
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aqueous core and hollow silica nanocapsules for confined enzyme modules†
Seong-Min
Jo
 a,
Shuai
Jiang
a,
Robert
Graf
a,
Frederik R.
Wurm
*ab and
Katharina
Landfester
*a
a,
Shuai
Jiang
a,
Robert
Graf
a,
Frederik R.
Wurm
*ab and
Katharina
Landfester
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128, Mainz, Germany. E-mail: frederik.wurm@utwente.nl; landfester@mpip-mainz.mpg.de
bSustainable Polymer Chemistry Group, MESA+ Institute for Nanotechnology, Faculty of Science and Technology, Universiteit Twente, PO Box 217, 7500 AE Enschede, The Netherlands
First published on 16th November 2020
Abstract
The development of enzyme modules by coupling several enzymes in confinement is of paramount importance to artificial biological reaction systems for efficient enzymatic reactions. Silica nanocapsules are ideal candidates for loading enzymes. Aqueous core silica nanocapsules have relatively been rarely reported due to the crux of difficulty in forming dense silica shells by interfacial sol–gel reactions. Herein we suggest a one-step synthesis of hollow silica nanocapsules with an aqueous core containing enzymes via a template-free and interfacial condensation method for developing enzyme modules with coupled enzymatic reactions. As a proof-of-concept, we developed enzyme modules for three useful purposes by encapsulating a couple of enzymes: (i) development of a miniature glucose sensor, (ii) protection of living cells, and (iii) regeneration of nicotinamide adenine dinucleotides (NADs). By the modulation of enzymes using silica nanocapsules, more efficient coupled reactions, separation of enzymatic reactions from surroundings, and easy handling of several enzymes by using a single module could be achieved. Therefore, our silica nanocapsules for enzyme modules can be promoted as general platforms for developing artificial nanoreactors.
Introduction
The development of enzyme modules by coupling several enzymes in confinement is of paramount importance to artificial biological reaction systems. Confining enzymes is significant to (i) maintain the high stability of the enzymes, (ii) decouple the reactions from surroundings, and (iii) locally enforce that several enzymes interact with each other.1,2 An enzymatic reaction is extremely sensitive and affected by the surroundings. Besides, many useful enzymatic reactions are carried out together as coupled reaction steps, including two or more sequential processes without the isolation of the intermediates.3 Thus, several coupled enzymes can work together more efficiently upon confinement as a single enzyme module.4–7As nanoplatforms for enzyme modules, silica nanocapsules are ideal candidates for loading enzymes compared to other containers, like polymersomes and metal–organic frameworks (MOFs). Polymersomes have the advantage of a large inner cavity to load hydrophilic enzymes,8–10 but they need specially synthesized designed copolymers, and the loading efficiency of enzymes is usually limited.11 Zeolitic imidazole frameworks (ZIFs) are widely used MOF-nanoparticles with loaded enzymes. The advantage here is that the particles are synthesized at ambient temperatures,12–14 but the interference of enzymatic reactions with metal ions or imidazole derivatives limits the choice of enzymes. Therefore, an encapsulation procedure is needed to encapsulate a wide range of enzymes and a combination of enzymes at room temperature with high efficiency without the formation of components that may inhibit the enzymatic reactions.
We encapsulated enzymes with high efficiency at room temperature inside one hollow cavity of silica nanocapsules, ensuring a higher activity of the enzymes after encapsulation. A silica shell is semi-permeable, meaning that it is impermeable for enzymes and permeable for the reactants and products of the reactions.15,16 Tremendous efforts have been devoted to developing hollow silica nanocapsules with an organic core, which are not favorable to load water-soluble enzymes.17–20 In contrast, aqueous core silica nanocapsules have relatively been less reported due to the crux of difficulty in forming dense silica shells by interfacial sol–gel reactions.21 The hard-templating synthesis of aqueous core silica nanocapsules is the most attempted route.22–29 Sacrificial template nanoparticles are utilized during the synthetic process, and then the core is removed via etching, solvation, calcination, or solubilization, leaving the enzymes entrapped in the nanocapsules. However, the hard template approach is performed under harsh conditions such as in acidic or alkaline solutions, in organic solvents, or at high temperatures to degrade the templates. In addition, a multi-step fabrication process is required. As most enzymes are highly sensitive and delicate, such harsh conditions decrease the activity and stability of enzymes. Even very robust enzymes, e.g., glucose oxidase (GOX), significantly lose their activity under such conditions. Thus, a more general, one-step, and mild synthetic strategy for the encapsulation of a wide variety of enzymes in silica shells is still demanded. Even though some studies reported the encapsulation of water-soluble substances in aqueous core silica nanocapsules by the sol–gel reactions of trichlorosilane precursors, the loading of enzymes has not yet been demonstrated possibly due to the formation of hydrochloric acid, which might denature the enzymes.30,31
Herein we propose a one-step synthesis of hollow silica nanocapsules with an aqueous core containing enzymes via a template-free and interfacial condensation method for developing enzyme modules by coupled enzymatic reactions. By combining two silica precursors, (3-aminopropyl)triethoxysilane (APTMS) and tetraethyl orthosilicate (TEOS), and based on a water-in-oil miniemulsion, we fabricated hollow and aqueous core silica nanocapsules through an interfacial sol–gel reaction. We showed that the sol–gel reaction by fluoride-catalysis allowed us to preserve the best enzyme due to the neutral synthetic conditions. Further steps were not necessary to remove any template. This approach allowed the encapsulation of enzymes by a simplified synthetic procedure and preservation of the high enzymatic activity. As a proof-of-concept, we developed enzyme modules for three useful purposes by encapsulating a couple of enzymes: (i) development of a miniature glucose sensor, (ii) protection of living cells, and (iii) regeneration of nicotinamide adenine dinucleotides (NADs). By the modulation of enzymes using silica nanocapsules, more efficient coupled reactions, separation of the enzymatic reactions from surroundings, and easy handling of several enzymes by using a single module could be achieved. Therefore, our silica nanocapsules can be promoted as general platforms for developing artificial nanoreactors.
Results and discussion
Synthesis of hollow silica nanocapsules with an aqueous core
To obtain core–shell structures with an aqueous core and a membrane that is permeable for the substrates of the enzymes but keeps the enzyme inside, we used the sol–gel chemistry of siloxanes at the interface of inverse (water-in-oil) miniemulsion droplets (Fig. 1b and c). Besides tetraethyl orthosilicate (TEOS), which was used as the matrix precursor for the sol–gel reaction, we added two interfacially active silica precursors to confine the sol–gel process at the interface and thus generate hollow silica nanocapsules instead of solid matrix particles. Octyl trimethoxysilane (octyl-TMS) and aminopropyl trimethoxysilane (APTMS) were used as comonomers, and their influence on the morphologies of the resulting nanocapsules was studied. In the first test reactions, we used ammonia as the catalyst for the sol–gel reaction, resulting in a pH value of 11 for the aqueous droplets of the emulsion. Polyglycerol polyricinoleate (PGPR) was used as the respective surfactant to stabilize the droplets in an inverse miniemulsion.32–35 The miniemulsion process involved a minimal amount of surfactant, which was expected to avoid the denaturation of the enzymes compared to microemulsion-based synthesis.36 The molar ratio of the silica precursors was varied, and it was found that TEOS alone did not lead to the formation of core–shell structures; additionally, APTMS was found to be a necessary ingredient to obtain core–shell structures (Fig. S1a, S1d and S1f†). With a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:2 = APTMS:TEOS:octyl-TMS, we successfully obtained hollow silica capsules with an aqueous core (Fig. S1d and S1e†). In addition, octyl-TMS was not found to be essential for the preparation of the capsules despite its amphiphilic nature (Fig. S1d to S1f†). Particularly, the combination of APTMS:TEOS = 1:5 generated core–shell structures effectively. Similar results were obtained when APTMS was substituted with the respective ethyl derivative, aminopropyl triethoxysilane (APTES) (Fig. S2†). We assumed that the amino group on APTMS (or APTES) acted as an additional catalyst directly at the interface of the miniemulsion, facilitating the polycondensation of the silica precursors at the interface, resulting in a core–shell morphology instead of a solid particle morphology.37 A molar ratio APTMS:TEOS = 1:5 was used for further studies. We also explored the optimal conditions to obtain the nanocapsules in terms of the total amount of the precursors and the volume of the dispersed phase. The decreased amount of the silica precursors tended to form crude silica shells (Fig. S3a to S3c†). An increased volume of the dispersed phase could generate cracked silica shells (Fig. S3d to S3f†).
:3:2 = APTMS:TEOS:octyl-TMS, we successfully obtained hollow silica capsules with an aqueous core (Fig. S1d and S1e†). In addition, octyl-TMS was not found to be essential for the preparation of the capsules despite its amphiphilic nature (Fig. S1d to S1f†). Particularly, the combination of APTMS:TEOS = 1:5 generated core–shell structures effectively. Similar results were obtained when APTMS was substituted with the respective ethyl derivative, aminopropyl triethoxysilane (APTES) (Fig. S2†). We assumed that the amino group on APTMS (or APTES) acted as an additional catalyst directly at the interface of the miniemulsion, facilitating the polycondensation of the silica precursors at the interface, resulting in a core–shell morphology instead of a solid particle morphology.37 A molar ratio APTMS:TEOS = 1:5 was used for further studies. We also explored the optimal conditions to obtain the nanocapsules in terms of the total amount of the precursors and the volume of the dispersed phase. The decreased amount of the silica precursors tended to form crude silica shells (Fig. S3a to S3c†). An increased volume of the dispersed phase could generate cracked silica shells (Fig. S3d to S3f†).
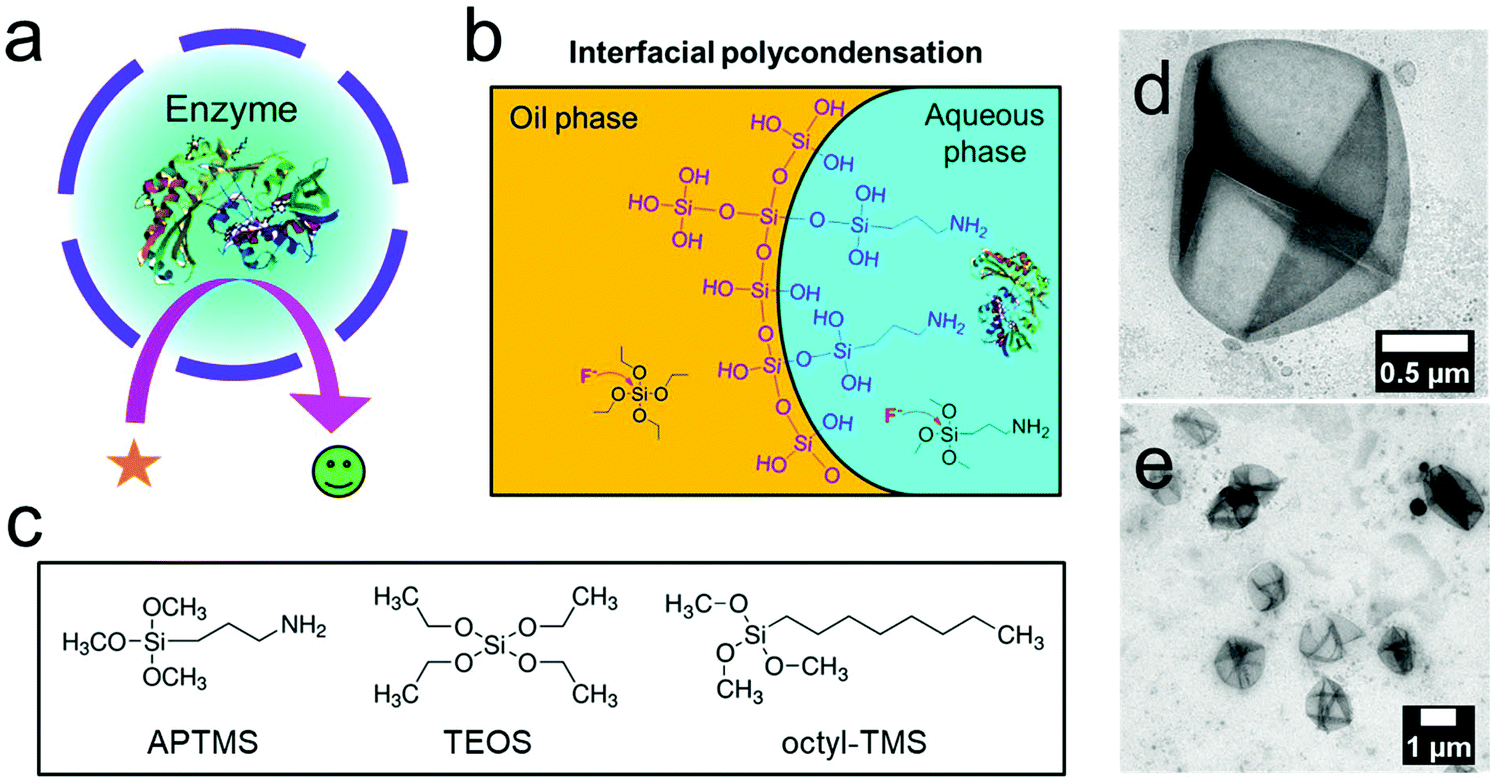 | ||
| Fig. 1 Silica nanocapsules with an aqueous core. (a) Scheme of an enzyme-loaded silica nanocapsule. (b) Interfacial sol–gel process in an inverse miniemulsion of silica precursors catalyzed by fluoride to form hollow nanocapsules. (c) Silica precursors used in this study. (d and e) TEM images of hollow silica nanocapsules prepared by the sol–gel process in an inverse miniemulsion (APTMS, TEOS, octyl-TMS at a molar ratio of 1:3:2 and ammonia (5%, 3 M) were used as the silica source). | ||
Based on the results above, we replaced ammonia with a fluoride anion as an alternative catalyst. As several enzymes are sensitive to basic conditions, their encapsulation at a neutral pH-value is desirable. With a ratio of APTMS:TEOS = 1:5 and a catalyst loading of ammonia (3 M) or potassium fluoride (0.1 mM), hollow silica nanocapsules were obtained (Fig. 2a and b). Besides, to prepare nanocapsules with a diameter of ca. 600 nm, we added sodium chloride (10 mM) to the dispersed phase. The addition of sodium chloride increased the osmotic pressure inside the emulsion droplets, preventing the fusion and growth of the droplets by Ostwald ripening. We also formulated tetrasulfide-bond containing nanocapsules by using APTMS:TEOS:TESPT (bis(triethoxysilylpropyl)tetrasulfide) at a ratio of 1:3.5:1.5 and 0.1 mM fluoride (Fig. 2c). The size distribution and zeta potential of the nanocapsules are presented in Table S1.† The thickness of the shell was determined by analysis of the TEM micrographs (Table S1†) using the ImageJ software to be between 20 and 35 nm, which allowed the diffusion of small molecules (e.g. glucose; MW: 180 Da) through the shells, as reported previously.15,38
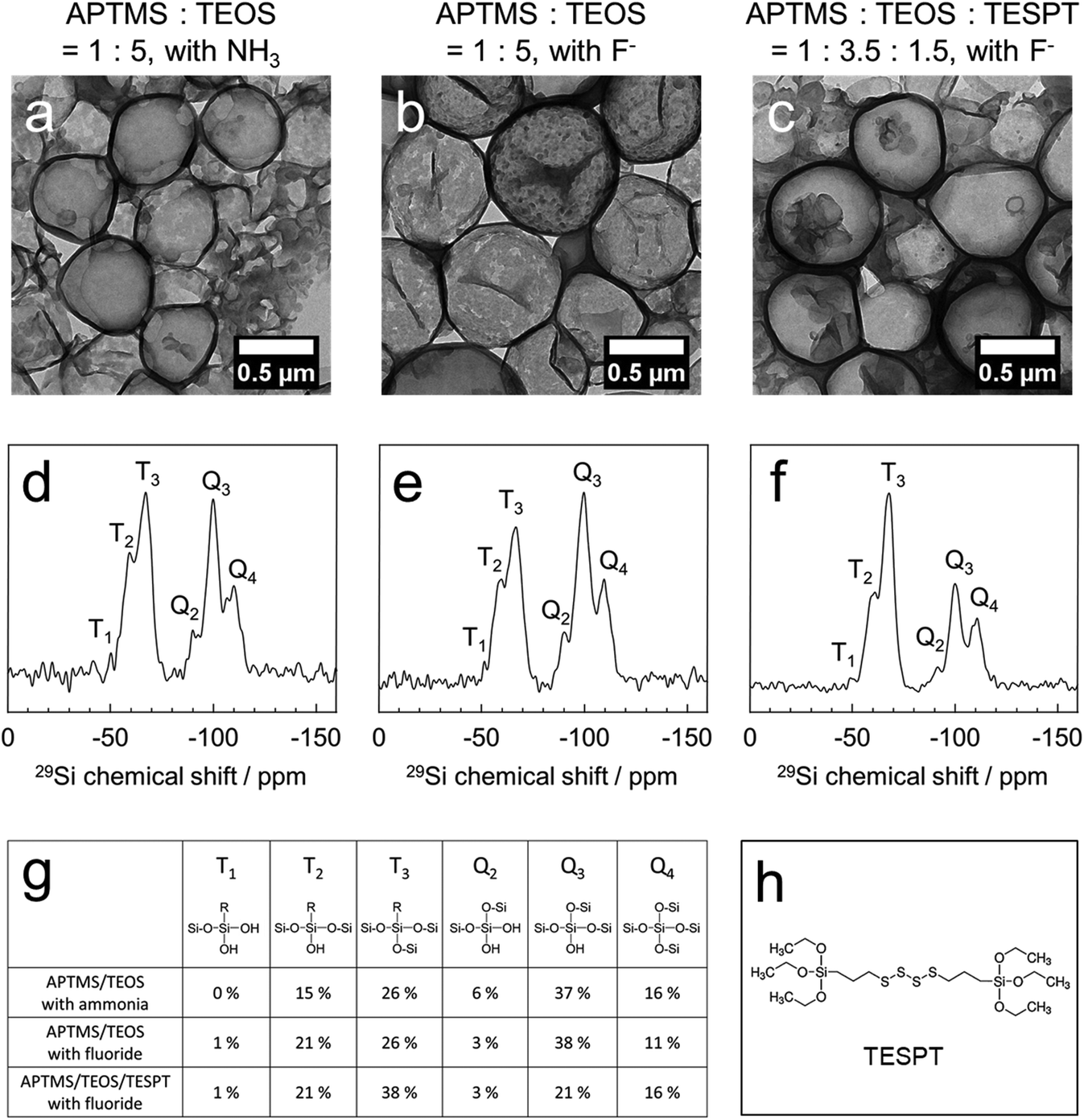 | ||
| Fig. 2 Silica nanocapsules prepared by using different catalysts and silica precursors. The TEM images of silica nanocapsules prepared by using APTMS and TEOS at a 1:5 molar ratio assisted by ammonia (3 M) (a), fluoride (0.1 mM) (b), and APTMS, TEOS, and TESPT at a 1:3.5:1.5 molar ratio assisted with fluoride (0.1 mM) (c). 29Si solid-state NMR spectra of the APTMS/TEOS silica nanocapsules with ammonia (d) and fluoride (e), and the APTMS/TEOS/TESPT nanocapsules with fluoride (f). (g) Summary of the quantitative analysis of silica compositions in each nanocapsule. (h) Chemical structure of TESPT. | ||
Solid-state 29Si magic-angle spinning (MAS) NMR spectroscopy revealed the composition of the silica nanocapsules as derived from the ratios of all the silica precursors, thereby proving the successful incorporation of all the precursors (Fig. 2d–f). In quantitative analysis, the APTMS/TEOS nanocapsules consisted of 41% APTMS (Ttotal) and 59% TEOS (Qtotal) when using the ammonia-catalyzed protocol, and 48% APTMS (Ttotal) and 52% TEOS (Qtotal) when using the fluoride-catalyzed protocol. Considering the silica composition of APTMS (16.7%) and TEOS (83.3%) in the reactant, APTMS showed a faster reaction kinetics than TEOS, followed by rapid polycondensation. The successful incorporation of TESPT in the APTMS:TEOS:TESPT nanocapsules was identified by an increased ratio of Ttotal (60%) generated from APTMS and TESPT. In our general protocol that intentionally omitted any porogen, the pore-size was further tuned by the copolymerization of a redox-cleavable silica precursor (TESPT; Fig. 2h). These silica nanocapsules carried additional tetrasulfide linkages in their shells, increasing the pore diameters under reductive conditions but keeping the core–shell morphology intact and the enzymes inside (see below).
In addition, solid-state 19F MAS NMR showed that no fluoride remained in the nanocapsules after purification. It was evident that fluoride did not react with the silica precursors to incorporate into the nanocapsules and only acted as a catalyst for the sol–gel reactions (Fig. S4†). The shell wall porosity was investigated by the Brunauer–Emmett–Teller (BET) gas adsorption technique. As we did not use a pore-forming agent to obtain a mesoporous silica structure, the N2 adsorption/dissociation curve showed a surface area of 58.1 m2 g−1 with a pore volume of 0.702 cc g−1. According to our previous reports, this degree of porosity is enough to allow the diffusion of small molecular substrates for enzymatic reactions, e.g., glucose (180 Da) or nicotinamide adenine dinucleotide (663.43 Da), through the silica matrix.15,39
Encapsulation of enzymes into silica nanocapsules
To use the semi-permeable nanocapsules as enzyme modules, the enzyme activity should remain as high as possible during the synthesis process. The emulsification step, use of surfactants and organic solvents, chemical reactions, pH-changes, and shear forces can lead to the denaturation of the enzymes. We prepared silica nanocapsules loaded with glucose oxidase (GOX) as a first model enzyme by ammonia or fluoride catalysis and compared the enzymatic activities of the resulting GOX-loaded nanocapsules. In both cases, core–shell structures with enzymes inside the aqueous core were obtained (Fig. 3a and b). No leakage of GOX from the nanocapsules was observed. Amplex red oxidation was used to assess the enzymatic activity of GOX. Compared to native GOX, the fluoride-catalyzed GOX nanocapsule showed higher remaining enzymatic activity (60%), whereas the ammonia-catalyzed GOX nanocapsule showed 9% of remaining enzymatic activity (Fig. 3c). This difference in the GOX activity loss can be explained by the changes in the secondary structure of GOX under basic conditions (3 M ammonia, pH 11), as proved by circular dichroism (Fig. S5†), while a fluoride-containing solution (0.1 mM fluoride, pH 7.4) did not affect the secondary structure of the enzymes.15 The size distribution and zeta potential of the nanocapsules are presented in Table S2.†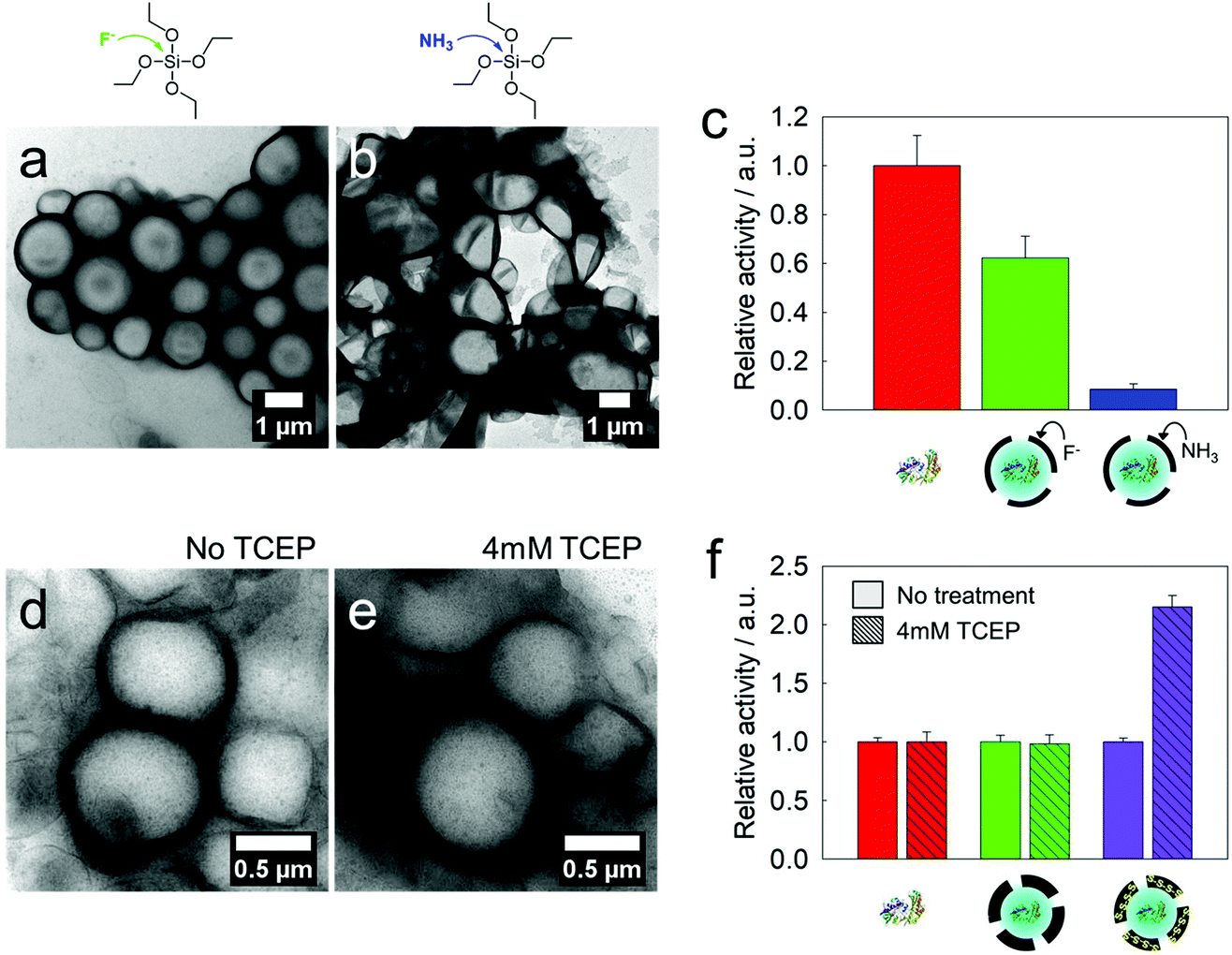 | ||
| Fig. 3 GOX-loaded silica nanocapsules: comparison of the catalysts and residual enzyme activities. GOX-loaded silica nanocapsules prepared by using APTMS and TEOS at a 1:5 molar ratio catalyzed by 0.1 mM fluoride (a) and 3 M ammonia (b). (c) Relative activities of native GOX and loaded GOX in the nanocapsules. A glucose concentration of 5 mM was used for this assay. (d and e) Tetra-sulfide-gated nanocapsules (molar ratio of APTMS:TEOS:TESPT = 1:3.5:1.5) before and after treatment with TCEP (4 mM). (f) Reduction-triggered enhancement of GOX reactions in tetra-sulfide-gated nanocapsules (purple) after treatment with TCEP (4 mM). | ||
As mentioned above, the incorporation of tetrasulfide groups was envisioned to tune the pores of the silica nanocapsules. When nanocapsules, which are composed of APTMS, TEOS, and TESPT (1:3.5:1.5 molar ratio) with loaded GOX, were treated with 4 mM tris(2-carboxyethyl)phosphine (TCEP), the strength of the tetrasulfide linkage had to be reduced. Even after the reduction, no leakage of the enzymes or collapse of the core–shell morphology was observed (Fig. 3d and e). However, the kinetics of the Amplex red oxidation increased by a factor of 2.2 compared to the capsules before reduction (Fig. 3f), which can be attributed to the widening of the pores in the shell.
Cascade reactions in micro-confinement: GOX to peroxidase
The protocol reported herein allows the simultaneous encapsulation of several enzymes to achieve enzymatic cascade reactions inside the confinement of the silica nanocapsules. As a first proof-of-concept, GOX and horseradish peroxidase (HRP) were loaded into the same nanocapsule (Fig. 4a). According to a thermogravimetric analysis (TGA), the encapsulation efficiency of the two enzymes in a silica nanocapsule was calculated to be ca. 80% (Fig. S6†). This set of enzymes could be used in colorimetric glucose sensors through a dye oxidation reaction, in which the non-fluorescent Amplex red is converted to fluorescent resorufin. The kinetics of the confined cascade reaction was compared to that of the same cascade reaction that occurred between the encapsulated GOX and free HRP; i.e. the latter dissolved in the continuous phase, requiring the transfer of the substrate through the shell. The cascade reaction with both enzymes confined inside the same nanocapsule proved to be ca. 2.5-fold faster compared to the separated reaction (Fig. S7†), indicating that the proximity of the two enzymes increased the reaction efficiency. The encapsulated enzymes were protected from the environment and could be easily separated from the reaction products by centrifugation. This dual-loaded nanocapsule could be utilized for developing a miniature glucose sensor (Fig. 4b). | ||
| Fig. 4 Cascade reaction from GOX to HRP. (a) Scheme of the GOX reaction consuming glucose and the HRP reaction to generate fluorescence in a nanocapsule. (b) Miniature glucose sensor: changes in the fluorescence at different concentrations of glucose. | ||
Cascade reactions in micro-confinement: GOX to catalase
GOX and catalase (CAT) were loaded into the same silica nanocapsule (Fig. 5a). According to the TGA measurement, the loading efficiency of these two enzymes into the silica nanocapsule was ca. 65% (Fig. S6†). Although GOX is a useful enzyme for developing glucose-responsive systems, one drawback is its generation of the toxic hydrogen peroxide as a byproduct. Using CAT can significantly increase the biocompatibility of a GOX-based system. Compared to single-loaded GOX, the co-loaded GOX and CAT showed slower reaction kinetics for the oxidation of Amplex red, as the redox process mediated by CAT removed H2O2. The GOX-loaded silica nanocapsules were treated with HeLa cells to observe H2O2-mediated cell death.40 The GOX-loaded nanocapsules led to a rapid cell death (Fig. 5b) due to H2O2 production. In contrast, when the nanocapsules were loaded with both GOX and CAT, the cytotoxicity was suppressed due to the competing disproportionation of H2O2 by CAT inside the nanocapsules. The simultaneous encapsulation of the two enzymes in the same nanocapsule showed a better efficiency in removing hydrogen peroxide, as the proximity of two enzymes is beneficial for an efficient cascade reaction from one enzyme to another.41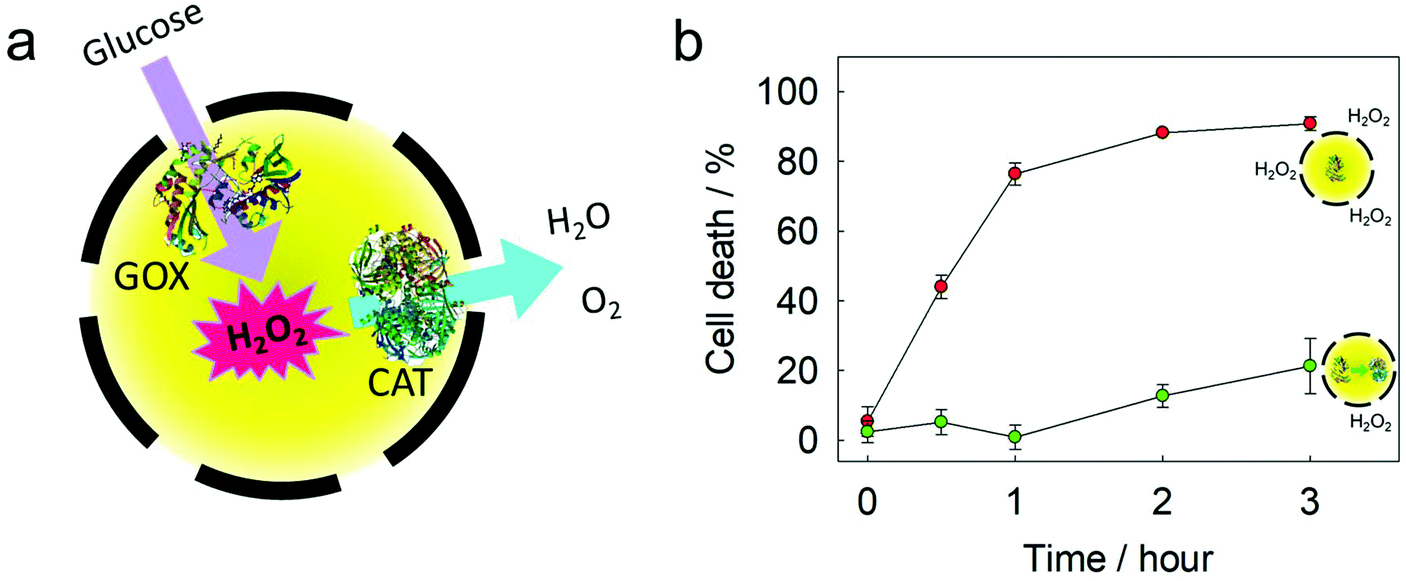 | ||
| Fig. 5 A cascade reaction from GOX to CAT. (a) Scheme of the GOX reaction to produce hydrogen peroxide, and the CAT reaction to degrade hydrogen peroxide in a nanocapsule. (b) Cell death caused by silica nanocapsules loaded with GOX (red) and loaded with both GOX and CAT (green). | ||
Cascade reactions in micro-confinement: an NAD+/NADH regeneration module
Lactate dehydrogenase (LDH) and glucose dehydrogenase (GDH) were encapsulated simultaneously in a silica nanocapsule (Fig. 6a). According to TGA measurements, the encapsulation efficiency of these two enzymes in a silica nanocapsule was ca. 85% (Fig. S6†). While LDH catalyzes the conversion of NADH and pyruvate to NAD+ and lactate, respectively, GDH converts NAD+ to NADH by consuming glucose. By the combination of these two enzymes, an efficient module for the production of either NAD+ or NADH, depending on the substrate feedstock, is provided and might be used in various NAD+- or NADH-dependent enzymatic reactions.42 The conversion of NAD+/NADH was monitored by UV/Vis spectroscopy as the absorbance at 340 nm changed during reduction/oxidation. We achieved a cycle of several regeneration processes of either NAD+ or NADH through adding pyruvate or glucose, respectively (Fig. 6b). This two-enzyme system did not generate any toxic or reactive byproducts (e.g., aldehydes, reactive oxygen species, or alcohols), making it attractive for biomimetic redox reactions. As gluconic acid is a byproduct, only a buffered medium is necessary to keep the module working, making it easy to combine with other enzymatic pathways.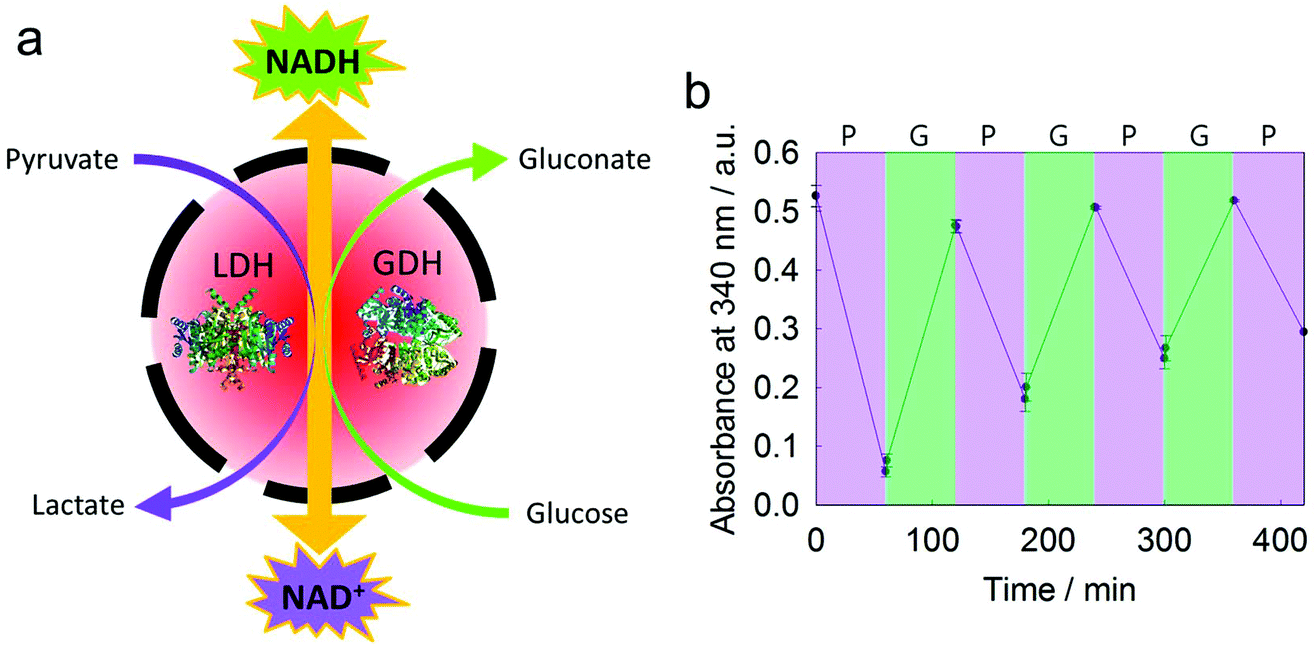 | ||
| Fig. 6 A cascade reaction between LDH and GDH. (a) Scheme of NAD+ and NADH regeneration in a nanocapsule. (b) Interconversion of NAD+ and NADH controlled by adding pyruvate and glucose. The increasing absorbance at 340 nm indicates the production of NADH by GDH. The decreasing absorbance indicates the production of NAD+ by LDH. | ||
Conclusions
We developed a general protocol for the preparation of hollow silica nanocapsules with an aqueous core, which was investigated for the confinement of enzymatic reactions. The combination of amphiphilic APTMS together with TEOS in an inverse miniemulsion forced the sol–gel-process to the interface of the droplets, resulting in hollow nanocapsules, without the need for a template. An efficient formation of silica nanocapsules was achieved at different molar ratios, and various concentrations of the silica precursors were used to tailor the membrane properties of the final nanocapsules. Ammonia and also fluoride were used as effective catalysts during the sol–gel-process. Fluoride catalysis enabled the sol–gel reactions at a neutral pH, and therefore the enzymatic activity after encapsulation was preserved. We explored three different cascade reactions in the silica nanocapsules. A cascade reaction from GOX to HRP could be used for a miniature glucose sensor. A cascade reaction from GOX to CAT proved to decrease the toxicity of the GOX reaction. A cascade reaction between LDH and GDH allowed a controlled interconversion between NAD+ and NADH. We believe that our protocol to develop silica nanocapsules can be a general strategy to load a wide range of enzymes because of the mild reaction conditions involved. Recently, enzymatic nanoreactors have not only been used for bio-production and pollutant removal but also showed potential for therapeutics, micromachines and designing synthetic cells. Our encapsulation technology is a versatile platform for enzyme-based systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is a part of the MaxSynBio consortium, which was jointly funded by the Federal Ministry of Education and Research of Germany and the Max Planck Society. We thank Petra Räder (MPIP) for the TGA measurements. This work is also supported by Open Access funding provided by the Max Planck Society.References
- A. Küchler, M. Yoshimoto, S. Luginbühl, F. Mavelli and P. Walde, Nat. Nanotechnol., 2016, 11, 409 CrossRef.
- J. Shi, Y. Wu, S. Zhang, Y. Tian, D. Yang and Z. Jiang, Chem. Soc. Rev., 2018, 47, 4295–4313 RSC.
- E. T. Hwang and S. Lee, ACS Catal., 2019, 9, 4402–4425 CrossRef CAS.
- M. Castellana, M. Z. Wilson, Y. Xu, P. Joshi, I. M. Cristea, J. D. Rabinowitz, Z. Gitai and N. S. Wingreen, Nat. Biotechnol., 2014, 32, 1011–1018 CrossRef CAS.
- X. Zhang, F. Zhang, Z. Lu, Q. Xu, C. Hou and Z. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 25565–25571 CrossRef CAS.
- T. Li, J. Li, Q. Pang, L. Ma, W. Tong and C. Gao, ACS Appl. Mater. Interfaces, 2019, 11, 6789–6795 CrossRef CAS.
- S.-M. Jo, K. A. I. Zhang, F. R. Wurm and K. Landfester, ACS Appl. Mater. Interfaces, 2020, 12, 25625–25632 CrossRef CAS.
- D. Gräfe, J. Gaitzsch, D. Appelhans and B. Voit, Nanoscale, 2014, 6, 10752–10761 RSC.
- J. Kim and K. T. Kim, ACS Appl. Mater. Interfaces, 2020, 12, 23502–23513 CrossRef CAS.
- R. J. R. W. Peters, M. Marguet, S. Marais, M. W. Fraaije, J. C. M. van Hest and S. Lecommandoux, Angew. Chem., Int. Ed., 2014, 53, 146–150 CrossRef CAS.
- L. D. Blackman, S. Varlas, M. C. Arno, A. Fayter, M. I. Gibson and R. K. O'Reilly, ACS Macro Lett., 2017, 6, 1263–1267 CrossRef CAS.
- X. Wu, J. Ge, C. Yang, M. Hou and Z. Liu, Chem. Commun., 2015, 51, 13408–13411 RSC.
- F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761–5765 CrossRef CAS.
- T.-H. Wei, S.-H. Wu, Y.-D. Huang, W.-S. Lo, B. P. Williams, S.-Y. Chen, H.-C. Yang, Y.-S. Hsu, Z.-Y. Lin, X.-H. Chen, P.-E. Kuo, L.-Y. Chou, C.-K. Tsung and F.-K. Shieh, Nat. Commun., 2019, 10, 5002 CrossRef.
- S.-M. Jo, F. R. Wurm and K. Landfester, Nano Lett., 2020, 20, 526–533 CrossRef CAS.
- S. Jiang, D. Prozeller, J. Pereira, J. Simon, S. Han, S. Wirsching, M. Fichter, M. Mottola, I. Lieberwirth, S. Morsbach, V. Mailänder, S. Gehring, D. Crespy and K. Landfester, Nanoscale, 2020, 12, 2626–2637 RSC.
- F. Maia, J. Tedim, A. D. Lisenkov, A. N. Salak, M. L. Zheludkevich and M. G. S. Ferreira, Nanoscale, 2012, 4, 1287–1298 RSC.
- S. Jiang, M. Mottola, S. Han, R. Thiramanas, R. Graf, I. Lieberwirth, V. Mailänder, D. Crespy and K. Landfester, Part. Part. Syst. Charact., 2020, 37, 1900484 CrossRef CAS.
- R. Vecchione, G. Luciani, V. Calcagno, A. Jakhmola, B. Silvestri, D. Guarnieri, V. Belli, A. Costantini and P. A. Netti, Nanoscale, 2016, 8, 8798–8809 RSC.
- Z. Teng, W. Li, Y. Tang, A. Elzatahry, G. Lu and D. Zhao, Adv. Mater., 2019, 31, 1707612 CrossRef.
- H. N. Yow and A. F. Routh, Soft Matter, 2006, 2, 940–949 RSC.
- C.-H. Lin, J.-H. Chang, Y.-Q. Yeh, S.-H. Wu, Y.-H. Liu and C.-Y. Mou, Nanoscale, 2015, 7, 9614–9626 RSC.
- F.-P. Chang, Y.-P. Chen and C.-Y. Mou, Small, 2014, 10, 4785–4795 CrossRef CAS.
- K. A. Whitaker and E. M. Furst, Langmuir, 2014, 30, 584–591 CrossRef CAS.
- X. W. Lou, L. A. Archer and Z. Yang, Adv. Mater., 2008, 20, 3987–4019 CrossRef CAS.
- Z.-Z. Li, L.-X. Wen, L. Shao and J.-F. Chen, J. Controlled Release, 2004, 98, 245–254 CrossRef CAS.
- Y. Nakashima, C. Takai, H. Razavi-Khosroshahi, W. Suthabanditpong and M. Fuji, Adv. Powder Technol., 2018, 29, 904–908 CrossRef CAS.
- Y. Bao, C. Shi, T. Wang, X. Li and J. Ma, Microporous Mesoporous Mater., 2016, 227, 121–136 CrossRef CAS.
- F.-P. Chang, Y. Hung, J.-H. Chang, C.-H. Lin and C.-Y. Mou, ACS Appl. Mater. Interfaces, 2014, 6, 6883–6890 CrossRef CAS.
- T. Okada, K. Miyamoto, T. Sakai and S. Mishima, ACS Catal., 2014, 4, 73–78 CrossRef CAS.
- T. Okada and T. Koide, Langmuir, 2018, 34, 9500–9506 CrossRef CAS.
- E. M. Alexandrino, M. Wagner, K. Landfester and F. R. Wurm, Macromol. Chem. Phys., 2016, 217, 1941–1947 CrossRef CAS.
- S. J. Beckers, I. A. Dallo, I. del Campo, C. Rosenauer, K. Klein and F. R. Wurm, ACS Sustainable Chem. Eng., 2019, 7, 6991–6998 CrossRef CAS.
- W. He, M. Parowatkin, V. Mailänder, M. Flechtner-Mors, R. Graf, A. Best, K. Koynov, K. Mohr, U. Ziener, K. Landfester and D. Crespy, Biomacromolecules, 2015, 16, 2282–2287 CrossRef CAS.
- M. A. Hood, U. Paiphansiri, D. Schaeffel, K. Koynov, M. Kappl, K. Landfester and R. Muñoz-Espí, Chem. Mater., 2015, 27, 4311–4318 CrossRef CAS.
- Y.-S. Lin, S.-H. Wu, C.-T. Tseng, Y. Hung, C. Chang and C.-Y. Mou, Chem. Commun., 2009, 3542–3544, 10.1039/B902681A.
- R. M. Ottenbrite, J. S. Wall and J. A. Siddiqui, J. Am. Ceram. Soc., 2000, 83, 3214–3215 CrossRef CAS.
- H. Kim, S.-M. Jo, F. Meng, Y. Guo, H. Thérien-Aubin, R. Golestanian, K. Landfester and E. Bodenschatz, Adv. Funct. Mater., 2020, 2006019 CrossRef CAS.
- S.-M. Jo, F. R. Wurm and K. Landfester, ACS Appl. Mater. Interfaces, 2018, 10, 34230–34237 CrossRef CAS.
- L. F. Barros, T. Kanaseki, R. Sabirov, S. Morishima, J. Castro, C. X. Bittner, E. Maeno, Y. Ando-Akatsuka and Y. Okada, Cell Death Differ., 2003, 10, 687–697 CrossRef CAS.
- C. Cui, Y. Fang, B. Chen and T. Tan, Catal. Sci. Technol., 2019, 9, 477–482 RSC.
- B. El-Zahab, H. Jia and P. Wang, Biotechnol. Bioeng., 2004, 87, 178–183 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr07148j |
| This journal is © The Royal Society of Chemistry 2020 |