One, 2013, 8, e66608 CrossRef CAS PubMed.
- J. Yu, J. Yuan, X. Zhang, J. Liu and X. Fang, Chin. Sci. Bull., 2013, 58, 4045–4050 CrossRef CAS.
- J. V. Chacko, B. Harke, C. Canale and A. Diaspro, J. Biomed. Opt., 2014, 19, 105003 Search PubMed.
- N. Curry, G. Ghézali, G. S. Kaminski Schierle, N. Rouach and C. F. Kaminski, Front. Cell. Neurosci., 2017, 11, 104 CrossRef.
- P. Mahou, N. Curry, D. Pinotsi, G. K. Schierle and C. Kaminski, in Proc. SPIE, 2015, vol. 9331 Search PubMed.
- G. Lukinavičius, L. Reymond, E. D'Este, A. Masharina, F. Göttfert, H. Ta, A. Güther, M. Fournier, S. Rizzo, H. Waldmann, C. Blaukopf, C. Sommer, D. W. Gerlich, H.-D. Arndt, S. W. Hell and K. Johnsson, Nat. Methods, 2014, 11, 731–733 CrossRef PubMed.
- M. Cosentino, C. Canale, P. Bianchini and A. Diaspro, Sci. Adv., 2019, 5, eaav8062 CrossRef PubMed.
- T. Deguchi, E. Fazeli, S. Koho, P. Pennanen, M. Alanne, M. Modi, J. E. Eriksson, K. V. Vienola, P. E. Hänninen, J. Peltonen and T. Näreoja, J. Phys. D: Appl. Phys., 2020, 53, 014003 CrossRef CAS.
- B.-J. Chang, L.-J. Chou, Y.-C. Chang and S.-Y. Chiang, Opt. Express, 2009, 17, 14710 CrossRef CAS PubMed.
- L. M. Hirvonen, K. Wicker, O. Mandula and R. Heintzmann, Eur. Biophys. J., 2009, 38, 807–812 CrossRef PubMed.
- A. Markwirth, M. Lachetta, V. Mönkemöller, R. Heintzmann, W. Hübner, T. Huser and M. Müller, Nat. Commun., 2019, 10, 4315 CrossRef.
- A. Sandmeyer, M. Lachetta, H. Sandmeyer, W. Hübner, T. Huser and M. Müller, bioRxiv, 2019, 797670 Search PubMed.
- P. T. Brown, R. Kruithoff, G. J. Seedorf and D. P. Shepherd, bioRxiv, 2020, 1–12 CAS.
- T. Woo, S. H. Jung, C. Ahn, B. Hwang, H. Kim, J. H. Kang and J.-H. Park, Optica, 2020, 7, 973 CrossRef.
- M. Lachetta, H. Sandmeyer, A. Sandmeyer, J. Schulte Am Esch, T. Huser and M. Müller, bioRxiv, 2020, 2020.10.02.323527 Search PubMed.
- M. G. L. Gustafsson, L. Shao, P. M. Carlton, C. J. R. Wang, I. N. Golubovskaya, W. Z. Cande, D. A. Agard and J. W. Sedat, Biophys. J., 2008, 94, 4957–4970 CrossRef CAS PubMed.
- L. Schermelleh, P. M. Carlton, S. Haase, L. Shao, L. Winoto, P. Kner, B. Burke, M. C. Cardoso, D. A. Agard, M. G. L. Gustafsson, H. Leonhardt and J. W. Sedat, Science, 2008, 320, 1332–1336 CrossRef CAS.
- D. Axelrod, J. Cell Biol., 1981, 89, 141–145 CrossRef CAS.
- A. L. Mattheyses, S. M. Simon and J. Z. Rappoport, J. Cell Sci., 2010, 123, 3621–3628 CrossRef CAS PubMed.
- E. J. Ambrose, Nature, 1956, 178, 1194–1194 CrossRef CAS PubMed.
- T. Funatsu, Y. Harada, M. Tokunaga, K. Saito and T. Yanagida, Nature, 1995, 374, 555–559 CrossRef CAS PubMed.
- K. N. Fish, Curr. Protoc. Cytom., 2009, 50, 12.18.1–12.18.13 Search PubMed.
- Y. Fu, P. W. Winter, R. Rojas, V. Wang, M. McAuliffe and G. H. Patterson, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4368–4373 CrossRef CAS PubMed.
- M. Guo, P. Chandris, J. P. Giannini, A. J. Trexler, R. Fischer, J. Chen, H. D. Vishwasrao, I. Rey-Suarez, Y. Wu, X. Wu, C. M. Waterman, G. H. Patterson, A. Upadhyaya, J. W. Taraska and H. Shroff, Nat. Methods, 2018, 15, 425–428 CrossRef CAS PubMed.
- L. Zhou, M. Cai, T. Tong and H. Wang, Sensors, 2017, 17, 938 CrossRef PubMed.
- S. Handschuh-Wang, T. Wang and X. Zhou, RSC Adv., 2017, 7, 47464–47499 RSC.
- A. E. X. Brown, A. Hategan, D. Safer, Y. E. Goldman and D. E. Discher, Biophys. J., 2009, 96, 1952–1960 CrossRef CAS.
- T. Gudzenko and C. M. Franz, Mol. Biol. Cell, 2015, 26, 3190–3204 CrossRef CAS.
- J. Oreopoulos and C. M. Yip, Biophys. J., 2009, 96, 1970–1984 CrossRef CAS PubMed.
- J. Oreopoulos, R. F. Epand, R. M. Epand and C. M. Yip, Biophys. J., 2010, 98, 815–823 CrossRef CAS.
- Y. Ebenstein, N. Gassman, S. Kim and S. Weiss, in Journal of Molecular Recognition, NIH Public Access, 2009, vol. 22, pp. 397–402 Search PubMed.
- H. Sanchez, A. Kertokalio, S. van Rossum-Fikkert, R. Kanaar and C. Wyman, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11385–11390 CrossRef CAS PubMed.
- H. Sánchez, M. W. Paul, M. Grosbart, S. E. Van Rossum-Fikkert, J. H. G. Lebbink, R. Kanaar, A. B. Houtsmuller and C. Wyman, Nucleic Acids Res., 2017, 45, 4507–4518 CrossRef PubMed.
- H. Sanchez, R. Kanaar and C. Wyman, Ultramicroscopy, 2010, 110, 844–851 CrossRef CAS.
- W. Frederickx, S. Rocha, Y. Fujita, K. Kennes, H. De Keersmaecker, S. De Feyter, H. Uji-I and W. Vanderlinden, ACS Nano, 2018, 12, 168–177 CrossRef CAS PubMed.
- H. Sánchez and C. Wyman, BMC Bioinf., 2015, 16, 27 CrossRef PubMed.
- M. Grosbart, D. Ristić, H. Sánchez and C. Wyman, in Methods in Molecular Biology, Humana Press Inc., 2018, vol. 1665, pp. 259–280 Search PubMed.
- A. Sidhu, D. Ristic, H. Sánchez and C. Wyman, in Methods in Enzymology, Academic Press Inc., 2018, vol. 600, pp. 347–374 Search PubMed.
- H. Sanchez, M. Reuter, M. Yokokawa, K. Takeyasu and C. Wyman, DNA Repair, 2014, 20, 110–118 CrossRef CAS PubMed.
- W. D. Heyer, K. T. Ehmsen and J. Liu, Annu. Rev. Genet., 2010, 44, 113–139 CrossRef CAS PubMed.
- S. Nishida, Y. Funabashi and A. Ikai, Ultramicroscopy, 2002, 91, 269–274 CrossRef CAS PubMed.
- M. S. Z. Kellermayer, Á. Karsai, A. Kengyel, A. Nagy, P. Bianco, T. Huber, Á. Kulcsár, C. Niedetzky, R. Proksch and L. Grama, Biophys. J., 2006, 91, 2665–2677 CrossRef CAS PubMed.
- A. B. Mathur, G. A. Truskey and W. M. Reichert, Biophys. J., 2000, 78, 1725–1735 CrossRef CAS PubMed.
- A. Trache and S.-M. Lim, J. Biomed. Opt., 2009, 14, 034024 CrossRef PubMed.
- S.-M. Lim, J. P. Trzeciakowski, H. Sreenivasappa, L. J. Dangott and A. Trache, Integr. Biol., 2012, 4, 615–627 CrossRef CAS PubMed.
- W. Christenson, I. Yermolenko, B. Plochberger, F. Camacho-Alanis, A. Ros, T. P. Ugarova and R. Ros, Ultramicroscopy, 2014, 136, 211–215 CrossRef CAS PubMed.
- A. Kumar and G. M. Whitesides, Appl. Phys. Lett., 1993, 63, 2002–2004 CrossRef CAS.
- A. Ortega-Esteban, K. Bodensiek, C. San Martín, M. Suomalainen, U. F. Greber, P. J. De Pablo and I. A. T. Schaap, ACS Nano, 2015, 9, 10571–10579 CrossRef CAS PubMed.
- A. Sarkar, R. B. Robertson and J. M. Fernandez, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12882–12886 CrossRef CAS PubMed.
- K. Maki, S. W. Han, Y. Hirano, S. Yonemura, T. Hakoshima and T. Adachi, Sci. Rep., 2018, 8, 1575 CrossRef.
- M. C. Harris, D. Cislo, J. S. Lenz, C. Umbach and M. Lindau, PLoS One, 2017, 12, e0173993 CrossRef PubMed.
- A. Bouhelier, in Reference Module in Materials Science and Materials Engineering, Elsevier, 2016 Search PubMed.
- A. Lewis, M. Isaacson, A. Harootunian and A. Muray, Ultramicroscopy, 1984, 13, 227–231 CrossRef.
- D. W. Pohl, W. Denk and M. Lanz, Appl. Phys. Lett., 1984, 44, 651–653 CrossRef.
- E. Betzig, A. Lewis, A. Harootunian, M. Isaacson and E. Kratschmer, Biophys. J., 1986, 49, 269–279 CrossRef CAS PubMed.
- Y. Inouye and S. Kawata, Opt. Lett., 1994, 19, 159–161 CrossRef CAS.
- F. Zenhausern, M. P. O'Boyle and H. K. Wickramasinghe, Appl. Phys. Lett., 1994, 65, 1623–1625 CrossRef CAS.
- S. Kawata, Y. Inouye and P. Verma, Nat. Photonics, 2009, 3, 388–394 CrossRef CAS.
- N. Hayazawa, K. Furusawa, A. Taguchi and S. Kawata, J. Appl. Phys., 2009, 106, 113103 CrossRef.
- A. Hartschuh, E. J. Sánchez, X. S. Xie and L. Novotny, Phys. Rev. Lett., 2003, 90, 095503 CrossRef.
- E. A. Muller, B. Pollard, H. A. Bechtel, P. van Blerkom and M. B. Raschke, Sci. Adv., 2016, 2, e1601006–e1601006 CrossRef.
- W. Bao, N. J. Borys, C. Ko, J. Suh, W. Fan, A. Thron, Y. Zhang, A. Buyanin, J. Zhang, S. Cabrini, P. D. Ashby, A. Weber-Bargioni, S. Tongay, S. Aloni, D. F. Ogletree, J. Wu, M. B. Salmeron and P. J. Schuck, Nat. Commun., 2015, 6, 7993 CrossRef CAS PubMed.
- V. Subramaniam, A. K. Kirsch and T. M. Jovin, Cell. Mol. Biol., 1998, 44, 689–700 CAS.
- P. Bazylewski, S. Ezugwu and G. Fanchini, Appl. Sci., 2017, 7, 973 CrossRef.
- S. Berweger, D. M. Nguyen, E. A. Muller, H. A. Bechtel, T. T. Perkins and M. B. Raschke, J. Am. Chem. Soc., 2013, 135, 18292–18295 CrossRef CAS PubMed.
- A. Rygula, T. Oleszkiewicz, E. Grzebelus, M. Z. Pacia, M. Baranska and R. Baranski, Spectrochim. Acta, Part A, 2018, 197, 47–55 CrossRef CAS PubMed.
- T. Umakoshi, S. Fukuda, R. Iino, T. Uchihashi and T. Ando, Biochim. Biophys. Acta, Gen. Subj., 2020, 1864, 129325 CrossRef CAS PubMed.
- S. Fukuda, T. Uchihashi, R. Iino, Y. Okazaki, M. Yoshida, K. Igarashi and T. Ando, Rev. Sci. Instrum., 2013, 84, 073706 CrossRef PubMed.
- T. Uchihashi, H. Watanabe, S. Fukuda, M. Shibata and T. Ando, Ultramicroscopy, 2016, 160, 182–196 CrossRef CAS PubMed.
- M. Shibata, H. Watanabe, T. Uchihashi, T. Ando and R. Yasuda, Biophys. Physicobiol., 2017, 14, 127–135 CrossRef CAS PubMed.
- M. G. L. Gustafsson, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13081–13086 CrossRef CAS PubMed.
- T. Ando, Biophys. Rev., 2018, 10, 285–292 CrossRef CAS PubMed.
- T. Ando, T. Uchihashi and T. Fukuma, Prog. Surf. Sci., 2008, 83, 337–437 CrossRef CAS.
- T. Ando, Nanotechnology, 2012, 23, 062001 CrossRef PubMed.
- H. Sanchez, Y. Suzuki, M. Yokokawa, K. Takeyasu and C. Wyman, Integr. Biol., 2011, 3, 1127 CrossRef CAS PubMed.
Footnote |
| † Contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |