
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-layer assembly of cellulose nanofibrils in a microfluidic device for the selective capture and release of viable tumor cells from whole blood†
Tharagan
Kumar‡
 a,
Ruben R. G.
Soares‡
a,
Leyla
Ali Dholey
a,
Harisha
Ramachandraiah§
a,
Negar Abbasi
Aval
b,
Zenib
Aljadi
a,
Torbjörn
Pettersson
*b and
Aman
Russom
*ac
a,
Ruben R. G.
Soares‡
a,
Leyla
Ali Dholey
a,
Harisha
Ramachandraiah§
a,
Negar Abbasi
Aval
b,
Zenib
Aljadi
a,
Torbjörn
Pettersson
*b and
Aman
Russom
*ac
aKTH Royal Institute of Technology, Division of Nanobiotechnology, Department of Protein Science, Science for Life Laboratory, Solna, Sweden. E-mail: aman.russom@scilifelab.se
bKTH Royal Institute of Technology, Department of Fibre and Polymer Technology, School of Engineering Sciences in Chemistry, Biotechnology and Health, Stockholm, Sweden. E-mail: torbj@kth.se
cAIMES - Center for the Advancement of Integrated Medical and Engineering Sciences at Karolinska Institutet and KTH Royal Institute of Technology, Stockholm, Sweden
First published on 20th October 2020
Abstract
According to reports by the World Health Organization (WHO), cancer-related deaths reached almost 10 million in 2018. Nearly 65% of these deaths occurred in low- to middle-income countries, a trend that is bound to increase since cancer diagnostics are not currently considered a priority in resource-limited settings (RLS). Thus, cost-effective and specific cancer screening and diagnostics tools are in high demand, particularly in RLS. The selective isolation and up-concentration of rare cells while maintaining cell viability and preventing phenotypic changes is a powerful tool to allow accurate and sensitive downstream analysis. Here, multi-layer cellulose nanofibril-based coatings functionalized with anti-EpCAM antibodies on the surface of disposable microfluidic devices were optimized for specific capture of target cells, followed by efficient release without significant adverse effects. HCT 116 colon cancer cells were captured in a single step with >97% efficiency at 41.25 μL min−1 and, when spiked in whole blood, an average enrichment factor of ∼200-fold relative to white blood cells was achieved. The release of cells was performed by enzymatic digestion of the cellulose nanofibrils which had a negligible impact on cell viability. In particular, >80% of the cells were recovered with at least 97% viability in less than 30 min. Such performance paves the way to expand and improve clinical diagnostic applications by simplifying the isolation of circulating tumor cells (CTCs) and other rare cells directly from whole blood.
1. Introduction
As healthcare and life expectancy improves in resource-limited settings (RLS) the focus on parasitic/infectious diseases and malnutrition will gradually shift towards cancer screening and management. Among 9.6 million annual cancer-related deaths reported in 2018, it is estimated that as high as 75% will be in low- and middle-income countries (LMC) by 2030.1 Due to the lack of state-of-the-art medical and laboratory facilities in RLS allowing routine cancer monitoring, the development of simple and cost-effective diagnostic devices, e.g. allowing direct targeting of potential cancer cells from biological samples,2 are critical to minimize its growing impact.1Tools allowing the isolation of rare cells, such as circulating tumor cells (CTCs) directly from blood provide key insights towards early disease diagnostics, drug development and therapeutic monitoring.2 The enrichment of CTCs, particularly in a viable state for subsequent expansion and functional/phenotypical analysis, is paramount to allow an accurate downstream analysis to elucidate the regulatory pathways underlying tumor growth and metastasis.2 Furthermore, these techniques known as liquid biopsies contrast with conventional tissue biopsies which are highly invasive and demand advanced medical facilities. Therefore, these can potentially be a holy grail for early cancer detection and are considered a key element for cancer control in LMCs by the WHO.1 The enrichment of rare cells today is routinely achieved using fluorescent assisted cell sorting (FACS),3 density gradient centrifugation, and immunomagnetic enrichment methods,2 the latter coupled for example with the FDA approved CellSearch™ equipment. Academic research has been active in improving liquid biopsy techniques towards enhanced separation efficiency and reduced method complexity and costs. In particular, recent developments using microfluidic platforms offer many advantages over conventional benchtop techniques such as low sample and reagent volume requirements, ease of use, improved separation efficiency and the possibility for integration with downstream processes.4 Among the microfluidic platforms, immunoaffinity-based cell separation is very attractive and it has been used extensively for cancer diagnostics, where CTCs have been isolated from whole blood.5–8 Affinity-based isolation utilizes antibodies that recognize and bind specifically to cell surface markers expressed by target cells, often resulting in high cell purity. Additionally, the demand for viable cell release for downstream analysis of the cells is increasing. Thus, several approaches have been reported in the past few years to capture and release potential CTCs in microfluidic devices, typically using anti-EpCAM antibodies. These strategies resort either to antibodies immobilized on “sensitive” substrates9–13 or to aptamers, the later prone to direct degradation by exonucleases.14,15 The term “sensitive” refers to the designed proneness of the substrate to release the antibodies under mild conditions via for example enzymatic degradation,11 competitive binding,9 cleavage of disulfide bridges13 or photocleavage.12 Particular examples of immunoaffinity capture and release strategies are (1) Strep-tag labelled antibodies immobilized on Strep-tactin, released by competition with D-biotin;9 (2) antibody immobilization on zinc-phosphate hierarchical nanostructures, dissolved with 1% sodium citrate at room temperature;10 (3) antibody immobilization on layer-by-layer (LbL) assemblies of biodegradable polymers such as alginate, amenable to degradation by alginate lyase enzymes;11 (4) anchoring of anti-EpCAM antibodies after CTC capture using click chemistry followed by release using DTT to reduce a disulfide bridge in the anchor group and; (5) immobilization of antibodies via a ssDNA tag by hybridization, subsequently released by photocleavage with UV irradiation.12 Among these methods, nanowire-based strategies,10,13,15 comprising dense arrays of nm-wide structures protruding several microns into the solution, provide the overall best capture and release efficiencies above 90% due to the increase in effective antibody-coated surface area available to capture the cells. However, this assembly requires lengthy, complex and expensive clean room-based fabrication processes. On the other hand, simpler surface modification methods based on aptamers, LbL assemblies, competitive-liable or photolabile linkers suffer from lower capture (e.g. 79%,9 80% (ref. 11) and 55% (ref. 12)) efficiency, despite achieving comparable or even superior cell purity, i.e. ratio of captured cancer cells relative to other blood cells.
Aiming at improving the cell capture/release efficiency, coupled with a simple, rapid and cost-effective surface functionalization, the engineering of surface coatings based on cellulose nanofibrils (CNF) is emerging as a relatively recent trend combining an extremely versatile and biodegradable material with dense three-dimensional features to increase biomolecular surface density.16 In particular, these nanometric fibrils with very high aspect ratios have a high density of hydroxyl groups which can be easily modified into carboxyl, epoxy or amine groups.17,18 These modifications allow a broad range of CNF coating and biomolecule functionalization strategies, which have been extensively explored for biosensing applications.19–23 Besides the widely explored covalent immobilization,24 CNF coatings can also be exploited using electrostatic immobilization,25 becoming particularly relevant to explore LbL assemblies to modulate the content and thickness of the coated layer, previously demonstrated to hold potential for CTC capture with other polymeric materials.11
Here, we demonstrate the generation of a LbL assembly comprising up to 5 layers of CNF inside a microfluidic channel, functionalized with anti-EpCAM antibodies for specific capture of potential cancer cells. Cellulase enzymes were then used to effectively degrade the CNF and release the captured cells within 30 min with minimal impact on cell viability.
2. Experimental methods
2.1. Design and fabrication of microfluidic devices
Two different types of microfluidic devices were used in this work, plain microchamber and herringbone chips. These will henceforth be referred to as P-Chip or HB-Chip, respectively. The P-Chip comprises a chamber with dimensions of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × 4000 × 50 μm (L × W × H). The HB-Chip comprises a chamber with 39000 × 22000 × 100 μm (L × W × H) containing microgrooves to promote convective mixing on the PDMS side of the chamber. The polydimethylsiloxane (PDMS) devices were fabricated using standard SU-8 mold replication with a 1:10 ratio of curing agent to PDMS oligomers (Sylgard 184 Silicone Elastomer) and baking for 8 h at 65 °C. After peeling the devices from the mold, access holes were punched using a 20-gauge biopsy puncher. All devices were then sealed against isopropanol rinsed glass slides (Corning microscope slides, plain) after subjecting both surfaces to an oxygen plasma treatment for 30 s (Femto Science CUTE, 100 W, 80 Pa O2).
000 × 4000 × 50 μm (L × W × H). The HB-Chip comprises a chamber with 39000 × 22000 × 100 μm (L × W × H) containing microgrooves to promote convective mixing on the PDMS side of the chamber. The polydimethylsiloxane (PDMS) devices were fabricated using standard SU-8 mold replication with a 1:10 ratio of curing agent to PDMS oligomers (Sylgard 184 Silicone Elastomer) and baking for 8 h at 65 °C. After peeling the devices from the mold, access holes were punched using a 20-gauge biopsy puncher. All devices were then sealed against isopropanol rinsed glass slides (Corning microscope slides, plain) after subjecting both surfaces to an oxygen plasma treatment for 30 s (Femto Science CUTE, 100 W, 80 Pa O2).
2.2. Preparation of cellulose nanofibrils and carboxymethylation
Carboxymethylated cellulose nanofibrils (CNF generation 2 from RISE Bioeconomy, Sweden) were used to build multilayers in the microfluidic devices. The dispersion of CNF was prepared as described earlier,26i.e. by ultra-sonication followed by being placed in a centrifuge for 1 h at 2250g This procedure typically generates CNF with a charge of 600 μeq g−1, diameters of 4 nm and lengths in the range of 1–5 μm.2.3. Surface structure characterization of LbL assembly of CNF
The surface structure of the CNF LbL assembly, sequentially added to the surface of a silica wafer, was measured with atomic force microscopy (AFM). Firstly, a poly(allylamine hydrochloride) (PAH) solution was prepared in MilliQ water at a concentration of 100 mg L−1 and placed on a silica wafer for 30 minutes. After washing the surface with MilliQ water for 3 times, CNF was dispersed in MilliQ water at a concentration of 100 mg L−1 and adsorbed on the PAH base for 10 minutes, followed by 3 subsequent washes with MilliQ water. Polyethyleneimine (PEI) at a concentration of 100 mg L−1 in MilliQ water was then added also for 10 minutes. The CNF and PEI layers were continuously adsorbed up to 5 bi-layers of CNF-PEI with MilliQ water washes in between each layer. Before the AFM measurements, having CNF as the outermost layer, the substrates were washed with MilliQ water and dried with a flow of nitrogen gas. The measurements were performed with MultiMode 8 AFM (Bruker, Santa Barbara, CA, USA) in ScanAsyst mode using SCANASYST-Air cantilevers. PAH and PEI stock solutions were purchased from Sigma-Aldrich.2.4. On-chip surface functionalization with LbL assembly of CNF and measurement of CNF buildup
Unless stated otherwise, all flow rate conditions in this section refer to the P-Chip. The flow rate conditions applied to the HB-Chip can be derived from those used on the P-Chip by multiplying the values by a factor of 8.3, i.e. the relative difference in channel cross-section area, to ensure a constant linear velocity of the liquid. All surface modification steps were performed by sequentially flowing solutions at specific flow-rates through the devices. As a base layer, PAH was first adsorbed on the surface by flowing a 200 mg L−1 PAH solution prepared in MilliQ water at 5 μL min−1 for 10–30 min, followed by washing with MilliQ water. Then, to generate the CNF multilayer, a suspension of CNF and PEI solutions, flowed at the conditions optimized for CNF and both prepared in MilliQ water were sequentially flowed and intercalated with washing steps. CNF solutions were tested in concentrations ranging between 76 mg L−1 and 2.3 g L−1 and flowed at 5 μL min−1 for 10–30 min. PEI was prepared at a concentration of 200 mg L−1 and flowed for 10 min at 5 μL min−1. All washing steps were performed by flowing MilliQ water at 25 μL min−1 for 2 min. The buildup of CNF on the surface was measured using fluorescence microscopy after flowing a commercial carbohydrate fluorescent tracer at 5 μL min−1 for 10 min on the channel (Carbotrace™ from Ebba Biotech, Solna, Sweden, diluted 1000× according to the manufacturer's instructions).2.5. Antibody functionalization on CNF
Anti-EpCAM mouse monoclonal antibodies (mAbs) purchased from R&D Systems (MAB960, clone #158210) were grafted on the cellulose via carboxymethyl groups via EDC-NHS chemistry. The NHS modification of the carboxymethyl groups was performed by filling the channel with 5 mM 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 5 mM N-hydroxysuccinimide (NHS) prepared in 25 mM 2-(N-morpholino)ethanesulfonic acid (MES) buffer at pH 4.7, followed by an incubation time of 30 min. Then, the channel was washed with MES buffer at 25 μL min−1 for 2 min and subsequently filled with a solution containing 100 μg mL−1 anti-EpCAM mAb prepared also in MES buffer, followed by a second incubation time of 2 h at room temperature at 4 °C overnight. Both steps were performed after filling the channel at 5 μL min−1 (P-Chip) or 41.25 μL min−1 (HB-Chip) for 2 min. To confirm the conjugation of the antibody on CNF using fluorescence microscopy, a batch of anti-EpCAM mAb was pre-labeled with Alexa Fluor 430. The labelling was performed using succinimidyl ester activated Alexa Fluor 430 (Thermo Fisher) according to the protocol described in detail elsewhere.27 EDC, NHS and MES buffer tabs were purchased from Sigma-Aldrich.2.6. HCT 116 colon cancer cell culture
A batch of HTC 116 colon cancer cells (ATCC® CCL-247™) stored at −80 °C was first thawed in a 37 °C water bath and the dimethyl sulfoxide (DMSO) was removed by centrifugation at 1000g for 5 min followed by replacement of the solution by McCoy's 5A cell medium containing glutamine, fetal bovine serum (FBS) (10% v/v) and antibiotics (2% v/v from a solution containing 10 kU mL−1 penicillin and 10 mg mL−1 streptomycin). The cells were incubated in T-flasks at 37 °C, 5% CO2 and split every 2–3 days, followed by staining with Trypan Blue and viable cell counting using a Bio-Rad TC20 cell counter. The cell medium was purchased from Thermo Fisher. FBS and antibiotics were purchased from Sigma-Aldrich.2.7. Cell capture and on-chip immunocytochemistry
To monitor cell capture in the microfluidic devices, fluorescence microscopy was used during the optimization experiments. To characterize and optimize the cell capture and subsequent release, the cells were pre-stained with the cell-permeant Calcein AM viability dye (Thermo Fisher) to facilitate cell visualization, except when immunocytochemistry was subsequently performed. After staining, the cells were washed 3 times with 1 mL phosphate buffered saline (PBS) and then suspended either in 4% bovine serum albumin (BSA) prepared in PBS or whole blood samples of healthy donors supplied by a blood bank (Blodcentralen) in Stockholm, Sweden. Informed consent was obtained from all human subjects prior to the collection of blood samples. All the experiments were performed in compliance with the relevant laws and institutional guidelines of Sweden and were approved and performed under the guidelines by the ethics committee at Karolinska Institute, Stockholm, Sweden (Dnr: 02-196). The blood was drawn into EDTA tubes and processed in the same day. The cell suspension was subsequently flowed into the chip at 5 μL min−1 for 10 min (P-Chip) or 41.25 μL min−1 for 2 min (HB-Chip). On-chip immunocytochemistry was performed on the HB-Chip by sequentially flowing the following solutions: (1) PBS for 2 min at 206.25 μL min−1; (2) 8% formaldehyde for 2 min at 41.25 μL min−1 followed by incubation for at least 10 min; (3) PBS for 2 min at 206.25 μL min−1; (4) 1 mM HCl at 41.25 μL min−1 for 45 s; (5) PBS for 2 min at 206.25 μL min−1; (6) 4% BSA in PBS at 41.25 μL min−1 for 10 min followed by incubation for 1 h. (7) PBS for 2 min at 206.25 μL min−1; (8) 10 μg mL−1 anti-CK18-Alexa 488 (Abcam, ab206091, clone #EPR1626) and 10 μg mL−1 anti-CD45-Alexa 647 (R&D Systems, FAB1430R, clone #2D1) antibodies in PBS for 2 min at 41.25 μL min−1 followed by incubation overnight at 4 °C; (9) PBS for 2 min at 206.25 μL min−1; (10) Hoechst reagent in PBS at 41.25 μL min−1 for 2 min; (11) PBS for 2 min at 206.25 μL min−1. Cell capture and immunocytochemistry experiments were carried out for two independent chips in parallel. The Hoechst reagent was purchased from Abcam and used according to the manufacturer's instructions. Other chemicals were purchased from Sigma-Aldrich unless stated otherwise.2.8. Enzymatic release of captured cells
The release of captured cells was performed by flowing a PBS solution containing 10% of a commercial Viscozyme L cellulolytic enzyme mixture (Sigma Aldrich). This solution was flowed at 5 μL min−1 for 30 min (P-Chip) or 41.25 μL min−1 for 30 min (HB-Chip) into the microchannels and the cells were collected at the outlet using a pipette tip as a reservoir. After collection, the cells were transferred to an Eppendorf tube, resuspended in PBS after centrifugation for 3 min at 1000g and stained with Calcein AM and propidium iodide to evaluate the ratio of viable to non-viable cells relative to the initial cell suspension before processing.2.9. Fluorescence microscopy and image analysis
All fluorescence microscopy measurements were performed using a motorized Nikon Ti Eclipse inverted microscope coupled with a Lumencor SOLA light as an excitation source. A DAPI filter cube was used for Hoechst DNA staining dyes, a FITC filter cube was used for Alexa 488 and Alexa 430 labeled antibodies, a TRITC filter cube was used for the cellulose fluorescent tracer and a Cy5 filter cube was used for Alexa 647 labeled antibodies. All images were processed and analyzed in terms of grayscale average/profile and total cell counting using the software ImageJ (NIH, MD, USA).3. Results and discussion
3.1. Conceptual overview and preliminary AFM characterization
The combination of the nanostructured features of CNF with microfluidic flow-cells has significant potential to provide highly efficient and specific cell capture and recovery. The capture efficiency and specificity are conferred by a dense 3D layer of antibodies functionalized on the CNF, while the recovery is achieved via enzymatic degradation of the cellulose to release the coupled antibodies. The success of this concept inherently depends on the density and uniformity of the coated CNF to avoid (1) non-specific interactions of the cell surface with unmodified surfaces i.e. bare glass/PDMS or (2) specific interactions with antibodies coated directly on the glass which cannot be subsequently released. To minimize these effects which negatively impact the purity and recovery yield of target cells, the assembly of CNF as multiple layers ensures full surface coverage on both PDMS and glass while simultaneously providing a high surface density of capture antibodies.The buildup of the CNF-PEI LbL is schematized in Fig. 1A. The number of layers refers to the number of CNF layers and up to 5 CNF layers were tested. A higher number of layers were not tested due to practical limitations related to microfluidic device handling and total preparation time. The first layer of CNF was adsorbed to a base layer of PAH on both the PDMS and glass microchannel surfaces, according to previous reports.28Fig. 1B shows a schematic representing the side view of a glass/PDMS hybrid microchannel and the buildup of the LbL. Anti-EpCAM antibodies were conjugated on the upmost layers of cellulose using EDC-NHS chemistry to couple primary amines in the antibodies to carboxyl groups in the CNF. After cell capture, cellulolytic enzymes digest the cellulose and release the surface-bound antibodies, here used as a strategy to release the captured cells in mild conditions and at neutral pH. A proof-of-concept of this LbL assembly was first tested on a silicon wafer and characterized for 1, 3 and 5-layers using AFM according to the results in Fig. 1C–F. AFM images over a scratch in the LbL coatings show that a dry layer thickness up to ∼14 nm could be achieved with 5 CNF layers.
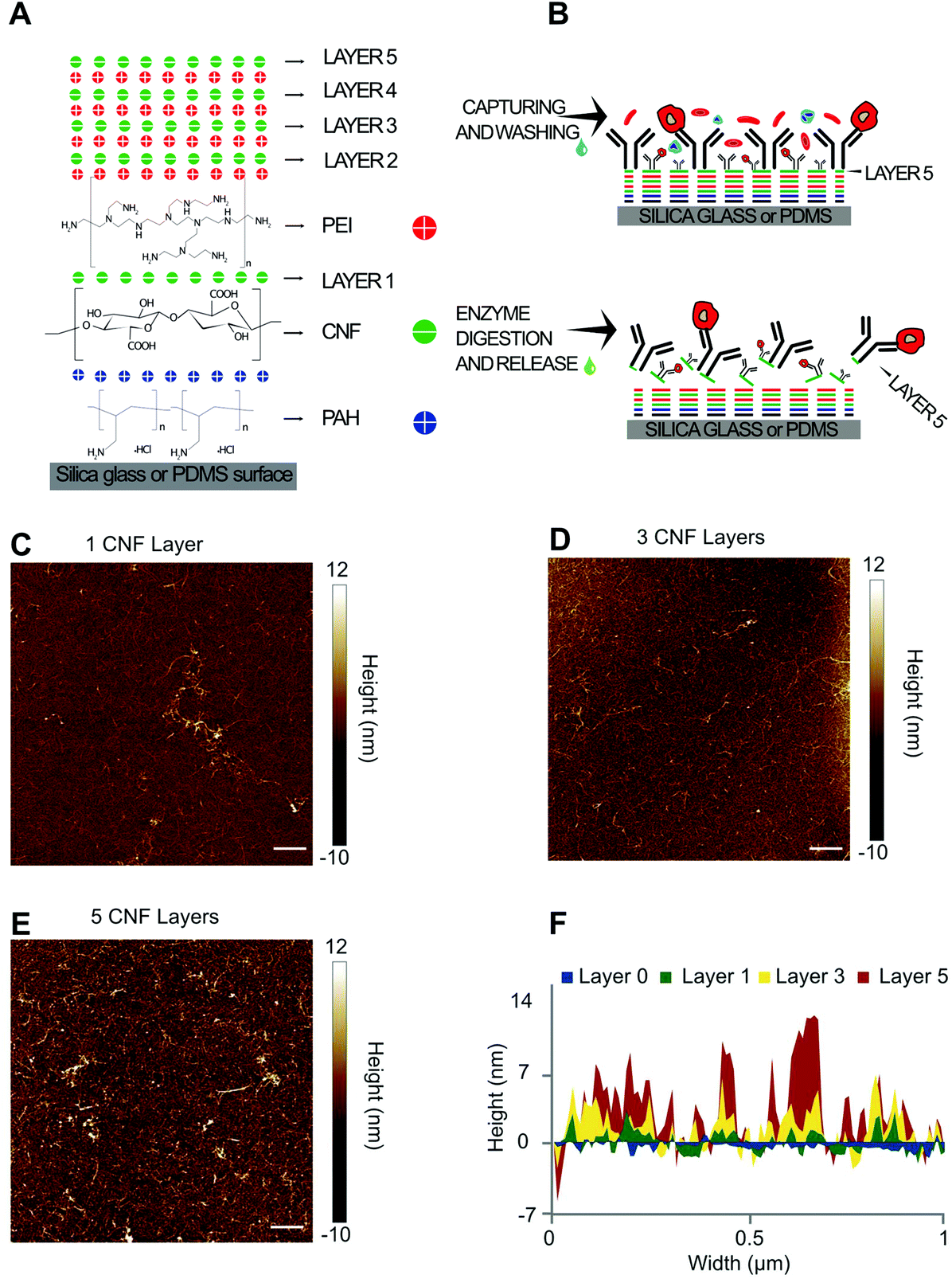 | ||
| Fig. 1 Schematics of LbL CNF assembly for affinity cell capture and enzymatic release and AFM characterization. (A) CNF LbL buildup at neutral pH on a surface modified with PAH. Both PEI and PAH have a global positive charge, while CNF has a global negative charge. (B) Affinity cell capture after conjugating anti-EpCAM antibodies on the upper layer of CNF (top), followed by digestion of the CNF layers using a cocktail of cellulolytic enzymes resulting in the release of trapped cells (bottom). (C–E) AFM characterization of 1, 3 or 5 CNF layers on a silicon wafer (10 × 10 μm). Scale bars refer to a width of 1 μm. (F) Height profile of the CNF LbL assembly for each of the tested number of layers. Layer 0 corresponds to the profile of the silicon wafer after scratching the CNF LbL assembly from the surface. | ||
3.2. Optimization of on-chip LbL CNF assembly using a fluorescent tracer
The on-chip coating of CNF was first optimized for flow rates using the microfluidic channel schematized in Fig. 2A, henceforth referred to as P-Chip. The optimization was performed for a single layer of CNF on a PAH-glass base layer. The analysis of the surface density and homogeneity of the CNF was performed using Carbotrace™ 680, a quantitative and non-destructive fluorescent tracer selective for cellulose29 with a high linear range up to ∼2.5 g L−1 cellulose in solution.30 The high specificity of the tracer for cellulose against the PAH base layer was first confirmed (Fig. S1†) before proceeding to optimize three key response parameters, i.e. average fluorescence intensity along the microchannel, depletion in fluorescence intensity along the microchannel and surface signal homogeneity measured perpendicular to the liquid flow. Under optimal conditions, the surface concentration of cellulose on the surface should be maximized while ensuring a homogeneous coating both along and across the microchannel to maximize capture efficiency. The results obtained for the optimization of the 3 response parameters against total CNF concentration (76 mg L−1–2.3 g L−1) in solution and total flow time (10–30 min) are shown in Fig. 2B–D. Focusing on the results obtained for the average fluorescence intensity along the microchannel, it could be observed that an increase in CNF concentration in solution above 230 mg L−1 did not translate into higher surface densities of adsorbed CNF. In fact, the highest tested CNF concentration resulted in the lowest intensity for 20 and 30 min flow time. While self-quenching of Carbotrace-derived fluorescence at high cellulose concentrations occurs, it was previously observed to result in a plateau rather than a decrease in measured fluorescence.30 This observation can instead be explained by the fact that the CNF are hindered to move and rotate freely in the flow at a higher concentration above its overlapping concentration (in the range of 0.4 to 0.7 g L−1),31 resulting in a lower effective concentration of free CNF in the appropriate conformation allowing adsorption on the surface. Concerning the depletion in fluorescence intensity along the microchannel, the lowest depletion (lower ΔRFU) along the channel was measured for the lowest concentration of CNF in solution for all tested flow times and the depletion value tends to increase with time. Combined with the observations made for the average fluorescence intensity along the microchannel, this increased intensity depletion result also suggests that the adsorption at the beginning of the channels are a consequence of the overlap concentration and a depletion of the fraction of CNF in appropriate conformation for adsorption is depleted at the beginning of the channel. Finally, regarding the surface signal homogeneity measured perpendicular to the liquid flow, the more homogeneous surface could be achieved at moderate CNF concentration in solution, irrespective of the total flow time. To derive the best overall condition from the data and strike an ideal balance between all three response parameters, weights of 1, 0.5 and 0.25 were attributed to the parameters average fluorescence intensity along the microchannel (1), depletion in fluorescence intensity along the microchannel (0.5) and surface signal homogeneity measured perpendicular to the liquid flow (0.25), respectively. Ideally, the best condition would provide the highest surface density of CNF, combined with the lowest heterogeneity along and across the channel. Considering the attributed weights, the best performing condition was a CNF concentration of 0.23 g L−1 flowed for 10 min, obtained by factoring the order value of each condition from best (1) to worse (9) with the attributed weights and selecting the condition with the lowest sum.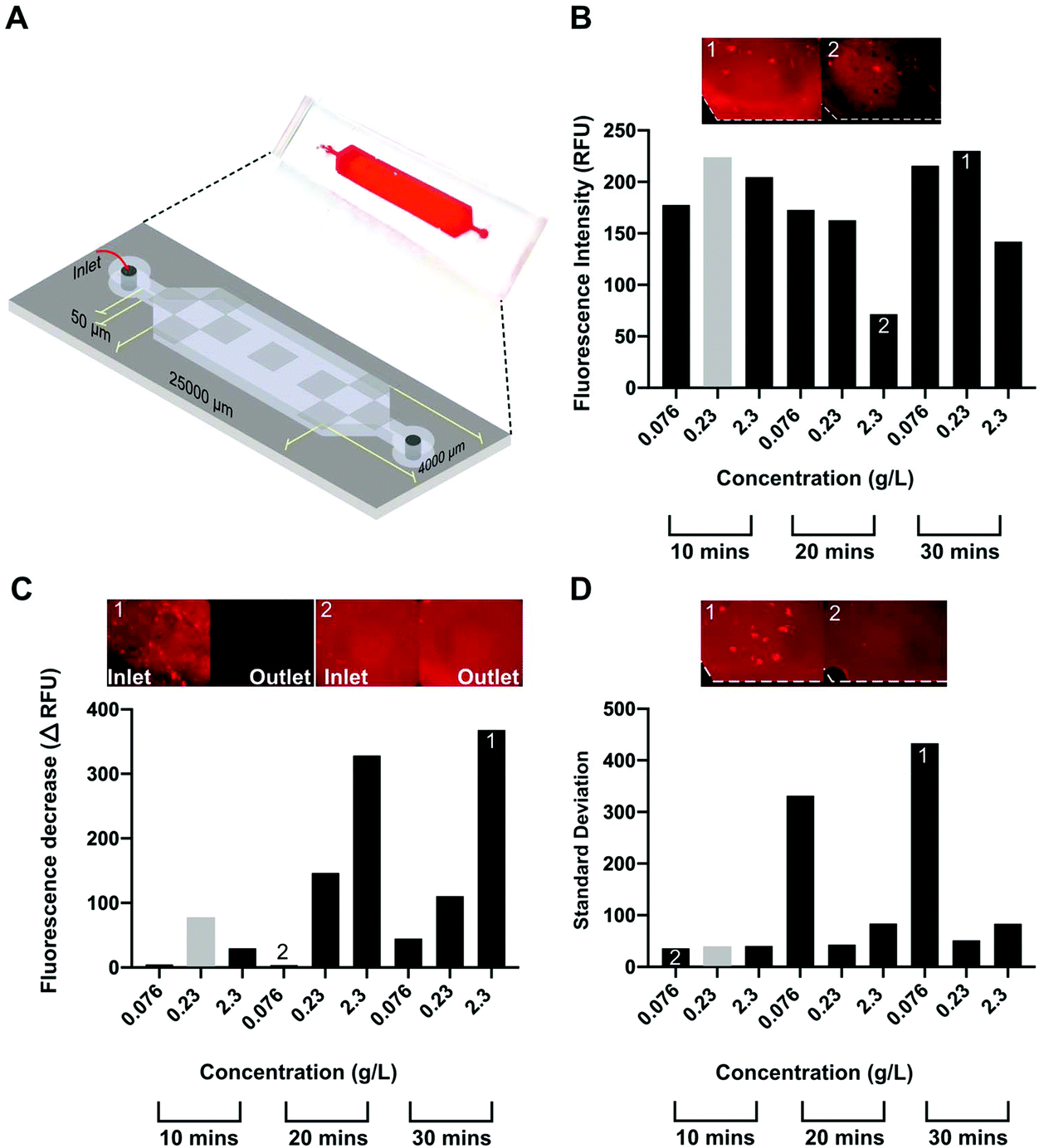 | ||
| Fig. 2 Optimization of CNF coating on P-Chips modified with PAH by changing the total CNF concentration in solution and total flow-time. (A) Photograph and schematics of the P-Chip used to optimize the CNF coating. The fluorescence signal, proportional to the surface density of CNF, was generated with Carbotrace according with section 2.4. The average fluorescence intensity along the microchannel was calculated as the average fluorescence intensity of 9 independent regions of interest (ROIs) along the channel (grey regions). The depletion in fluorescence intensity along the microchannel was calculated as the difference in fluorescence intensity on the inlet and outlet sides of the channel. The surface signal homogeneity measured perpendicular to the liquid flow was calculated as the average standard deviation of the fluorescence intensity profile perpendicular to the flow direction in 3 regions along the channel. The resulting responses for the 3 parameters with each combination of CNF concentration and flow time are shown in (B), (C) and (D), respectively. The columns highlighted in grey correspond to the overall optimal condition attributing a relative weight of 1, 0.5 and 0.25 to responses average fluorescence intensity along the microchannel, depletion in fluorescence intensity along the microchannel and surface signal homogeneity measured perpendicular to the liquid flow, respectively. The plotted data corresponds to 9 independent experiments measured according to each of the aforementioned parameters. | ||
3.3. Effect of number of layers on total CNF surface density, antibody immobilization and cell capture/release
After optimizing conditions to coat a single layer of CNF on the microfluidic channel, the impact of multiple layers on the (1) CNF surface density, (2) antibody concentration, (3) release of CNF using cellulolytic enzymes and (4) HCT 116 cell capture and enzymatic release were tested with the P-Chip and the results are shown in Fig. 3. Fig. 3A highlights the impact of 1-, 3- or 5-layers on the CNF surface density and homogeneity, demonstrating the effective buildup of a multilayer measured here by the increase in measured fluorescence signal. The results also show that a homogeneous coating can still be achieved with multiple layers. Differently, according to the results in Fig. 3B, the density of immobilized antibody follows an inverse trend. This observation, combined with an increase in surface homogeneity with 5-layers, suggests that for a single layer of CNF, a significant number of antibodies seem to be adsorbed to the non-CNF coated parts of the modified surface. Before testing the capture and release of cells, the efficiency of the cellulolytic enzymes in degrading 1 or 5 layers of CNF was shown (Fig. 3C), where a comparable final fluorescence was achieved in both cases after digestion for 30 min. A digestion time of 30 min was deemed necessary to maximize CNF degradation (Fig. S2†). Finally, the capture and release of HCT 116 cells spiked in 4% BSA were tested with both 1 and 5 CNF layers and the results are shown in Fig. 3D. The presence of a protein matrix in solution as is the case of BSA was first found to be necessary to reduce the extent of non-specific adsorption of the cells to the PAH layer (Fig. S3†). Concerning the observations of cell capture and release efficiency in the presence of 1 or 5 layers of CNF, two key conclusions were obtained, i.e. (1) fewer cells were captured with a higher number of CNF layers but, most importantly, (2) the release of captured cells is more efficient (94.5% of captured cells were released) with 5 layers of CNF. Both these conclusions further support the hypothesis that with a single layer of CNF, the uncoated surfaces can adsorb a higher surface density of antibodies, i.e. having more available non-specific binding sites compared to the lower number of carboxyl groups on the CNF, resulting in a large fraction of these antibodies not being released with the subsequent enzymatic treatment. These observations are also in agreement with previous reports highlighting lower antigen-capture efficiencies per surface area obtained with antibody densities above monolayer conditions.32 Finally, to further confirm the effectiveness of the method in the presence of lower target cell titers, the release efficiency was further observed to be >80% for input cell concentrations as low as 100 cells per mL (Fig. S4†).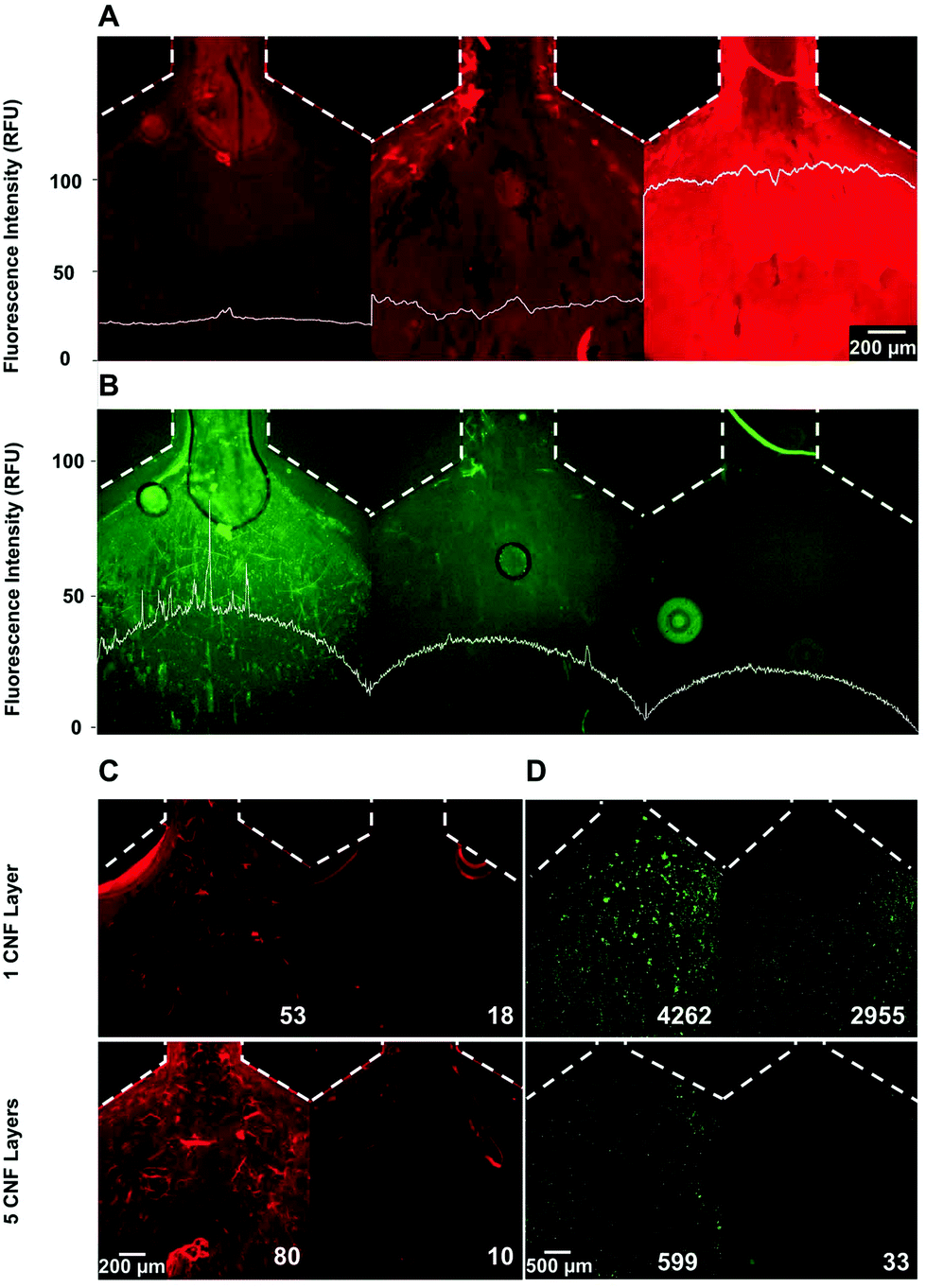 | ||
| Fig. 3 Effect of increasing numbers of CNF layers on CNF surface density, antibody immobilization, enzymatic CNF release and cell capture/release. Dotted lines indicate the border of the microchannel. The plots superimposed with the fluorescence microscopy images quantify the average lateral fluorescent intensity across the channel. (A) Cellulose surface density and homogeneity measured with Carbotrace™ 680 after coating 1, 3 or 5 CNF layers, from left to right, respectively. (B) Alexa 430 labeled anti-EpCAM antibody surface density with 1, 3 or 5 CNF layers from left to right, respectively. The results in A and B were measured in 3 independent devices, each coated with 1, 3 or 5 CNF layers. (C) Cellulose after coating (left) and after 30 min of enzyme digestion for 1 (top) and 5 (bottom) layers of CNF. The values indicate the background subtracted average fluorescence on the channel surface. (D) HCT 116 cell capture (left) and subsequent release (right) for 1 (top) and 5 (bottom) layers of CNF. The values indicate the total cell count in each image. | ||
3.4. Capture efficiency and selectivity of HCT 116 cell capture directly from undiluted whole blood
The optimized conditions were subsequently translated from the P-Chip to the HB-Chip (Fig. 4) by adjusting the flow rates to maintain the same residence time. The increased surface area provided by the larger footprint of the HB-Chip allows not only a higher sample processing throughput, but also improved capture efficiencies due to the convective flow induced by the surface grooves.6 The cell capture efficiency in the HB-Chip was first characterized and compared with the P-Chip (Table S1†). The capture yield with a HCT 116 cell input of ∼6 × 104 cells per mL was 97.2 ± 1.3% (mean ± SD, n = 2) for HB-Chip and 73.8 ± 7.9% (mean ± SD, n = 2) for the P-Chip, estimated by a balance of counted input cells vs. output cells (flow through plus PBS wash, i.e. non-captured cells). Comparing the obtained capture yield with previous reports using a similar HB architecture functionalized with a planar anti-EpCAM layer (∼92% at 20 μL min−1 (ref. 6)) not functionalized with any tridimensional functional material, the increase in capture efficiency at higher flow-rates allowed by the 3D features of the CNF assembly becomes evident. | ||
| Fig. 4 Schematics of the HB-Chip and HCT 116 cell capture directly from whole blood. On-chip immunocytochemistry was performed after cell capture. All images were acquired in a single device after flowing whole blood for 2 min at 41.25 μL min−1. | ||
The HB-Chip was subsequently tested for the direct capture of HCT 116 cells spiked in whole blood. Two independent chips were tested by flowing spiked blood and the results are compiled in Fig. 4 after performing on-chip immunocytochemistry to discriminate HCT 116 cells (CK18+, CD45−) from white blood cells (CK18−, CD45+). To determine the selectivity of cancer cell capture, 8 independent regions of interest (ROIs) with a cumulative cell count of 141 ± 37 cells were imaged with a 40× objective throughout the capture area. The relative fraction of HCT 116 to white blood cells was determined to be 3.07 ± 0.27 based on the data in Fig. 4. Considering that typically 1 mL of human blood contains 4 × 106–1.1 × 107 white blood cells per mL (ref. 33) and 105 HCT 116 cells per mL were spiked, the achieved enrichment factor ranges between ∼60-fold to ∼340-fold, assuming that for non-specific capture alone (no enrichment) the expected relative fraction of HCT 116 to white blood cells would be 0.05–0.009. In conclusion, the device allowed an increase in HCT 116 cell purity of at least 60-fold directly from unprocessed blood. However, considering the heterogeneity of blood composition in terms of cell, protein and metabolite titers, as well as the possible biological variations between different healthy donors or healthy vs. patient donors, the capture efficiency and absolute selectivity of the device are expected to vary significantly. In general, the obtained results qualitatively highlight the high specificity of the LbL assembly towards EpCAM positive vs. EpCAM negative cells. Finally, concerning the suitability of the method to process higher volumes of blood and tailor the device to capture lower cell titers, an emphasis should be placed in maximizing the width and length of the HB-Chip to avoid saturation with non-specifically bound cells. Nevertheless, accounting for the low number of captured white blood cells in Fig. 4 and the high densities of specific cells captured with higher input titers (Fig. S4†), the capacity of the proposed HB-Chip architecture is expected to be suitable to process a volume of blood at least 10-fold higher than the ∼80 μL demonstrated here.
3.5. Cell viability after enzymatic release
After characterizing the cell capture performance, the cell viability after enzymatic digestion of CNF was tested using Calcein AM as a marker of live cells and propidium iodide (PI) as a marker of dead cells. DAPI was used as an internal control. The results are shown in Fig. 5 and highlight the fraction of viable cells before on-chip capture and release (control) and after on-chip processing using two independent devices (samples 1 and 2). Both devices were loaded with the sample characterized in the control experiment. It was possible to observe that the cell viability after processing 97.7 ± 0.3% (mean ± SD, n = 2) for both sample 1 and 2 was not significantly different from the viability before processing in the microfluidic chips, i.e. control measurement (97.5%).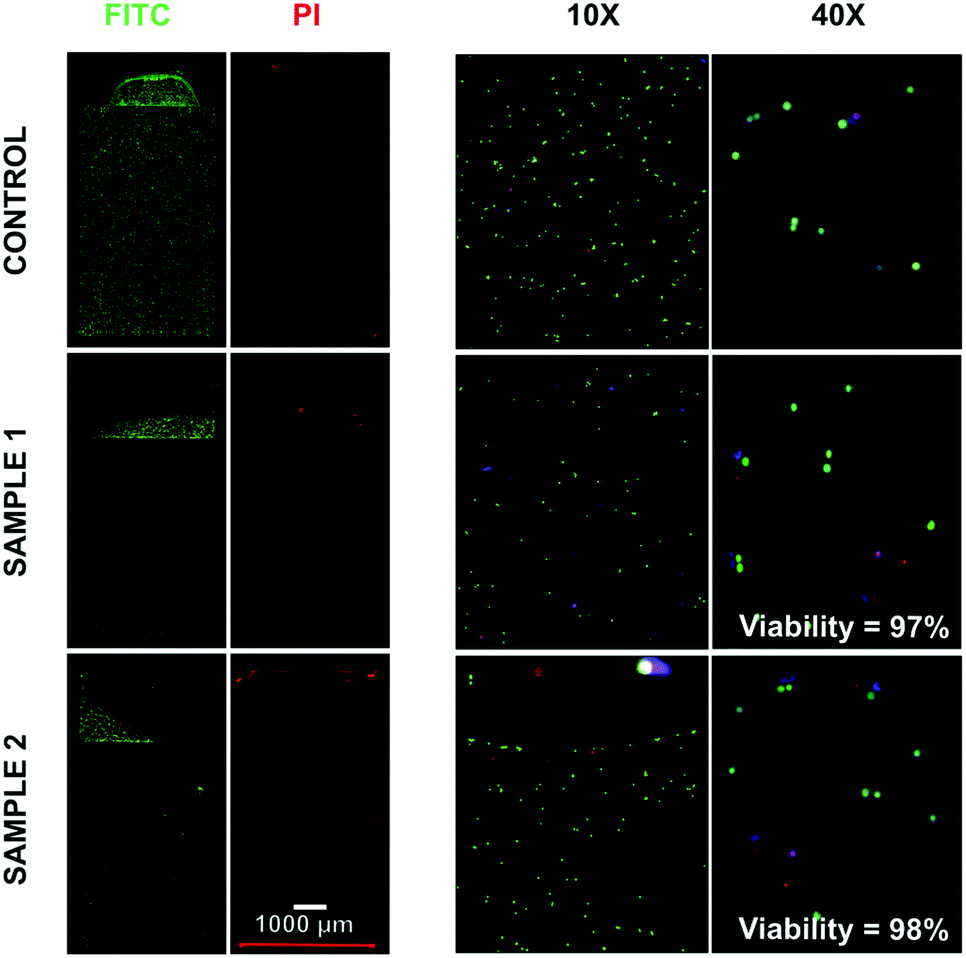 | ||
| Fig. 5 Evaluation of cell viability before (Control) and after capture/release on the HB-Chip. Samples 1 and 2 refer to two independent capture and release experiments performed on the HB-Chip. Prior to insertion into the counting slides, cells were stained with Calcein-AM and propidium iodide (PI). Viability values are calculated as the ratio of live (Calcein AM stained) to dead cells (PI stained). | ||
4. Conclusions
Towards the development of simple and affordable CTC enrichment tools to bring accurate cancer diagnostics closer to resource-limited settings, a microfluidic chip modified with a multilayer CNF assembly was developed for efficient immunoaffinity cell capture and enzymatic release. The device comprising 5 layers of CNF and coated with anti-EpCAM antibodies allowed the direct capture of HCT 116 cells from whole blood and provided an enrichment factor of ∼200-fold relative to white blood cells. Furthermore, a capture efficiency of >97% at 41.25 μL min−1 was achieved, i.e. approximately 1 hour required to process 2.5 mL whole blood thus duplicating the throughput of herringbone microchannels alone.6 This capture performance was combined with an average release efficiency of 88.2 ± 6.5% (mean ± SD, n = 4) (Fig. S4†) of the trapped cells having at least 97% viability after enzymatic digestion of CNF for 30 min. These figures of merit, comparable to state-of-the-art nanowire-based capture that require clean-room microfabrication and complex fabrication procedures with high cost,10,15 were achieved with a simple, cost-effective, versatile and bio-based surface modification approach. Finally, considering the affinity-based interactions provided by the specific immobilized antibody, the developed method is envisioned as a general strategy for the simple and effective enrichment of rare cells prior to further expansion or multiomic analysis in the context of cancer diagnostics.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the Swedish Childhood Cancer Foundation (Barncancerfonden) and European Commission through the EU FP7 CanDo (ID: 610472) project for funding this research.References
- S. C. Shah, V. Kayamba, R. M. Peek Jr. and D. Heimburger, J. Global Oncol., 2019, 1–8, DOI:10.1200/jgo.18.00200
.
- E. Lin, T. Cao, S. Nagrath and M. R. King, Annu. Rev. Biomed. Eng., 2018, 20, 329–352 CrossRef CAS
- N. Bhagwat, K. Dulmage, C. H. Pletcher Jr., L. Wang, W. DeMuth, M. Sen, D. Balli, S. S. Yee, S. Sa, F. Tong, L. Yu, J. S. Moore, B. Z. Stanger, E. P. Dixon and E. L. Carpenter, Sci. Rep., 2018, 8, 5035 CrossRef
- H. Cho, J. Kim, H. Song, K. Y. Sohn, M. Jeon and K.-H. Han, Analyst, 2018, 143, 2936–2970 RSC
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 CrossRef CAS
- S. L. Stott, C.-H. Hsu, D. I. Tsukrov, M. Yu, D. T. Miyamoto, B. A. Waltman, S. M. Rothenberg, A. M. Shah, M. E. Smas, G. K. Korir, F. P. Floyd, A. J. Gilman, J. B. Lord, D. Winokur, S. Springer, D. Irimia, S. Nagrath, L. V. Sequist, R. J. Lee, K. J. Isselbacher, S. Maheswaran, D. A. Haber and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18392 CrossRef CAS
- V. Murlidhar, M. Zeinali, S. Grabauskiene, M. Ghannad-Rezaie, M. S. Wicha, D. M. Simeone, N. Ramnath, R. M. Reddy and S. Nagrath, Small, 2014, 10, 4895–4904 CrossRef CAS
- G. Galletti, M. S. Sung, L. T. Vahdat, M. A. Shah, S. M. Santana, G. Altavilla, B. J. Kirby and P. Giannakakou, Lab Chip, 2014, 14, 147–156 RSC
- N. N. Lu, M. Xie, J. Wang, S. W. Lv, J. S. Yi, W. G. Dong and W. H. Huang, ACS Appl. Mater. Interfaces, 2015, 7, 8817–8826 CrossRef CAS
- S. Guo, J. Xu, M. Xie, W. Huang, E. Yuan, Y. Liu, L. Fan, S. Cheng, S. Liu, F. Wang, B. Yuan, W. Dong, X. Zhang, W. Huang and X. Zhou, ACS Appl. Mater. Interfaces, 2016, 8, 15917–15925 CrossRef CAS
- W. Li, E. Reategui, M. H. Park, S. Castleberry, J. Z. Deng, B. Hsu, S. Mayner, A. E. Jensen, L. V. Sequist, S. Maheswaran, D. A. Haber, M. Toner, S. L. Stott and P. T. Hammond, Biomaterials, 2015, 65, 93–102 CrossRef CAS
- Y. Deng, Y. Zhang, S. Sun, Z. Wang, M. Wang, B. Yu, D. M. Czajkowsky, B. Liu, Y. Li, W. Wei and Q. Shi, Sci. Rep., 2014, 4, 7499 CrossRef CAS
- J. Dong, Y. J. Jan, J. Cheng, R. Y. Zhang, M. Meng, M. Smalley, P.-J. Chen, X. Tang, P. Tseng, L. Bao, T.-Y. Huang, D. Zhou, Y. Liu, X. Chai, H. Zhang, A. Zhou, V. G. Agopian, E. M. Posadas, J.-J. Shyue, S. J. Jonas, P. S. Weiss, M. Li, G. Zheng, H.-H. Yu, M. Zhao, H.-R. Tseng and Y. Zhu, Sci. Adv., 2019, 5(7), eaav9186 CrossRef CAS
- X. Yu, B. Wang, N. Zhang, C. Yin, H. Chen, L. Zhang, B. Cai, Z. He, L. Rao, W. Liu, F. B. Wang, S. S. Guo and X. Z. Zhao, ACS Appl. Mater. Interfaces, 2015, 7, 24001–24007 CrossRef CAS
- J. Li, C. Qi, Z. Lian, Q. Han, X. Wang, S. Cai, R. Yang and C. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 2511–2516 CrossRef CAS
- H. Golmohammadi, E. Morales-Narváez, T. Naghdi and A. Merkoçi, Chem. Mater., 2017, 29, 5426–5446 CrossRef CAS
- K. Missoum, M. N. Belgacem and J. Bras, Materials, 2013, 6, 1745–1766 CrossRef CAS
- Y. Habibi, Chem. Soc. Rev., 2014, 43, 1519–1542 RSC
- S. Rajala, T. Siponkoski, E. Sarlin, M. Mettanen, M. Vuoriluoto, A. Pammo, J. Juuti, O. J. Rojas, S. Franssila and S. Tuukkanen, ACS Appl. Mater. Interfaces, 2016, 8, 15607–15614 CrossRef CAS
- S. Shin and J. Hyun, ACS Appl. Mater. Interfaces, 2017, 9, 26438–26446 CrossRef CAS
- B. Wu, G. Zhu, A. Dufresne and N. Lin, ACS Appl. Mater. Interfaces, 2019, 11, 16048–16058 CrossRef CAS
- Y. Zhang and O. J. Rojas, Biomacromolecules, 2017, 18, 526–534 CrossRef CAS
- K. M. A. Uddin, V. Jokinen, F. Jahangiri, S. Franssila, O. J. Rojas and S. Tuukkanen, Glob. Chall., 2019, 3, 1800079 CrossRef
- E. Karabulut, T. Pettersson, M. Ankerfors and L. Wågberg, ACS Nano, 2012, 6, 4731–4739 CrossRef CAS
- A. Chakrabarty and Y. Teramoto, Polymers, 2018, 10(5), 517 CrossRef
- J. Erlandsson, T. Pettersson, T. Ingverud, H. Granberg, P. A. Larsson, M. Malkoch and L. Wågberg, J. Mater. Chem. A, 2018, 6, 19371–19380 RSC
- I. F. Pinto, C. R. F. Caneira, R. R. G. Soares, N. Madaboosi, M. R. Aires-Barros, J. P. Conde, A. M. Azevedo and V. Chu, Methods, 2017, 116, 112–124 CrossRef CAS
- E. Gustafsson, E. Johansson, L. Wagberg and T. Pettersson, Biomacromolecules, 2012, 13, 3046–3053 CrossRef CAS
- F. X. Choong, L. Lantz, H. Shirani, A. Schulz, K. P. R. Nilsson, U. Edlund and A. Richter-Dahlfors, Cellulose, 2019, 26, 4253–4264 CrossRef CAS
- F. X. Choong, M. Back, S. E. Steiner, K. Melican, K. P. Nilsson, U. Edlund and A. Richter-Dahlfors, Sci. Rep., 2016, 6, 35578 CrossRef
- A. Naderi, T. Lindström and T. Pettersson, Cellulose, 2014, 21, 2357–2368 CrossRef CAS
- B. Saha, P. Songe, T. H. Evers and M. W. J. Prins, Analyst, 2017, 142, 4247–4256 RSC
- J. G. Hollowell, O. W. van Assendelft, E. W. Gunter, B. G. Lewis, M. Najjar and C. Pfeiffer, Vital Health Stat., 2005, 11, 1–156 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr05375a |
| ‡ These authors contributed equally. |
| § Current address: Biopromic AB, Tomtebodavägen 23A, Solna, Sweden. |
| This journal is © The Royal Society of Chemistry 2020 |