
Isolation of DNA aptamers targeting N-cadherin and high-efficiency capture of circulating tumor cells by using dual aptamers†

Abstract
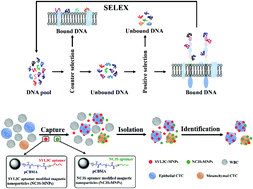
Circulating tumor cells (CTCs) acquire mesenchymal markers (e.g., N-cadherin) and lose epithelial markers (e.g., epithelial cell adhesion molecule, EpCAM) during the epithelial-mesenchymal transition (EMT) and are therefore ideal biomarkers of tumor metastasis. However, it is still a challenge to efficiently capture and detect circulating tumor cells with different phenotypes simultaneously. In this work, to obtain aptamers targeting N-cadherin in the native conformation on live cells, we established stable N-cadherin overexpressing cells (N-cadherin cells) and used these cells to identify a panel of N-cadherin-specific aptamers through the cell-SELEX approach. Two aptamer candidates obtained after 12 rounds of selection showed a low equilibrium dissociation constant in the nanomolar range, indicating high binding affinity. The truncated aptamer candidate NC3S showed the highest binding affinity to N-cadherin cells with a low Kd value of 20.08 nM. The SYL3C aptamer was reported to target cancer cell surface biomarker EpCAM. Then, we synthesized two kinds of aptamer-modified magnetic nanoparticles (SYL3C-MNPs and NC3S-MNPs). Both SYL3C and NC3S aptamers possess excellent capture specificity and efficiency for the target cells. The aptamer–MNP cocktail exhibits a considerable capture efficiency and sensitivity for rare cancer cells of epithelial and mesenchymal phenotypes. Furthermore, no CTCs were found in blood samples from healthy donors, while CTCs were successfully isolated by using the aptamer–MNP cocktail for 15 out of 16 samples collected from patients. In summary, the two kinds of aptamer-modified MNPs could be utilized as a promising tool for capturing CTCs from clinical samples.
- This article is part of the themed collections: Editor’s Choice: Recent breakthroughs in nanobiotechnology and 2020 Nanoscale HOT Article Collection
 Please wait while we load your content...
Please wait while we load your content...

