
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fractionation of block copolymers for pore size control and reduced dispersity in mesoporous inorganic thin films†
Alberto
Alvarez-Fernandez
 a,
Barry
Reid
a,
Jugal
Suthar
ab,
Swan Yia
Choy
a,
Maximiliano
Jara Fornerod
a,
Niamh
Mac Fhionnlaoich
a,
Lixu
Yang
a,
Benjamin
Schmidt-Hansberg
c and
Stefan
Guldin
*a
a,
Barry
Reid
a,
Jugal
Suthar
ab,
Swan Yia
Choy
a,
Maximiliano
Jara Fornerod
a,
Niamh
Mac Fhionnlaoich
a,
Lixu
Yang
a,
Benjamin
Schmidt-Hansberg
c and
Stefan
Guldin
*a
aDepartment of Chemical Engineering, University College London, Torrington Place, London, WC1E 7JE, UK. E-mail: s.guldin@ucl.ac.uk
bUCL School of Pharmacy, University College London, 29-39 Brunswick Square, Bloomsbury, London, WC1N 1AX, UK
cBASF SE, Process Research & Chemical Engineering, Coating & Film Processing, Carl-Bosch-Strasse 38, 67056 Ludwigshafen am Rhein, Germany
First published on 24th August 2020
Abstract
Mesoporous inorganic thin films are promising materials architectures for a variety of applications, including sensing, catalysis, protective coatings, energy generation and storage. In many cases, precise control over a bicontinuous porous network on the 10 nm length scale is crucial for their operation. A particularly promising route for structure formation utilizes block copolymer (BCP) micelles in solution as sacrificial structure-directing agents for the co-assembly of inorganic precursors. This method offers pore size control via the molecular weight of the pore forming block and is compatible with a broad materials library. On the other hand, the molecular weight dependence impedes continuous pore tuning and the intrinsic polymer dispersity presents challenges to the pore size homogeneity. To this end, we demonstrate how chromatographic fractionation of BCPs provides a powerful method to control the pore size and dispersity of the resulting mesoporous thin films. We apply a semi-preparative size exclusion chromatographic fractionation to a polydisperse poly(isobutylene)-block-poly(ethylene oxide) (PIB-b-PEO) BCP obtained from scaled-up synthesis. The isolation of BCP fractions with distinct molecular weight and narrowed dispersity allowed us to not only tune the characteristic pore size from 9.1 ± 1.5 to 14.1 ± 2.1 nm with the identical BCP source material, but also significantly reduce the pore size dispersity compared to the non-fractionated BCP. Our findings offer a route to obtain a library of monodisperse BCPs from a polydisperse feedstock and provide important insights on the direct relationship between macromolecular characteristics and the resulting structure-directed mesopores, in particular related to dispersity.
Introduction
Structural control over a bicontinuous porous inorganic network on the 10 nm length scale is crucial for the functioning of many mesoporous thin film architectures, as demonstrated for mesoporous sensors,1–3 (photo)catalysis,4,5 photovoltaics, including perovskite6 and dye-sensitized solar cells,7 optical coatings,8 protective barriers9 and separation and purification membranes.10,11 Besides the mean pore size, the resulting pore dispersity is another important material property, as evidenced in examples related to electrochemistry,2 sensing,12,13 and controlled release.14,15 This is best showcased in mass transport-limited scenarios governed by the Knudsen diffusion.16,17To this end, block copolymer (BCP) based co-assembly represents a promising bottom-up approach to create ordered mesoporous structures with tunable size and morphology.18–21 In the so-called persistent micelle templating method, inorganic precursors (typically sol–gel derived) are embedded into the corona of BCP micelles via preferential supramolecular interactions.22 In a subsequent step, the hybrid composite can be transformed into an ordered inverse opal-type mesoporous structure by either thermal calcination,22 or various physical or chemical treatments including UV-ozone degradation,23 oxygen etching24 or photocatalytic reactions.25
The use of BCP as structure-directing agent offers reliable control over porosity and pore size. While porosity is commonly tuned by the mixing ratio between organic and inorganic precursor, the pore size is predominantly determined by the molecular weight of the sacrificial block forming the micelle core,26,27 leading to final pore dimensions in the range of 5 to 50 nm.
Several recent studies have identified the close relationship between the BCP dispersity and the size distribution of the obtained co-assembled features,28–30 highlighting the requirement of precise control over the macromolecular characteristics of the structure-directing agent. In general, the synthesis of BCPs with bespoke molecular weight and narrow dispersity (Đ) is an elaborate process that involves multiple steps, some of which are furthermore difficult to govern. Synthesis is either conducted by sequential addition of monomers via living/controlled chain-growth polymerization procedures30,31 or relies on coupling reactions between different end-functionalized chains segments. For the former, close control must be achieved over reaction dynamics and conversion efficiency in order to obtain defined molecular weights and low polydispersity.32 Anionic and cationic polymerization33 as well as some types of controlled radical polymerization34 have been successfully employed in the synthesis of low polydisperse BCPs. The second strategy requires end-group functionalization for effective coupling, e.g. via Diels–Alders, thiol–ene or cycloaddition reactions.35 These coupling reaction must be rapid and selective to ensure efficient linking between the chains.36 In all cases, synthesis is highly sensitive to impurities and precise reactions conditions. Furthermore, adequate purification processes are necessary for the employed reagents, making the synthesis procedure overall very sensitive and time consuming.32
Size exclusion chromatographic (SEC) fractionation is routinely applied for purifying BCPs from their side reaction products, mainly unreacted precursors or unlinked homopolymers.37–40 More recently, Park et al. have extended liquid chromatography to the separation of BCPs in terms of their molecular weights.41 Fractions containing polystyrene-block-polyisoprene copolymers with narrower distribution in molecular weight and chemical composition were collected using a HPLC with two separation stages, showing the great potential of this approach to obtain an extensive BCP library with reduced dispersity and synthetic effort. Further experiments have established chromatography fractionation as a reliable and effective technique to provide systematic control over the macromolecular parameters (i.e. molecular weight and polydispersity) of the BCP.28,29,42,43
In this work, we present the application of BCP chromatographic fractionation to create tailored inorganic mesoporous thin films. Crucially, we identify the role of molecular weight, composition and dispersity on the resulting pore size distribution. We first isolate different BCP fractions from a polydisperse amphiphilic poly(isobutylene)-block-poly(ethyleneoxide) (PIB-b-PEO) sample and characterize their molecular weight and composition via gel permeation chromatography (GPC) and nuclear magnetic resonance (NMR). Different BCP fractions then serve for formulation with aluminosilicate sol. After deposition, the obtained thin films are characterized in both hybrid and calcined form via grazing incidence small angle X-ray scattering (GISAXS). Atomic force microscopy (AFM) and ellipsometric porosimetry (EP) to enable a full validation of this approach.
Experimental
Materials
PIB39-b-PEO36 BCP (Mn 4.85 kg mol−1; polydispersity index 1.26) was supplied by BASF following a previously reported synthetic route.44 Size exclusion chromatography (SEC) was chosen as the isolation technique for the different molecular weight (Mw) BCPs and was conducted with an Akta Explorer instrument (GE Life Sciences, Sweden). A semipreparative column (Tricorn 10/300, GE Life Sciences, Sweden) packed with Toyopearl HW-50F (Tosoh, Japan) served as the stationary phase and methanol was used as mobile phase. The sample concentration was set to 200 mg mL−1 and the flowrate of the mobile phase at 1 mL min−1. Samples were injected manually using an INV-907 injection valve equipped with a 500 μl sample loop. The chromatograms were recorded with a UV-900 UV absorption detector operating at a wavelength of 280 nm. Fractions of 1 ml in volume were collected with a fraction collector (Frac-950, GE Life Sciences, Sweden). The solvent was removed from the fractions by evaporation.BCP characterization
1H-NMR spectra were recorded on either a Bruker Advance III (600 MHz) or Bruker Advance Neo (700 MHz) NMR spectrometer at room temperature with CDCl3 as solvent. GPC was conducted at room temperature, using 5 μm PSS SDV medium combination columns (300 mm × 9 mm) with THF as solvent. Measurements were performed at a flow rate of 1 mL min−1 with a differential refractometer RI and a multi-angle light scattering detector (Brookhaven Instruments Bi-MwA). The elution times were converted into molecular weights via a calibration curve based on low dispersity polystyrene standards.Preparation of mesoporous aluminosilicate films
Inorganic sol material and mesoporous inorganic aluminosilicate films were prepared as described elsewhere.8,25 BCP samples were first dissolved in a toluene/1-butanol azeotrope solution before mixing with the inorganic sol in an organic/inorganic ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8. All samples were spin-coated at 2000 rpm for 20 seconds and immediately annealed on a programmable hot plate using a ramp rate of 1 °C per min to a final temperature of 130 °C for 30 minutes. To remove the organic structure-directing agent, samples were calcined in a muffle furnace at 450 °C for 1 hour.
:8. All samples were spin-coated at 2000 rpm for 20 seconds and immediately annealed on a programmable hot plate using a ramp rate of 1 °C per min to a final temperature of 130 °C for 30 minutes. To remove the organic structure-directing agent, samples were calcined in a muffle furnace at 450 °C for 1 hour.
Characterization of mesoporous aluminosilicate films
A Semilab SE2000 spectroscopic ellipsometer was used to perform spectroscopic ellipsometry (SE) and ellipsometric porosimetry (EP) measurements. All SE and EP data analysis was carried out on the Semilabs SEA software (v1.6.2). Prior to EP measurements, samples were placed on a hotplate at 120 °C for 10 minutes. This was to ensure that no residual atmospheric water molecules remained in the pores prior to measurement. EP porosity isotherms were derived from the evolution of the refractive index value as a function of relative humidity in the sample chamber.45 Application of the Lorentz-Lorentz effective medium approximation allowed accurate determination of the overall porosity of each sample based on the change in refractive index as a consequence of capillary condensation inside the mesopores.46 The incremental onset of capillary condensation was related to the pore diameter of the material via the Kelvin Equation.45,47 All pore size and pore volume measurements were derived from the adsorption branches of the EP porosity isotherm and adjusted based on the assumption of an ellipsoidal pore shape due to film contraction during template removal.45GISAXS data were acquired on a SAXSLab Ganesha wit at an incidence angle of 0.2. GISAXS data analysis was performed using FitGISAXS. Atomic Force Microscopy (AFM) images were obtained from a Bruker Dimension Icon atomic force microscope with a Bruker ScanAsyst Air probe (nominal tip radius 2 nm) in ScanAsyst mode.
Results and discussion
Several molecular weight (Mw) BCP were isolated following the methodology sketched in Fig. 1. In a first step, a concentrated solution of the polydisperse BCP was injected into the system. Steric and chemical interactions between the hydroxylated methacrylate-based column and the polydisperse BCP resulted in a fractionation of the feedstock. These then served as sacrificial host for the co-assembly of inorganic guest material. 0.5 ml of a 200 mg ml−1 solution of the PIB-b-PEO BCP in MeOH was first injected into the SEC system and subsequently eluded using MeOH with a flowrate of 1 ml min−1. Fractions were taken automatically every minute. Fig. 2A displays the SEC chromatogram of the polydisperse PIB-b-PEO recorded by a UV detector. The elution was divided into twenty-three fractions as shown in Fig. 2A by segmenting the retention volume every 1 ml. The BCP appeared between retention volume VR = 16 to 21 ml. Only one broad peak was observed. The obtained fractions were heated at 65 °C overnight in order to evaporate the MeOH. Note that the fractions A16 and A20 at the respective tails of the elution peak did generally not provide sufficient material to perform the macromolecular characterization, hence only fractions A17, A18 and A19 were considered for further experiments. In order to determine the composition and molecular weight of the isolated fractions 1H NMR and GPC experiments were carried out. Fig. 2B shows the GPC traces of the as-synthesized PIB-b-PEO BCP (black), and the three fractions studied during this work A17 (blue), A18 (green) and A19 (red).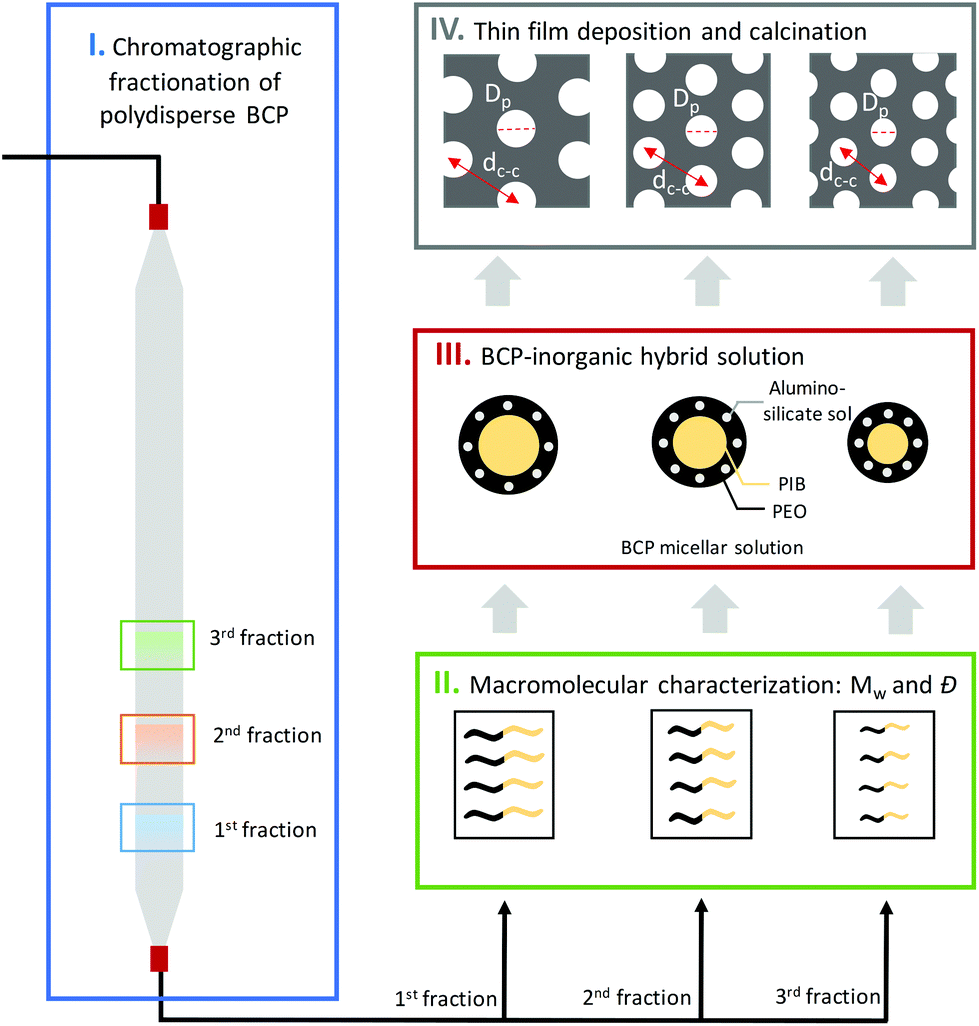 | ||
| Fig. 1 Schematic illustration of the BCP fractionation process. A polydisperse BCP is split into several molecular weight fractions by size exclusion chromatography. In a subsequent step, the BCP fractions serve as structure-directing agent to create mesoporous materials with tuneable pore sizes. | ||
 | ||
| Fig. 2 (A) Isothermal SEC profile of the as-synthesized BCP. (B) Overlay of the GPC chromatograms of the non-fractionated PIB-b-PEO block copolymer starting material (black line) and the three fractions isolated during the fractionation process, A17 (blue); A18 (green) and A19 (red). (C) PIB:PEO composition of each fraction from 1H NMR. | ||
Table 1 lists all the important macromolecular characteristics of the samples studied during this work. The molecular weight (Mw) of the non-fractionated BCP was determined to be 7.2 kg mol−1 (via a calibration curve based on low dispersity polystyrene standards), with a dispersity Đ = 1.25. In comparison, the three fractions exhibited a Mw of 8.3 kg mol−1 (A17), 7.4 kg mol−1 (A18) and 6.6 kg mol−1 (A19), respectively. It is important to highlight that Đ ≈ 1.1 was calculated for each of the collected fractions, a significantly narrower distribution than the as-synthesized BCP (Đ ≈ 1.25).
The degree of polymerization of the two blocks (PIB and PEO), obtained by 1H NMR with taking the signal of the phenyl linkage as reference (see Fig. S1 and Table S1, ESI†), was found to vary between the several collected fractions. The subsequently calculated PIB:PEO molar ratio corresponds to 1.7 (A17), 1.5 (A18) and 1.3 (A19), respectively. This compares to a molar ratio of 1.5 determined for the as-synthesized BCP. One may argue whether the increasing relative amount of PEO may be related to preferential interactions via hydrogen bonding between the PEO block and the hydroxylated methacrylate column packing material. However, when calculating the absolute molecular weight, Mw of the PEO block remained somewhat constant between the fractions, while the Mw of the PIB varied significantly. It is therefore reasonable to assume that the observed polydispersity is related to the PIB phenol precursor material rather than the subsequent grafting of polyethylene oxide.8,48 Four BCPs served as structure-directing agents during this work. The three monodisperse BCP fractions (A17, A18, A19) were prepared alongside the polydisperse, non-fractionated BCP for comparison. To this end, BCP samples were first dissolved in a toluene/1-butanol azeotrope solution before mixing with the inorganic sol. In a subsequent step, samples were spin-coated at 2000 rpm for 30 s. The hybrid aluminosilicate-BCP thin films obtained were first studied by 2D GISAXS (see ESI, Fig. S2† for diffraction patterns and line-cuts along qy integrated around the Yoneda band). The diffraction ring found in all cases is consistent with the presence of poly-oriented domains. The position of the first Bragg peak (q*) enabled to calculate the centre-to-centre pore distance (dc–c), with 29.3 nm, 25.2 nm and 23.8 nm obtained for samples co-assembled by A17, A18 and A19, respectively. Hence, the decreasing Mw of the pore forming block directly manifested in a reduced feature size in the hybrid film.
Fig. 3A–D shows the topographical AFM images of the mesoporous aluminosilicate films after BCP removal in a calcination step. The centre-to-centre distance (dc–c) was calculated for each sample using the correlation peak of the power spectral density (PSD) (see Fig. S3, ESI†). Again, a clear relationship between the Mw of the BCP and the resulting dc–c was found, with dc–c = 26.1 nm for the non-fractionated BCP and dc–c = 31.9 nm, 26.6 nm and 23.8 nm for the three fractions A17, A18 and A19, respectively.
 | ||
| Fig. 3 AFM topographical images of the mesoporous films obtained using the non-fractionated BCP (A) and the three different BCP fractions isolated during the fractionation process A19 (B), A18 (C) and A17 (D). (E–H) Image overlay with pore recognition.(I) Corresponding pore diameter histograms obtained by image analysis. | ||
Image analysis of the topographic AFM images was carried out using the software Pebbles49 to provide quantitative information about the in-plane pore dimension (Di–p) on the sample surface. The obtained pore size distribution histograms showed a constant increase with the molecular weight of the Di–p for the different BCP isolated fractions, i.e. 10.9 ± 1.4 nm for A19, 12.3 ± 1.5 nm for A18 and 14.8 ± 1.6 nm for A17, and with significantly reduced pore dispersity when compared to the non-fractionated BCP (13.0 ± 3.6). Sample analysis by AFM is limited to the surface topography and, thus, alternative characterization techniques are required to evaluate the obtained results.50
To this end, ellipsometric porosimetry (EP) has emerged as a reliable characterization technique to probe porosity, pore size and pore morphology of thin films.50Fig. 4 (left column) shows the EP adsorption isotherms obtained for the respective samples. Please note that the films exhibited a porosity in the range of 47% to 54%. Analysis of the adsorption isotherms allowed to determine the pore size distribution of the mesoporous thin films via a modified Kelvin equation.45 The Isotropic Inorganic Pore Contraction model45 was applied to calculate the in-plane (Di–p) and out-of-plane (Do–p) pore diameter distribution (PDD) data based on the EP measurements. Fig. 4 (right column) shows the PDD for the mesoporous aluminosilicate samples created using different Mw BCP. In line with AFM observations, a notable increase in Di–p was found with larger BCP molecular weight. With the lower Mw BCP (A19) exhibited a Di–p of 9.1 ± 1.5 nm, the medium BCP (A18) displayed a Di–p of 11.9 ± 1.9 nm, and the largest BCP (A17) resulted in a Di–p of 14.1 ± 2.1 nm. The Do–p dimensions scaled accordingly with 4.7 ± 0.9 nm (A19); 8.3 ± 1.1 nm (A18) and 9.2 ± 1.3 nm (A17). These results provide a clear indication of the correlation between the pore size and the Mw of the BCP used as structure-directing agent. In order to study the dependence between both parameters, the results were fitted to the following scaling law: Rp = CNαPEONβPIB, where R is the pore radius, C is a constant, and N represents the degree of polymerization of each block.51 Since NPEO was found approximately constant for the three different fractions, the relation can be simplified as Rp = C′[NβPIB]. Indeed, a linear variation with a β = 0.8 was previously found for excluded-volume BCP micelles,52,53 confirming the above-mentioned scaling law (see Fig. 5A). An analogous dependence of the resulting pore size with the molecular weight of one of the BCP blocks was recently observed in TiO2 mesoporous films made from a library of poly(N,N-dimethylacrylamide)-b-polystyrene BCP synthesized via reversible addition–fragmentation chain transfer polymerization.54 However, while previous approaches required extensive synthetic efforts to obtain a BCP library, the herein presented chromatographic fractionation enables systematic studies on structure–function relationships from a single polydisperse feedstock with little preparative work (see ESI, Fig. S4†).
 | ||
| Fig. 4 EP adsorption isotherms for the as-synthesized BCP and the different fractions with correlated pore size distributions: non-fractionated BCP (A, B), A19 (C, D), A18 (E, F) and A17 (G, H). | ||
 | ||
| Fig. 5 (A) Logarithmic variation of the pore radius as a function of NPIB for the three different fractions studied in this work. (B) Correlation between BCP Mw and pore size dispersity Đ for the different polymers studied in this work. (C) Correlation between BCP Mw and pore size entropy for the different polymers studied in this work. | ||
Moreover, analysis of EP desorption isotherms provided information about the size of the pore necks, i.e. the narrow restrictions that are connecting pores within the network.45,55,56 As shown in the ESI (Fig. S5†), the mesoporous films prepared using the lowest Mw BCP as structure-directing agent (A19) exhibited pore necks of ≈4.5 nm, which expanded to ≈5.8 nm and ≈7.3 nm for the medium (A18) and larger (A17) sized structure-directing agent, respectively. To summarize, all structural parameters obtained by the different techniques are listed in Table 2.
| d c–c AFM (nm) | d c–c GISAXS (nm) | D i–p AFM (nm) | D i–p EP (nm) | D o–p EP (nm) | Porosity EP (%) | PND EP (nm) | |
|---|---|---|---|---|---|---|---|
| A17 | 31.9 | 29.3 | 14.8 ± 1.4 | 14.1 ± 2.1 | 9.2 ± 1.3 | 54 | 7.3 ± 2.0 |
| A18 | 26.6 | 25.2 | 12.3 ± 1.5 | 11.9 ± 1.9 | 7.3 ± 1.1 | 51 | 5.8 ± 1.5 |
| A19 | 23.8 | 23.8 | 10.9 ± 1.6 | 9.1 ± 1.5 | 4.7 ± 0.9 | 48 | 4.5 ± 1.1 |
| BCPref | 26.2 | 26.8 | 13.0 ± 3.6 | 10.7 ± 4.8 | 7.7 ± 3.7 | 47 | 4.6 ± 3.1 |
Notably, when comparing the fractionated to the non-fractionated results, a clear narrowing of the pore size distribution is apparent. For AFM, the dispersity Đ of the surface-accessible mesopores, defined as (coefficient of variance)2 + 1,57 was found to be 1.08 for the non-fractionated case, which reduced as a consequence of fractionation to 1.02 (A19), 1.01 (A18) and 1.01 (A17), respectively. Alongside, EP of the non-fractionated BCP (Fig. 4A and B) provided a less defined adsorption isotherm, indicating a more polydisperse porous structure. Indeed, when comparing the results obtained for the non-fractionated BCP (Di–p of 10.7 ± 4.8 nm and Do–p of 7.7 ± 3.7 nm) with the fractionated samples, a clear improvement in the pore size dispersity was observed. In this case, the pore size dispersity Đ, reduced from 1.20 to 1.03 (A19), 1.03 (A18) and 1.02 (A17) as a consequence of fractionation (see Fig. 5B).
While reporting of Đ is common practice in the community, it requires the population to follow a prescribed distribution. This may not be the case for mesopores, where poly- or heterodisperse pore sizes are often found.9,58 Indeed, the pore sizes obtained by EP herein could not be precisely described by normal distributions (see ESI, Fig. S6†). We have recently introduced the concept of information entropy to report on nanoparticle dispersity.59 Crucially, this approach is assumption-free and allows a reliable comparison of dispersity between different populations via a user-friendly macro. While this approach was first presented on colloidal populations, it can be equally applied to pore size or molecular weight distributions. To this end, the absolute entropy (E) results obtained herein are shown in Table S2.† In the case of the BCP molecular weight, the absolute entropy value reduced from 8150 (non-fractionated BCP) to 6090 (A17), 3760 (A18) and 5490 (A19) after the fractionation. A similar tendency was found for the pore size distribution. EP analysis provided an entropy value E for the non-fractionated BCP of 28.1, which decreased as a consequence of fractionation to 14.0 (A19), 16.5 (A18) and 23.1 (A17), respectively. In comparison, AFM image analysis led to entropy values of 8.16 (as-synth), 5.00 (A19), 5.40 (A18) and 5.42 (A17), respectively. Linear regression of both variables (Fig. 5C) confirms the direct relationship between the dispersity of the BCP structure-directing agent Mw and the dispersity in pore size of the final inorganic porous structure. This correlation was found to be more pronounced in the case of the EP measurements (p value = 0.049) than in the case of the AFM data (p-value = 0.205). While the different techniques are consistent in their findings, some deviation may be related to the fact that AFM refers to the topology of a limited number of pores on the sample surface, while EP determines pore size by capillary condensation occurring in the bulk of the film. Moreover AFM lateral resolution can be affected by tip convolution effects, and therefore some discrepancies between the data obtained by the different techniques may be expected.50 Nevertheless, a narrower Mw distribution of the BCP induced in all cases a lower pore size dispersity in the final mesoporous film. This provides further evidence that the herein presented approach of BCP chromatographic fractionation not only offers a straightforward method to obtain an extensive BCP library with little synthetic effort but also reduced dispersity in inorganic mesoporous films.
We note that out approach is fundamentally different to the more common route of pore expansion by supramolecular co-assembly with swelling agents that selectively interact with the pore-forming block, e.g. benzene derivatives,60,61 homopolymers,55,62 carboxylic acids,63,64 or solvents such as toluene or xylene.65,66 In contrast to the herein presented strategy, swelling agents present multiple challenges, i.e. macroscopic phase separation, increase in the pore size dispersity, multimodal pore size distribution and a decrease in the long-range order of the structure,67 which limits their applicability in particular when both mean pore diameter and dispersity are important. Our findings are equally relevant for other BCP techniques, such as sequential infiltration synthesis68,69 or aqueous metal reduction,70,71 where nanostructured BCP films serve as scaffolds for multistep fabrication procedures post self-assembly.
Conclusions
Herein, we establish a route to pore size control in mesoporous inorganic thin film architectures based on fractionation by size exclusion chromatography of a polydisperse BCP. The various Mw BCP fractions served for fabrication of aluminosilicate mesoporous architectures via co-assembly. The combination of characterization by AFM, GISAXS and EP allowed the establishment of a close relationship between the molecular properties of the structure-directing BCP and the resulting mesoporous network, with significant variations in the center-to-center distance and pore sizes. Importantly, a close relationship between the entropic dispersity calculations of the BCP and the formed mesopores underlines the importance of polymer dispersity in the initial feedstock and highlights the relevance of the herein presented approach.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AAF, MJF and SG acknowledge funding by EPSRC New Investigator Award (EP/R035105/1). BR is grateful for an EPSRC Industrial Case Award (EP/M506448/1) in support of BASF. JS acknowledges the EPSRC (EP/L01646X), the MRC (MR/R000328/1), University College London and University of Nottingham CDT in Advanced Therapeutics and Nanomedicines for financial support. LY is grateful for an Inspiration Grant by the UCL Centre for Nature Inspired Engineering (EPSRC Award, EP/K038656/1). The authors thank Dr Han Wu and the EPSRC CNIE research facility service (EPSRC Award, EP/K038656/1) at University College London for the collection of GISAXS data and Dr Alex Obiakor from Tosoh Bioscience for technical advice on column fractionation.Notes and references
- C. Roychaudhuri, Sens. Actuators, B, 2015, 210, 310–323 CrossRef CAS
.
- S. Alberti, P. Y. Steinberg, G. Giménez, H. Amenitsch, G. Ybarra, O. Azzaroni, P. C. Angelomé and G. J. A. A. Soler-Illia, Langmuir, 2019, 35, 6279–6287 CrossRef CAS
- N. Reta, C. P. Saint, A. Michelmore, B. Prieto-Simon and N. H. Voelcker, ACS Appl. Mater. Interfaces, 2018, 10, 6055–6072 CrossRef CAS
- W. Kim, S. Y. Choi, Y. M. Jeon, S. K. Lee and S. H. Kim, ACS Appl. Mater. Interfaces, 2014, 6, 11484–11492 CrossRef CAS
- E. Doustkhah, J. Lin, S. Rostamnia, C. Len, R. Luque, X. Luo, Y. Bando, K. C.-W. Wu, J. Kim, Y. Yamauchi and Y. Ide, Chem. – Eur. J., 2019, 25, 1614–1635 CrossRef CAS
- J. Shao, S. Yang, L. Lei, Q. Cao, Y. Yu and Y. Liu, Chem. Mater., 2016, 28, 7134–7144 CrossRef CAS
- P. Docampo, S. Guldin, T. Leijtens, N. K. Noel, U. Steiner and H. J. Snaith, Adv. Mater., 2014, 26, 4013–4030 CrossRef CAS
- B. Reid, A. Taylor, Y. Chen, B. Schmidt-Hansberg and S. Guldin, ACS Appl. Mater. Interfaces, 2018, 10, 10315–10321 CrossRef CAS
- C. Sanchez, C. Boissière, D. Grosso, C. Laberty and L. Nicole, Chem. Mater., 2008, 20, 682–737 CrossRef CAS
- P. Wang, M. Wang, F. Liu, S. Ding, X. Wang, G. Du, J. Liu, P. Apel, P. Kluth, C. Trautmann and Y. Wang, Nat. Commun., 2018, 9, 569 CrossRef
- E. A. Jackson and M. A. Hillmyer, ACS Nano, 2010, 4, 3548–3553 CrossRef CAS
- J. F. Angiolini, M. Stortz, P. Y. Steinberg, E. Mocskos, L. Bruno, G. Soler-Illia, P. C. Angelomé, A. Wolosiuk and V. Levi, Phys. Chem. Chem. Phys., 2017, 19, 26540–26544 RSC
- P. Y. Steinberg, M. M. Zalduendo, G. Giménez, G. J. A. A. Soler-Illia and P. C. Angelomé, Phys. Chem. Chem. Phys., 2019, 21, 10347–10356 RSC
- H. Y. Lian, Y. H. Liang, Y. Yamauchi and K. C.-W. Wu, J. Phys. Chem. C, 2011, 115, 6581–6590 CrossRef CAS
- R. K. Kankala, Y. Han, J. Na, C. Lee, Z. Sun, S. Wang, T. Kimura, Y. S. Ok, Y. Yamauchi, A. Chen and K. C. –W. Wu, Adv. Mater., 2020, 32, 1907035 CrossRef CAS
-
E. Nagy, Basic Equations of Mass Transport Through a Membrane Layer, Elsevier, 2019 Search PubMed
- A. Walcarius, Electroanalysis, 2015, 27, 1303–1340 CrossRef CAS
- P. C. A. Alberius, K. L. Frindell, R. C. Hayward, E. J. Kramer, G. D. Stucky and B. F. Chmelka, Chem. Mater., 2002, 14, 3284–3294 CrossRef CAS
- E. L. Crepaldi, G. J. A. A. Soler-Illia, D. Grosso, F. Cagnol, F. Ribot and C. Sanchez, J. Am. Chem. Soc., 2003, 125, 9770–9786 CrossRef CAS
- M. Nedelcu, J. Lee, E. J. W. Crossland, S. C. Warren, M. C. Orilall, S. Guldin, S. Hüttner, C. Ducati, D. Eder, U. Wiesner, U. Steiner and H. J. Snaith, Soft Matter, 2009, 5, 134–139 RSC
- H. N. Lokupitiya, A. Jones, B. Reid, S. Guldin and M. Stefik, Chem. Mater., 2016, 28, 1653–1667 CrossRef CAS
- M. Templin, A. Franck, A. Du Chesne, H. Leist, Y. Zhang, R. Ulrich, V. Schadler and U. Wiesner, Science, 1997, 278, 1795–1798 CrossRef CAS
- A. Hozumi, H. Sugimura, K. Hiraku, T. Kameyama and O. Takai, Chem. Mater., 2000, 12, 3842–3847 CrossRef CAS
- S. Guldin, P. Kohn, M. Stefik, J. Song, G. Divitini, F. Ecarla, C. Ducati, U. Wiesner and U. Steiner, Nano Lett., 2013, 13, 5329–5335 CrossRef CAS
- B. Reid, A. Taylor, A. Alvarez-Fernandez, M. H. Ismael, S. Sharma, B. Schmidt-Hansberg and S. Guldin, ACS Appl. Mater. Interfaces, 2019, 11, 19308–19314 CrossRef CAS
- J. Noolandi and K. M. Hong, Macromolecules, 1983, 16, 1443–1448 CrossRef CAS
- E. Bloch, P. L. Llewellyn, T. Phan, D. Bertin and V. Hornebecq, Chem. Mater., 2009, 21, 48–55 CrossRef CAS
- S. Park, D. Y. Ryu, J. K. Kim, M. Ree and T. Chang, Polymer, 2008, 49, 2170–2175 CrossRef CAS
- N. A. Lynd, A. J. Meuler and M. A. Hillmyer, Prog. Polym. Sci., 2008, 33, 875–893 CrossRef CAS
- H. Feng, X. Lu, W. Wang, N. G. Kang and J. W. Mays, Polymer, 2017, 9, 494 Search PubMed
-
M. Zhang, S. M. June, T. E. Long and J. Kong, in Reference Module in Materials Science and Materials Engineering, Elsevier, 2016 Search PubMed
- N. Hadjichristidis, M. Pitsikalis and H. Iatrou, Adv. Polym. Sci., 2005, 189, 1–124 CrossRef CAS
-
N. Hadjichristidis and A. Hirao, Anionic Polymerization, Springer Japan, Tokyo, 2015
- A. Mühlebach, S. G. Gaynor and K. Matyjaszewski, Macromolecules, 1998, 31, 6046–6052 CrossRef
- T. P. Lodge, Macromol. Chem. Phys., 2003, 204, 265–273 CrossRef CAS
- A. B. Chang and F. S. Bates, Macromolecules, 2020, 53, 2765–2768 CrossRef CAS
- M. Schollenberger and W. Radke, J. Chromatogr. A, 2011, 1218, 7828–7831 CrossRef CAS
- W. Kim, J. Han, C. Y. Ryu and H. Yang, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 3612–3620 CrossRef CAS
- J. Lee, K. Lee, J. Park, J. K. Kim and T. Chang, Polymer, 2015, 80, 46–51 CrossRef CAS
- M. Janco, D. Berek, A. Önen, C.-H. Fischer, Y. Yagci and W. Schnabel, Polym. Bull., 1997, 38, 681–688 CrossRef CAS
- S. Park, D. Cho, J. Ryu, K. Kwon, W. Lee and T. Chang, Macromolecules, 2002, 35, 5974–5979 CrossRef CAS
- S. Park, K. Kwon, D. Cho, B. Lee, M. Ree and T. Chang, Macromolecules, 2003, 36, 4662–4666 CrossRef CAS
- A. C. Makan, P. Sinha, N. Ngaza, W. Van Aswegen and H. Pasch, Anal. Bioanal. Chem., 2013, 405, 9041–9047 CrossRef CAS
- T. von Graberg, P. Hartmann, A. Rein, S. Gross, B. Seelandt, C. Röger, R. Zieba, A. Traut, M. Wark, J. Janek and B. M. Smarsly, Sci. Technol. Adv. Mater., 2011, 12, 025005 CrossRef
- C. Boissiere, D. Grosso, S. Lepoutre, L. Nicole, A. B. Bruneau and C. Sanchez, Langmuir, 2005, 21, 12362–12371 CrossRef CAS
- W. Li, J. Liu and D. Zhao, Nat. Rev. Mater., 2016, 1, 16023 CrossRef CAS
- M. R. Baklanov, K. P. Mogilnikov, V. G. Polovinkin and F. N. Dultsev, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct., 2000, 18, 1385–1391 CrossRef CAS
- E. L. Malins, C. Waterson and C. R. Becer, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 634–643 CrossRef CAS
- S. Mondini, A. M. Ferretti, A. Puglisi and A. Ponti, Nanoscale, 2012, 4, 5356 RSC
- A. Alvarez-Fernandez, B. Reid, M. J. Fornerod, A. Taylor, G. Divitini and S. Guldin, ACS Appl. Mater. Interfaces, 2020, 12, 5195–5208 CrossRef CAS
- E. Bloch, P. L. Llewellyn, T. Phan, D. Bertin and V. Hornebecq, Chem. Mater., 2009, 21, 48–55 CrossRef CAS
- C. Svaneborg and J. S. Pedersen, Macromolecules, 2002, 35, 1028–1037 CrossRef CAS
- M. Gordon, Polymer, 1981, 22, 565 CrossRef
- J. Billet, S. Vandewalle, M. Meire, N. Blommaerts, P. Lommens, S. W. Verbruggen, K. De Buysser, F. Du Prez and I. Van Driessche, J. Mater. Sci., 2020, 55, 1933–1945 CrossRef CAS
- B. Reid, A. Alvarez-Fernandez, B. Schmidt-Hansberg and S. Guldin, Langmuir, 2019, 35, 14074–14082 CrossRef CAS
- M. Thommes, Chem. Ing. Tech., 2010, 82, 1059–1073 CrossRef CAS
- S. Harrisson, Polym. Chem., 2018, 9, 1366–1370 RSC
-
P. Innocenzi, S. Costacurta, T. Kidchob, L. Malfatti, P. Falcaro and G. Soler-Illia, in Thin Films, Springer, Dordrecht, 2008, pp. 105–123 Search PubMed
- N. Mac Fhionnlaoich and S. Guldin, Chem. Mater., 2020, 32, 3701–3706 CrossRef CAS
- J. Yi and M. Kruk, Langmuir, 2015, 31, 7623–7632 CrossRef CAS
- J. Fan, C. Yu, J. Lei, Q. Zhang, T. Li, B. Tu, W. Zhou and D. Zhao, J. Am. Chem. Soc., 2005, 127, 10794–10795 CrossRef CAS
- A. Sarkar, A. Thyagarajan, A. Cole and M. Stefik, Soft Matter, 2019, 15, 5193–5203 RSC
- A. Álvarez-Fernández, F. Valdés-Bango, R. Losada-Ambrinos, J. I. Martín, M. Vélez, J. M. Alameda and F. J. García Alonso, Polym. Int., 2018, 67, 393–398 CrossRef
- A. Alvarez–Fernandez, F. Valdes–Vango, J. I. Martín, M. Vélez, C. Quirós, D. Hermida–Merino, G. Portale, J. M. Alameda and F. J. García Alonso, Polym. Int., 2019, 68, 1914–1920 CrossRef
- Y. Li and M. Kruk, RSC Adv., 2015, 5, 69870–69877 RSC
- L. Huang and M. Kruk, Chem. Mater., 2015, 27, 679–689 CrossRef CAS
- Q. L. Wu and S. E. Rankin, J. Phys. Chem. C, 2011, 115, 11925–11933 CrossRef CAS
- C. Zhou, T. Segal-Peretz, M. E. Oruc, H. S. Suh, G. Wu and P. F. Nealey, Adv. Funct. Mater., 2017, 27, 1701756 CrossRef
- Q. Peng, Y. C. Tseng, S. B. Darling and J. W. Elam, ACS Nano, 2011, 5, 4600–4606 CrossRef CAS
- A. Alvarez-Fernandez, K. Aissou, G. Pécastaings, G. Hadziioannou, G. Fleury and V. Ponsinet, Nanoscale Adv., 2019, 1, 849–857 RSC
- J. Chai, D. Wang, X. Fan and J. M. Buriak, Nat. Nanotechnol., 2007, 2, 500–506 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Additional structural analysis results using GISAXS, AFM and EP. See DOI: 10.1039/d0nr05132b |
| This journal is © The Royal Society of Chemistry 2020 |