
Synthesis of lead-free Cs4(Cd1−xMnx)Bi2Cl12 (0 ≤ x ≤ 1) layered double perovskite nanocrystals with controlled Mn–Mn coupling interaction†

Abstract
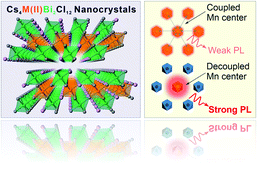
Lead-free perovskites and their analogues have been extensively studied as a class of next-generation luminescent and optoelectronic materials. Herein, we report the synthesis of new colloidal Cs4M(II)Bi2Cl12 (M(II) = Cd, Mn) nanocrystals (NCs) with unique luminescence properties. The obtained Cs4M(II)Bi2Cl12 NCs show a layered double perovskite (LDP) crystal structure with good particle stability. Density functional theory calculations show that both samples exhibit a wide, direct bandgap feature. Remarkably, the strong Mn–Mn coupling effect of the Cs4M(II)Bi2Cl12 NCs results in an ultra-short Mn photoluminescence (PL) decay lifetime of around 10 μs, around two orders of magnitude faster than commonly observed Mn2+ dopant emission in NCs. Diluting the Mn2+ ion concentration through forming Cs4(Cd1−xMnx)Bi2Cl12 (0 < x < 1) alloyed LDP NCs leads to prolonged PL lifetimes and enhanced PL quantum yields. Our study provides the first synthetic example of Bi-based LDP colloidal NCs with potential for serving as a new category of stable lead-free perovskite-type materials for various applications.
- This article is part of the themed collection: Nanoscale 2021 Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

