
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced charge storage of nanometric ζ-V2O5 in Mg electrolytes†
Ian D.
Johnson
 abc,
Gene
Nolis
cde,
Liang
Yin
cf,
Hyun Deog
Yoo
cdg,
Prakash
Parajuli
ch,
Arijita
Mukherjee
ch,
Justin L.
Andrews
i,
Mario
Lopez
cd,
Robert F.
Klie
ch,
Sarbajit
Banerjee
i,
Brian J.
Ingram
bc,
Saul
Lapidus
cf,
Jordi
Cabana
*cd and
Jawwad A.
Darr
*a
abc,
Gene
Nolis
cde,
Liang
Yin
cf,
Hyun Deog
Yoo
cdg,
Prakash
Parajuli
ch,
Arijita
Mukherjee
ch,
Justin L.
Andrews
i,
Mario
Lopez
cd,
Robert F.
Klie
ch,
Sarbajit
Banerjee
i,
Brian J.
Ingram
bc,
Saul
Lapidus
cf,
Jordi
Cabana
*cd and
Jawwad A.
Darr
*a
aDepartment of Chemistry, University College London, London WC1H 0AJ, UK. E-mail: j.a.darr@ucl.ac.uk; Tel: +44 (0)20 7679 4345
bChemical Sciences and Engineering Division, Argonne National Laboratory, Lemont, Illinois 60439, USA
cJoint Center for Energy Storage Research, Argonne National Laboratory, Lemont, IL 60439, USA
dDepartment of Chemistry, University of Illinois at Chicago, Chicago, IL 60607, USA. E-mail: jcabana@uic.edu; Tel: (+1) 312-355-4309
eCICenergiGUNE, Parque Tecnológico de Álava, Albert Einstein 48, ED.CIC, 01510, Miñano, Spain
fX-ray Science Division, Argonne National Laboratory, Lemont, Illinois 60439, USA
gDepartment of Chemistry and Chemical Institute for Functional Materials, Pusan National University, Busan 46241, Republic of Korea
hDepartment of Physics, University of Illinois at Chicago, Chicago, IL 60607, USA
iDepartment of Chemistry, Texas A&M University, College Station, TX 77843, USA
First published on 30th October 2020
Abstract
V2O5 is of interest as a Mg intercalation electrode material for Mg batteries, both in its thermodynamically stable layered polymorph (α-V2O5) and in its metastable tunnel structure (ζ-V2O5). However, such oxide cathodes typically display poor Mg insertion/removal kinetics, with large voltage hysteresis. Herein, we report the synthesis and evaluation of nanosized (ca. 100 nm) ζ-V2O5 in Mg-ion cells, which displays significantly enhanced electrochemical kinetics compared to microsized ζ-V2O5. This effect results in a significant boost in stable discharge capacity (130 mA h g−1) compared to bulk ζ-V2O5 (70 mA h g−1), with reduced voltage hysteresis (1.0 V compared to 1.4 V). This study reveals significant advancements in the use of ζ-V2O5 for Mg-based energy storage and yields a better understanding of the kinetic limiting factors for reversible magnesiation reactions into such phases.
Introduction
There is an increasing societal need for reliable and improved energy storage. Li-ion batteries currently possess the highest energy density of all commercially available rechargeable batteries,1 and are currently the power source of choice for mobile devices and Electric Vehicles (EVs). However, Li-ion batteries are already highly optimized and are approaching their theoretical maximum storage capability.2 Therefore, there is a clear need for the development of “beyond Li-ion” rechargeable battery technologies.3 Of these, Mg batteries could potentially offer ca. twice the energy-density of current Li-ion batteries.4 In part, this would be enabled by their incipient capability to safely and reversibly plate Mg metal at the anode,5–7 especially if remaining challenges at high current densities can be resolved.8,9 Metallic Mg anodes offer a much greater volumetric energy density (up to 3833 mA h cm−3) than current Li-ion graphite anodes (∼800 mA h cm−3).6 Nevertheless, a key limitation currently impeding the development of an energy-dense Mg battery is the dearth of high redox-potential electrode materials that are able to reversibly store Mg2+ with acceptable efficiency. Electrode materials that demonstrate efficient Mg2+ storage and suitable Mg transport properties, typically contain heavy chalcogenides such as Mo6S8 or TiS2, whose voltages are too low to realize any improvement over advanced Li-ion batteries.10–13 In contrast, oxide materials can offer much higher potentials vs. Mg/Mg2+, e.g. 2.5 V for V2O5,14,15 2.9 V for MgMn2O4,16 and 3.5 V for MgCr2O4,14,15,17–19 although these materials typically exhibit Mg insertion and removal kinetics that are moderate-to-poor (e.g. overpotentials of >0.7 V for V2O5, corresponding to ∼70% energy efficiency). While there have been numerous studies of alternate cathode chemistries,20–23 none have yet yielded a high-energy density Mg cathode with good kinetics.Recently, Andrews et al. reported reversible intercalation of Mg2+ into micron-sized ζ-V2O5.14 While ζ-V2O5 only achieved a stable capacity of ca. 60 mA h g−1 after 10 cycles, with a large voltage hysteresis of ∼1.5 V, the authors suggested that the metastable tunnel structure of ζ-V2O5 had relatively low Mg diffusion activation energy barriers, especially compared to the thermodynamically stable layered α-V2O5 polymorph (in the range of 600 to 900 meV versus 1200 meV, respectively).14 Therefore, if the capacity and voltage hysteresis can be improved, ζ-V2O5 is a strong candidate for a high-voltage Mg electrode material. One strategy for this endeavor is nanosizing ζ-V2O5, which might mitigate the effects of diffusion barriers by reducing the required diffusion length for Mg to access the core of the particle. Furthermore, nanosizing increases the ζ-V2O5 specific surface area, which will increase the reaction area for transfer of Mg to and from the electrolyte to the electrode. These beneficial effects should not only increase capacity, but also reduce the observed overpotentials of cycling.24
Herein, we utilized a multistep process beginning with a Continuous Hydrothermal Flow Synthesis (CHFS) process to obtain nanometric ζ-V2O5 for assessment in Mg electrolytes. The use of CHFS enabled the production of nano-sized crystallites via highly supersaturating conditions and a short reaction time (residence time on the order of a few seconds),25,26 and the process has previously produced a wide variety of high-performance nanosized electrode materials for Mg batteries, Li-ion batteries and supercapacitors.17,27–31
Experimental section
Continuous Hydrothermal Flow Synthesis
Two precursor solutions were prepared; the first was an aqueous solution of V4+ dissolved in Deionised (DI) water, which was prepared by combining V2O5 (Sigma-Aldrich, Gillingham, UK) and oxalic acid (Sigma-Aldrich, Gillingham, UK) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 molar ratio and stirring overnight to produce a 0.2 M dark blue solution. The second solution was 0.033 M AgNO3 (Fisher Scientific, Loughborough, UK) dissolved in DI water. Each solution was pumped via two separate precursor pumps (P2 and P3, Fig. S1a†) into a ¼′′ stainless steel T-piece mixer at a flow rate of 40 mL min−1 each. This combined mixture was fed into the Confined Jet Mixer (CJM, Fig. S1b†) at a total flow rate of 80 mL min−1, where it combined with 80 mL min−1 of supercritical water at 450 °C and 24.1 MPa from pump P1. The mixing dynamics of combination was in the turbulent regime, with a Reynolds number of >6900, resulting in rapid formation of nanoparticles in flow (mixing temperature of 335 °C). The nanoparticle slurry flowed along an outlet pipe at this temperature with a residence time of 7.2 s before passing through a pipe-in-pipe countercurrent heat exchanger that cooled it to ca. 40 °C. Finally, the cooled slurry passed through a back-pressure regulator (Tescom model 26-1762-24194, Emerson Process Management Regulator Technologies, Inc., South Lanarkshire, UK), which maintained a pressure of 24 MPa throughout the flow reactor. The slurry was collected in a beaker open to the atmosphere and allowed to settle; after three hours, the supernatant was siphoned off. The slurry was placed in dialysis bags (Visking Dialysis Tubing, Medicell Membranes Ltd, London, UK), which were suspended in stirred DI water, which was replaced at regular intervals. The ions in the slurry (unreacted precursor and waste counterions) were removed until the conductivity of the supernatant was below 100 μS m−1.
:4 molar ratio and stirring overnight to produce a 0.2 M dark blue solution. The second solution was 0.033 M AgNO3 (Fisher Scientific, Loughborough, UK) dissolved in DI water. Each solution was pumped via two separate precursor pumps (P2 and P3, Fig. S1a†) into a ¼′′ stainless steel T-piece mixer at a flow rate of 40 mL min−1 each. This combined mixture was fed into the Confined Jet Mixer (CJM, Fig. S1b†) at a total flow rate of 80 mL min−1, where it combined with 80 mL min−1 of supercritical water at 450 °C and 24.1 MPa from pump P1. The mixing dynamics of combination was in the turbulent regime, with a Reynolds number of >6900, resulting in rapid formation of nanoparticles in flow (mixing temperature of 335 °C). The nanoparticle slurry flowed along an outlet pipe at this temperature with a residence time of 7.2 s before passing through a pipe-in-pipe countercurrent heat exchanger that cooled it to ca. 40 °C. Finally, the cooled slurry passed through a back-pressure regulator (Tescom model 26-1762-24194, Emerson Process Management Regulator Technologies, Inc., South Lanarkshire, UK), which maintained a pressure of 24 MPa throughout the flow reactor. The slurry was collected in a beaker open to the atmosphere and allowed to settle; after three hours, the supernatant was siphoned off. The slurry was placed in dialysis bags (Visking Dialysis Tubing, Medicell Membranes Ltd, London, UK), which were suspended in stirred DI water, which was replaced at regular intervals. The ions in the slurry (unreacted precursor and waste counterions) were removed until the conductivity of the supernatant was below 100 μS m−1.
The cleaned and concentrated slurry was freeze-dried by heating from −60 °C to 25 °C over 24 h under vacuum (<13 Pa, VirTis Genesis 35 XL, SP Scientific, New York, U.S.). The initial product (a mixture of Ag metal and VOx) was converted to Ag0.33V2O5 by annealing at 400 °C for 5 hours (ramp rate 1 °C min−1) in air. Finally, ζ-V2O5 was prepared by chemically leaching the Ag0.33V2O5 product in 0.71 M HCl in DI water at 210 °C in an autoclave for 24 h. The crude product was subsequently washed with 3 × 50 mL 5 wt% Na2S2O3 in DI water (with centrifugation at 4000 rpm for 5 minutes after each wash), and a final wash of 50 mL 5 wt% Na2S2O3 for 36 hours to completely remove the AgCl byproduct. The resulting brown sludge was centrifuged in DI water (3 × 10000 rpm for 10 minutes) to yield clean ζ-V2O5.
Physical characterization of powders and electrodes
XRD patterns of the as-synthesized VO2:Ag mixture, Ag0.33V2O5, and Nano ζ-V2O5 were collected using a Stoe StadiP diffractometer in transmission mode (coupled θ–2θ geometry), using Mo-Kα radiation, with the sample sandwiched between two plastic foil disks held together with a thin layer of silicon grease. A pre-sample Ge (111) monochromator selected the Mo Kα1 radiation only (λ = 0.709 Å) and included a 0.5 mm collimator restricted to 3 mm height. The sample was rotated in the beam, and the diffraction intensity recorded using a Dectris Mython 1k silicon strip detector covering 18° 2θ. Patterns were collected between 2–40° 2θ, with a step size of 0.5° 2θ and a collection time of 20 s per step.
High resolution synchrotron X-ray powder diffraction data were collected at 11-BM beamline at the Advanced Photon Source (APS) of Argonne National Laboratory (ANL) with the wavelengths of 0.412799 Å or 0.457854 Å. Samples were loaded in 1.1 mm Kapton capillaries. Structures were refined using the Rietveld method as implemented in the TOPAS software package (Bruker-AXS, version 6) across a d-spacing range of 5.0 to 0.5 Å.27
V L2,3-edge and O K-edge XAS was performed at beamline 4-ID-C, Advanced Photon Source, Argonne National Laboratory, USA. At 4-ID-C, spectra were collected simultaneously in both Total-Electron-Yield (TEY) and Total-Fluorescence Yield (TFY) mode utilizing photocurrent for the TEY and a silicon drift diode detector for the TFY, in order to make direct surface to bulk comparisons. Data were obtained at a spectral resolution of ∼0.2 eV, with a 2 s dwell time. Three scans were performed on each sample, at each absorption edge, and scans were averaged in order to maximize the signal to noise ratio. The V L2,3- and O K-edges were scanned in the range 500 to 560 eV. The V and O energy scales were normalized using a SrTiO3 standard.
Scanning Transmission Electron Microscopy (STEM) imaging, Energy Dispersive X-ray (EDX) and Electron Energy Loss (EEL) spectroscopy were performed on an aberration-corrected JEOL JEM-ARM200CF, equipped with a cold field emission gun operated at 200 kV, which allows a 73 pm spatial resolution and a 0.35 eV energy resolution. The microscope is equipped with High Angle Annular Dark Field and Low Angle Annular Dark Field (HAADF and LAADF) detectors, Bright Field (BF) detector, post-column Gatan Continuum spectrometer and an Oxford XMAX100TLE Silicon Drift Detector (SDD). Imaging of the V2O5 samples was done along specific zone axes, which show unmixed atomic columns. STEM images were acquired simultaneously in HAADF, LAADF and ABF modes to identify both heavy elements, as well as light elements. The collection angles for HAADF, LAADF and ABF detectors were set at 90–370, 40–160 and 14–28 mrad, respectively. The EELS spectra were collected using a Gatan Quantum imaging filter with a convergence angle of 30 mrad and a collection angle of 35 mrad. The TEM samples were prepared in a glovebox under an argon environment to prevent any changes to the sample structure as the result of exposure to oxygen.
Scanning Electron Microscopy (SEM) images were collected using a JEOL JSM-7500 FE instrument equipped with a high-brightness conical field-emission gun and a low aberration conical objective lens. Images were collected at an accelerating voltage of either 3 or 5 kV and a working distance of ∼5 and ∼15 mm, respectively. Prior to imaging, powdered ζ-V2O5 samples were fixed to aluminum sample plates using conductive carbon tape and were subsequently imaged without further manipulation.
Electrochemical characterization
Electrodes were prepared by hand-grinding the ζ-V2O5 powder samples, carbon black (Denka), and polyvinylidene difluoride (PVDF) (Kynar) in N-methylpyrrolidone (NMP) [Sigma–Aldrich, Dorset, UK] in a 60:20:20 wt% ratio, which were then cast on 1/2′′ stainless steel 316 mesh current collectors and allowed to dry overnight in air. The active material loading of 60 wt%, while lower than typical loadings for Li-ion electrode testing, is common for Mg-ion testing given the sluggish reaction kinetics of Mg insertion and removal.15,32,33 These were pressed (8 tons) and dried at 100 °C overnight under vacuum in the glovebox antechamber. Cells that were cycled repeatedly, possessed active material loadings in the range 1.5 to 2.5 mg cm−2, whereas charged or discharged electrodes used for post-mortem analysis were in the range 0.7 to 1.5 mg cm−2.
Li-ion coin cells were assembled using CR2032 components (Hohsen, Japan) with an Li foil anode (Alfa Aesar, Massachusetts, USA), a glass fiber separator (VWR, grade 691, 28297-289) and 1 M LiPF6 in a mixture of Ethylene Carbonate (EC) and Diethylcarbonate (DEC) [1:1, v/v, Novolyte Technologies, China] as the electrolyte. The active material loading was 2.5 mg cm−2. Cells were cycled at a charge/discharge rate of C/10, where 1C rate = 300 mA g−1 (at room temperature).
The Mg-ion coin cells contained an Activated Carbon Cloth (ACC) [model ACC-5092-20, American Technical Trading Inc., Pleasantville, New York, USA] as a counter electrode, a glass fiber separator (grade 691, 28297-289, VWR International, USA) and 0.5 M Mg[N(SO2)2((CF3)2)2–(C9H20N)(N(SO2)2(CF3)2] ionic liquid electrolyte (abbreviated as MgTFSI2–PY14TFSI) with low H2O content (∼43 ppm). The electrolyte was made by dissolving MgTFSI2 (M1208c, Solvionic, France) in PY14TFSI (Pyr0408a, Solvionic, France). The potential of cathode was calibrated by considering the ACC anode potential, which was originally 2.2 V vs. Mg/Mg2+ and linearly proportional to the state-of-charge (SoC).15 The polarization of the ACC electrode was quantified by measuring the voltage change of a symmetric ACC|ACC cell with State of Charge (SoC), using the same electrolyte and cycling temperature used for the ACC|ζ-V2O5 cells within this manuscript. This allowed the unambiguous determination of the cathodic voltage as a function of the SoC.
Electrochemistry was carried out at 110 °C in the potential ranges 1.2 to −1.7 V vs. ACC as indicated in the text. The charge/discharge rate (15 mA g−1) was galvanostatically controlled by a Bio-Logic VMP3 potentiostat. After magnesiation and demagnesiation of the ζ-V2O5 samples, the electrodes were recovered, rinsed in acetonitrile five times, and dried at room temperature under vacuum for 1 minute before characterization. To generate the di/dV profiles, the voltage profiles were smoothed using a Loess function (with point windows in the range of 100–350) before differentiation.
Results and discussion
The nanometric ζ-V2O5 sample, henceforth referred to as “Nano ζ-V2O5”, was synthesized via a multi-step process involving CHFS, as detailed in the Experimental section and the ESI (Fig. S1 and S2†).25,26 An acid leaching step was incorporated to remove Ag+ from the Ag0.33V2O5 intermediate. For purposes of comparison, the larger micrometric ζ-V2O5 (henceforth referred to as “Bulk ζ-V2O5”) was synthesized using a batch hydrothermal procedure.14,34 Scanning Transmission Electron Microscope (STEM) and Scanning Electron Microscope (SEM) analysis revealed the Nano ζ-V2O5 formed ∼100 nm crystallites (Fig. 1a and Fig. S3a†), with a small population of faceted rods up to 500 nm in length. In contrast, the Bulk ζ-V2O5 sample took the form of micrometer-long wires (ca. 150 nm in diameter, Fig. S3b†).14 High-Angle Annular Dark Field (HAADF) imaging revealed the atomic arrangement of V within Nano ζ-V2O5 along the [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)
![[5 with combining macron]](https://www.rsc.org/images/entities/char_0035_0304.gif) 0] zone axis (Fig. 1b), where the tunnel structure of ζ-V2O5 was clearly visible. Electron Energy-Loss Spectroscopy (EELS, Fig. 1c) of the Nano ζ-V2O5 revealed a similar fine-structure of the V L-edges to that reported previously for ζ-V2O5, and EDS of these particles (Fig. S7†) revealed significant intrusion of Na from the Na2S2O3 solution (used to remove the AgCl byproduct generated in the acid washing step). It should be noted that a small amount of Ag is present in the Bulk ζ-V2O5 structure following washing, with nominal stoichiometry Ag0.06V2O5.34 As the same washing protocol was used for both the Nano and Bulk ζ-V2O5, it is suggested that the smaller particle sizes enabled the enhanced substitution of Ag with Na due to the increased accessibility to the ζ-V2O5 tunnels caused by the larger surface area.
0] zone axis (Fig. 1b), where the tunnel structure of ζ-V2O5 was clearly visible. Electron Energy-Loss Spectroscopy (EELS, Fig. 1c) of the Nano ζ-V2O5 revealed a similar fine-structure of the V L-edges to that reported previously for ζ-V2O5, and EDS of these particles (Fig. S7†) revealed significant intrusion of Na from the Na2S2O3 solution (used to remove the AgCl byproduct generated in the acid washing step). It should be noted that a small amount of Ag is present in the Bulk ζ-V2O5 structure following washing, with nominal stoichiometry Ag0.06V2O5.34 As the same washing protocol was used for both the Nano and Bulk ζ-V2O5, it is suggested that the smaller particle sizes enabled the enhanced substitution of Ag with Na due to the increased accessibility to the ζ-V2O5 tunnels caused by the larger surface area.
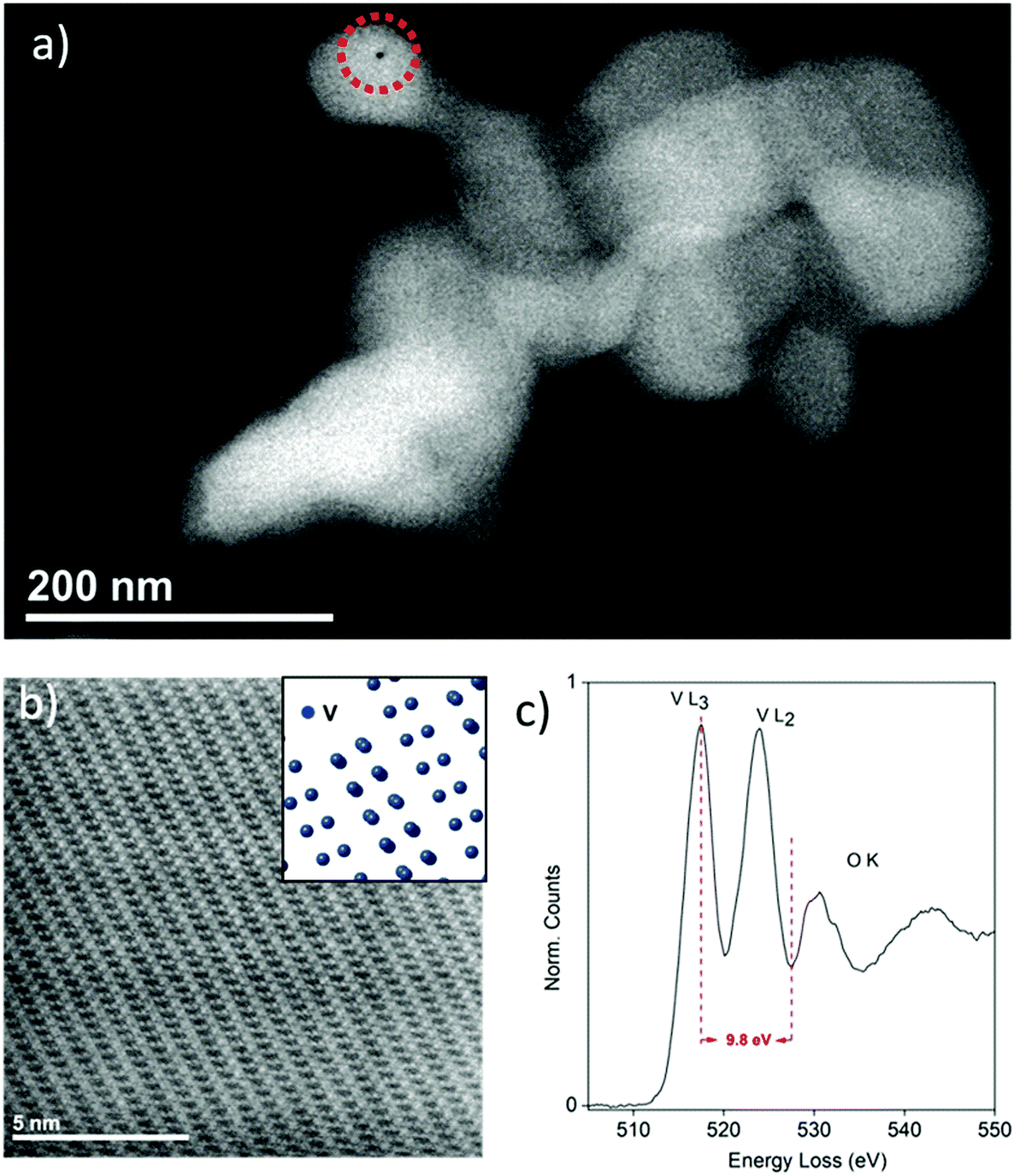 | ||
| Fig. 1 STEM imaging and spectroscopy analysis of Nano ζ-V2O5, showing (a) a representative particle cluster composed of crystallites of ∼100 nm width, where the location of the hole drilled during EDX analysis is indicated with a red dotted circle, (b) an atomic-resolution HAADF image of the particle along the [ 0] zone axis, where the tunnel structure is clearly visible, with the arrangement of V atoms of the HAADF image provided as an inset. (c) EEL spectrum of the V L- and O K-edges of the Nano ζ-V2O5 sample with the energy difference indicated by the dotted lines. | ||
Electrochemical cycling, conducted at 110 °C using Mg(TFSI)2–PY14TFSI electrolyte (details in the Experimental section), revealed key similarities and differences between the cycling properties of Bulk and Nano ζ-V2O5. In Bulk ζ-V2O5, the electrochemistry resembled that previously observed by Andrews et al. (Fig. 2a); a specific capacity of 114 mA h g−1 was observed for the first discharge, followed by rapid discharge capacity decay to below 80 mA h g−1 at cycle 10.14 Nano ζ-V2O5 had a similar discharge capacity at comparable electrochemical potentials to Bulk ζ-V2O5 on the first cycle (103 mA h g−1, Fig. 2b). This implied that the reaction responsible for charge storage was Mg intercalation, as conclusively demonstrated previously for Bulk ζ-V2O5.14 Following discharge, a larger charge capacity (146 mA h g−1) was observed for Nano ζ-V2O5 (completing the first discharge/charge cycle). This suggested that some of the trapped Na-ions in the one-dimensional tunnels of Nano ζ-V2O5 were extracted during charging on the first cycle. Nano ζ-V2O5 sustained a high average capacity of 130 mA h g−1 over 15 subsequent cycles at C/20, with no obvious capacity fade observed over the 15 cycles, whereas Bulk ζ-V2O5 rapidly degraded to below <70 mA h g−1 in the same cycle range (Fig. 2c). This observation revealed that reducing the ζ-V2O5 particle size had a significant beneficial impact on the reversibility and kinetics of the electrochemical reactions, with more surface access points to the tunnel structure per unit volume. It should be noted that the capacities for cycles 6 and 15 were anomalously low; as the voltage hysteresis is extremely sensitive to cell temperature and internal pressure, the authors believe that that a minor fluctuation in room temperature and/or cell pressure resulted in the premature termination of the discharge step by reaching the lower cut-off voltage. Given the slow cycling rates (C/20), examining long-term cycling (>50 cycles) was impractical. However, the authors suggest that further studies of these materials should examine the long-term cycle stability of nano ζ-V2O5.
 | ||
| Fig. 2 Electrochemical measurements performed at an elevated temperature of 110 °C: (a) the first, second and tenth cycles of Bulk ζ-V2O5 cycled in the range 1.2 to −1.7 V (vs. ACC) in an Mg-ion cell, (b) the first, second and tenth cycles of Bulk ζ-V2O5 cycled in the range 1.2 to −1.7 V (vs. ACC) in an Mg-ion cell, (c) comparison of the charge and discharge capacities of Bulk and Nano ζ-V2O5 in Mg-ion cells as a function of cycle number, (d) the first two cycles of Nano ζ-V2O5 cycled in the range 4.0 to 2.2 V (vs. Li/Li+) at a C/10 charge/discharge rate in a Li-ion cell at room temperature. | ||
The overpotentials of discharge and charge were significantly reduced in Nano ζ-V2O5; a hysteresis of 1.15, 0.94 and 0.98 V were observed on the first, second and tenth cycles, respectively, which was in contrast with 1.65, 1.29 and 1.43 V, respectively, for similar cycles for Bulk ζ-V2O5. The voltage hysteresis was determined by calculating the difference in average charge or discharge voltage as a function of capacity according to eqn (1), where ΔV is the voltage hysteresis, CChar is the charge capacity, and CDis is the discharge capacity.
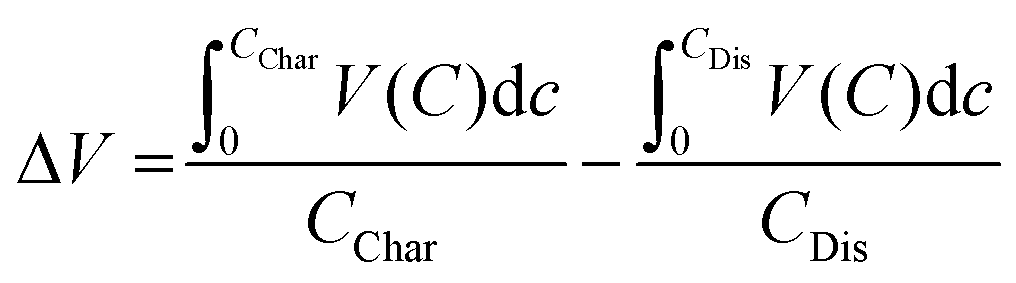 | (1) |
The improved cycling kinetics were further evidenced by significant differences between the dQ/dV behavior of the materials (Fig. S4†): bulk ζ-V2O5 showed the majority of charge/discharge behavior occurring at the extremes of the cycling window, i.e. <1.25 V and >2.75 V vs. Mg/Mg2+ (Fig. S4a†), whereas Nano ζ-V2O5 possessed significant redox activity in the range 2.0 to 2.7 V vs. Mg/Mg2+ (Fig. S4b†). However, a slight voltage fade was noticeable in Nano ζ-V2O5 between consecutive cycles, resulting in an overall voltage drop of ∼0.25 V over 10 cycles. Despite the voltage fade, the Nano ζ-V2O5 electrode clearly displayed favourable energetics of Mg insertion and removal compared to Bulk ζ-V2O5. As a result, Nano ζ-V2O5 possessed a higher energy density than Bulk ζ-V2O5 (250 and 140 W h kg−1, respectively, on the second cycle), which compares favorably with state-of-the-art sulfide materials such as Chevrel Mo6S8 (77 W h kg−1),10 Spinel Ti2S4 (228 W h kg−1),11 and oxides such as Mo2.48VO9.93 (∼250 W h kg−1),34 and MoO3 (270 W h kg−1),35 although it is still somewhat short of the highest energy density recorded for an oxide (α-V2O5, 660 W h kg−1).15 Clearly, nanosizing ζ-V2O5 increased its obtainable energy density by mitigating diffusive limitations on cycling kinetics. Unfortunately, the direct probing of Mg diffusion using standard techniques such as the Galvanostatic Intermittent Titration Technique (GITT) and Electrochemical Impedance Spectroscopy (EIS) are not yet suitable for these prototype systems, given the prevalent side reactions present between the electrodes and the electrolyte. However, it is plausible that ion conduction could be probed by performing solid-state impedance on a chemically magnesiated bulk ζ-V2O5 sample. It is suggested that this would be a fruitful avenue for future research to understand transport properties in these materials.
As EDS analysis (discussed later) was unable to confirm the extraction and possible re-insertion of Na from the Nano ζ-V2O5 electrode, electrochemical cycling of the Nano ζ-V2O5 in an Li-ion cell (Fig. 2d) was performed at room temperature. As electrolyte degradation and side-reactions are expected to be minimal in the potential range used in the Li-ion cycling tests, this meant that the measured capacity could be directly related to electrochemical activity and extraction of Na. The coulometric measurements revealed the existence of a discrepancy between discharge and charge capacities in the Li-ion cell on the first cycle (60 mA h g−1), similar to that observed in the first cycle of the Mg-ion cell, indicating some Na was removed after the first charge (Fig. 2d). Moreover on the subsequent cycle, the discharge capacity increased by 26 mA h g−1 in the Li-ion cell, similar to the increased capacity observed in Mg-ion testing (Fig. 2b), further implying that Na removal enabled greater Li (and Mg) insertion. This observation suggested that Na occupation in the tunnel structure could hinder or block intercalation of other ions (such as Li+ or Mg2+), and this effect was reduced by removing Na on charge. The re-insertion of Na into Nano ζ-V2O5 was unlikely given the relative dilution of Na within the electrolyte. Assuming all of the Na was removed on charge, and given the active electrode mass was 2.5 mg, the maximum amount of Na extracted into the Li or Mg electrolytes was ca. 1 × 10−5 mol. As the cells were flooded with electrolyte (300 μL), the overall concentration of Na in the electrolytes following charge, would be a maximum of 0.01 M. This was significantly lower than the concentrations of Li and Mg in their respective electrolytes (in the range 0.5 to 1 M), and therefore, Na most likely remained dissolved in the electrolyte on subsequent discharges, rather than re-inserting into the structure.
Rietveld refinement of high-resolution synchrotron XRD patterns revealed systematic changes in the lattice parameters between the pristine, discharged and charged electrodes of Nano ζ-V2O5 (Fig. 3a, Table 1 and Fig. S6†). ζ-V2O5, with monoclinic symmetry C2/m, forms chains of VOx distorted octahedra and square pyramids along the b-axis, which enclose tunnel sites within which the intercalated ions reside. The Na content and atomic position within the pristine sample could be extracted from Rietveld refinement, yielding a stoichiometry of Na0.283(1)V2O5 (Na0.3(2)V2O5, Table S1†). The larger unit cell volume of Nano ζ-V2O5 compared to literature reports arose mainly from a comparatively increased a parameter (Table 1), and it is suggested that the presence of ca. 0.3 equivalents of Na as opposed to 0.06 equivalents of Ag, may have accounted for the larger unit cell volume of the pristine sample in this study compared to those found in the literature.36 The increased a parameter also resulted in wider diffusion tunnels in Nano ζ-V2O5 compared to literature reports, which may have provided another benefit to Mg diffusion and therefore partially explain the improved electrochemical performance of Nano ζ-V2O5.36 Unfortunately, Mg and Na contents could not be extracted for the discharged and charged electrodes reliably due to peak broadening effects and the inherently low signal-to-noise ratio in XRD experiments of thin electrode samples, although insights into unit cell parameters could still be extracted. The lattice parameter a contracted upon reduction, and parameters b and c expanded, with significant peak broadening and peak splitting evident in certain diffraction peaks, e.g. the (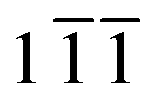 ) peak (Fig. 3b). These peaks were well indexed with two C2/m phases with two sets of increased lattice parameters compared to that of pristine sample, as shown in Fig. S6† and Table 1. These can be explained by two discharged states; a Mg-rich C2/m phase (∼60 vol%) and a Mg-poor C2/m phase (∼40 vol%), which may result from heterogeneous electrode reactions or increased relative reactivity of the ζ-V2O5 surface. These changes were effectively reversed upon charge, with only a 0.15% divergence in unit cell volume between the pristine state and the charged electrode, which suggested that the ion insertion and associated changes to the crystal structure, were largely reversible with discharge and charge, contributing to the stable electrochemical cycling capacity observed in Nano ζ-V2O5.
) peak (Fig. 3b). These peaks were well indexed with two C2/m phases with two sets of increased lattice parameters compared to that of pristine sample, as shown in Fig. S6† and Table 1. These can be explained by two discharged states; a Mg-rich C2/m phase (∼60 vol%) and a Mg-poor C2/m phase (∼40 vol%), which may result from heterogeneous electrode reactions or increased relative reactivity of the ζ-V2O5 surface. These changes were effectively reversed upon charge, with only a 0.15% divergence in unit cell volume between the pristine state and the charged electrode, which suggested that the ion insertion and associated changes to the crystal structure, were largely reversible with discharge and charge, contributing to the stable electrochemical cycling capacity observed in Nano ζ-V2O5.
 | ||
| Fig. 3 Synchrotron powder X-ray diffraction patterns of the pristine, discharged and charged electrodes. (a) The diffraction patterns shown over a wide Q range, where the Al peak from the electrode substrate is indicated. (b) The same data shown over a narrow Q range to highlight the shift and splitting of the (1 ) peak. | ||
| Material | a/Å | b/Å | c/Å | V/Å3 | β/° | R wp |
|---|---|---|---|---|---|---|
| Ref. 32 | 15.2750(2) | 3.60386(2) | 10.09771(7) | 522.271(6) | 110.0222(6) | 9.87 |
| Pristine | 15.40841(4) | 3.610815(6) | 10.078048(20) | 528.342(2) | 109.56366(17) | 7.14 |
| Discharged (Mg-poor) | 15.303(2) | 3.6566(3) | 10.155(1) | 535.6(1) | 109.52(1) | 11.60 |
| Discharged (Mg-rich) | 15.270(2) | 3.686(2) | 10.1924(8) | 541.4(1) | 109.322(8) | 11.60 |
| Charged | 15.4174(2) | 3.61270(2) | 10.0803(1) | 529.04(1) | 109.563(1) | 11.42 |
X-ray Absorption Spectroscopy data were collected using both total electron and fluorescence yield detectors on the Bulk ζ-V2O5 sample, and the pristine, discharged, and charged electrodes of Nano ζ-V2O5. Signals from electron detection mode (Total Electron Yield, TEY, Fig. 4a) correspond to the chemical state of the surface layer of the electrode, whereas fluorescence yields (Total Fluorescence Yield, TFY, Fig. 4b) correspond to approximately 100 nm into the sample, thus having a notable contribution from the bulk crystal structure, especially considering the particle size of the materials. According to the spectra shown in Fig. 4a (TEY detection mode), the shape and energy positions of V L2 (525 eV) and L3 (519 eV) of pristine Nano ζ-V2O5 were in good agreement with the standard material shown, and the previously reported Bulk ζ-V2O5.15,20,36–40 Upon discharge to −1.7 V (vs. ACC) at 110 °C, the V L2 and L3 spectral events red-shifted. For instance, the centers of gravity of the V L3 moved to 518 eV in both TEY and TFY detection modes. These changes are consistent with formation of V4+ in Mg-intercalated ζ-V2O5 upon discharge.15 Upon charge to 1.2 V (vs. ACC) the centers of gravity of the V L2 and L3 features blue-shifted in both TEY and TFY detection modes, indicating that V4+ re-oxidized, and that the redox activity of V was reversible throughout the particle.
 | ||
| Fig. 4 Integrated X-ray Absorption Spectra of the V L2,3 and O K edges of the pristine Bulk and Nano ζ-V2O5 electrodes, the discharged Nano ζ-V2O5 electrode, and the charged Nano ζ-V2O5, showing (a) electron yields, corresponding to surface chemistry and (b) fluorescence yields, corresponding to bulk properties. | ||
O K-edge features of pristine ζ-V2O5 were also evaluated in conjunction with V L-edges (shown in Fig. 4) owing to the close overlap of the two edges. The O K-edge spectra reflect transitions of O 1s core electrons to 2p states. In the “pre-edge” region (530–535 eV) O(2p) states are hybridized with V(3d) states and reflect their crystal field splitting. This region showed two absorption events that are attributed to t2g (530 eV) and eg (532 eV) V(3d)–O(2p) hybrid states corresponding to π and end-on σ interactions of [VO6] octahedra. In TEY detection mode (Fig. 4a), the pristine Nano ζ-V2O5t2g and eg peaks were similar to the pristine Bulk ζ-V2O5, indicating V5+ existed at the surface. In TFY detection mode (Fig. 4b), the pristine Nano ζ-V2O5t2g and eg peaks were of nearly similar intensity to one another, which suggested a partially reduced V core. This was consistent with EDS measurements that detected residual sodium after washing to remove Ag. Upon discharging to −1.7 V vs. ACC, the t2g peaks lost intensity in both the TEY and TFY spectra relative to eg, consistent with increasing electron density within the system. These changes were also observed in the electrochemical intercalation of Mg2+ into α-V2O5 by Yoo et al., providing strong evidence that, upon discharge, Mg2+ intercalated into Nano ζ-V2O5 and V reduced throughout the particle.15 Additionally, the onset of the main edge region (above 535 eV) red-shifted by 2 eV after discharge in both the TEY and TFY spectra, consistent with increasing electron density within the system, and a prominent feature formed at 535.3 eV. Upon recharging the cathode, the peak intensity ratios and energy positioning of t2g and eg features were recovered close to the pristine state. Each of these observations was consistent with oxidation of the V–O framework and provide unambiguous evidence for redox intercalation chemistry with cycling.41
EDS analysis was performed on the pristine, discharged and charged Nano ζ-V2O5 electrodes to investigate changes in stoichiometry with electrochemical cycling. The materials were found to be highly beam-sensitive, as evidenced by the circular beam damage evident after EDS spot analysis in Fig. 1a. To mitigate beam-damage, the imaging, EELS and EDS analysis reported herein were performed using electron dose rates that did not alter the particle structures. Quantification of the elemental ratios, averaged over a minimum of five particles for each electrode, revealed significant variances in Mg and Na content from particle-to-particle, manifesting as large error bars (Table S2†). While some variability in Mg and Na content was expected within the electrodes, given the differing structures and redox states observed in the XRD and XAS analysis, it was difficult to ascertain whether the observed elemental ratios and their variation adequately described the element quantity and distribution in the sample (Table S2†), due to the relative error in detection of light elements, such as Na and Mg, by EDS and the close proximity of their Kα-lines (Na Kα = 1.040 kV; Mg Kα = 1.250 kV). Overall, it was observed that the Mg content of Nano ζ-V2O5 increased on discharge, and decreased on charge, as expected from the electrochemistry. Furthermore, EDS mapping revealed a uniform dispersion of Mg within individual particles upon discharge, consistent with Mg intercalation (Fig. S7–S9†). EELS analysis of a particle region with stoichiometry Mg0.29Na0.26V2O4.32, similar to that predicted from the electrochemistry of the electrode, revealed changes in the V L-edge spectra which were consistent with the observations within the XAS study (Fig. S10†). Therefore, it is surmised that significant Mg was intercalated within specific ζ-V2O5 particles in the electrode, and that further studies are required to better understand distribution of Mg intercalation (and therefore reactivity) of individual particles within cycled Mg-ion electrodes.
Conclusions
In summary, ζ-V2O5 was synthesized by two separate methods; a standard batch hydrothermal method, and a method incorporating a Continuous Hydrothermal Flow Synthesis (CHFS) process. The former method produced micrometer-long wires ca. 150 nm in diameter, whereas the CHFS-based method enabled access to much smaller semi-spherical crystallite sizes of ca. 100 nm. The Nano-sized ζ-V2O5 showed improved electrochemical activity in Mg electrolytes, as evidenced by lower hysteresis, greater capacity, and improved capacity retention observed compared to the bulk ζ-V2O5. Synchrotron XRD and XAS verified reversible structural and redox changes in the nano-sized ζ-V2O5 electrode during cycling, with EDS confirming significant Mg intercalation within specific electrode particles.This study revealed that diffusive processes impose a limit on the degree and reversibility of the observed Mg electrochemistry in ζ-V2O5, and that nanosizing can significantly improve electrode kinetics, revealing a key contribution to the voltage hysteresis that is pervasive to oxides used as Mg intercalation electrodes.14,19,42,43 However, questions remain regarding the uniformity of magnesiation of cathode particles throughout the electrode. Therefore, it is suggested that future efforts should not only target ultrafine particles of candidate oxide electrode materials, but also probe their activity as a function of electrode depth, to elucidate kinetic limitations imposed by electrolyte permeation in the electrode and bulk electrode resistance. Such studies would further enrich our understanding of the intercalation electrochemistry of Mg2+ in oxide electrodes that generate true prospects for batteries with high energy density.
Author contributions
IDJ wrote the manuscript. IDJ, JLA and SB synthesized the samples reported in the manuscript. GN performed the XAS analysis. LY and SHL performed the X-Ray and Rietveld analysis. IDJ, HDY, and ML performed the Li-ion and Mg-ion electrochemistry. PP, AM and RFK performed the STEM-HAADF and EELS analysis. BI, JC and JAD edited the manuscript. JC leads the Mg battery research team at the University of Illinois at Chicago. JAD is academic lead of the synthesis team at University College London, co-developed the current CHFS process, and is a co-inventor of the CJM mixer that was used in this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
IDJ and JAD would like to thank the EPSRC for funding the JUICED project (EP/R023662/1). IDJ, GN, LY, HDY, PP, AM, RK, BI, SL and JC were supported as part of the Joint Center for Energy Storage Research (JCESR, a U.S. Department of Energy, Energy Innovation Hub). IDJ would also like to thank the Materials Modelling and Molecular Doctoral Training Centre (EP/G036675/1) and the STFC for providing funding support for travel within the collaboration (STFC/MDC Futures Early Career Award, ST/N002385/1). JLA and SB acknowledge support from the National Science Foundation under NSF 1809866. HDY acknowledges support from the National Research Foundation (NRF-2018R1C1B6004808 and NRF-2018R1A5A1025594) of the Korean Ministry of Science and ICT. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, under Contract No. DE-AC02-06CH11357. IDJ would like to thank Dr Tim T. Fister for support with SEM imaging.References
- M. Armand and J.-M. Tarascon, Building Better Batteries, Nature, 2008, 451, 652–657 CrossRef CAS.
- M. S. Whittingham, Ultimate Limits to Intercalation Reactions for Lithium Batteries, Chem. Rev., 2014, 114, 11414–11443 CrossRef CAS.
- X.-P. Gao and H.-X. Yang, Multi-Electron Reaction Materials for High Energy Density Batteries, Energy Environ. Sci., 2010, 3, 165–240 RSC.
- P. Canepa, G. Sai Gautam, D. C. Hannah, R. Malik, M. Liu, K. G. Gallagher, K. A. Persson and G. Ceder, Odyssey of Multivalent Cathode Materials: Open Questions and Future Challenges, Chem. Rev., 2017, 117, 4287–4341 CrossRef CAS.
- C. Ling, D. Banerjee and M. Matsui, Study of the Electrochemical Deposition of Mg in the Atomic Level: Why It Prefers the Non-Dendritic Morphology, Electrochim. Acta, 2012, 76, 270–274 CrossRef CAS.
- M. Matsui, Study on Electrochemically Deposited Mg Metal, J. Power Sources, 2011, 196, 7048–7055 CrossRef CAS.
- Z. Takehara, Future Prospects of the Lithium Metal Anode, J. Power Sources, 1997, 68, 82–86 CrossRef CAS.
- R. Davidson, A. Verma, D. Santos, F. Hao, C. Fincher, S. Xiang, J. V. Buskirk, K. Xie, M. Pharr, P. P. Mukherjee and S. Banerjee, Formation of Magnesium Dendrites during Electrodeposition, ACS Energy Lett., 2019, 4, 375–376 CrossRef CAS.
- H. D. Yoo, S.-D. Han, I. L. Bolotin, G. M. Nolis, R. D. Bayliss, A. K. Burrell, J. T. Vaughey and J. Cabana, Degradation Mechanisms of Magnesium Metal Anodes in Electrolytes Based on (CF3SO2)2N – at High Current Densities, Langmuir, 2017, 33, 9398–9406 CrossRef CAS.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Prototype Systems for Rechargeable Magnesium Batteries, Nature, 2000, 407, 724–727 CrossRef CAS.
- X. Sun, P. Bonnick, V. Duffort, M. Liu, Z. Rong, K. A. Persson, G. Ceder and L. F. Nazar, A High Capacity Thiospinel Cathode for Mg Batteries, Energy Environ. Sci., 2016, 9, 2273–2277 RSC.
- D. Aurbach, I. Weissman, Y. Gofer and E. Levi, Nonaqueous Magnesium Electrochemistry and Its Application in Secondary Batteries, Chem. Rec., 2003, 3, 61–73 CrossRef CAS.
- E. Levi, Y. Gofer and D. Aurbach, On the Way to Rechargeable Mg Batteries: The Challenge of New Cathode Materials, Chem. Mater., 2010, 22, 860–868 CrossRef CAS.
- J. L. Andrews, A. Mukherjee, H. D. Yoo, A. Parija, P. M. Marley, S. Fakra, D. Prendergast, J. Cabana, R. F. Klie and S. Banerjee, Reversible Mg-Ion Insertion in a Metastable One-Dimensional Polymorph of V2O5, Chem, 2018, 4, 564–585 CAS.
- H. D. Yoo, J. R. Jokisaari, Y. S. Yu, B. J. Kwon, L. Hu, S. Kim, S. D. Han, M. Lopez, S. H. Lapidus, G. M. Nolis, B. J. Ingram, I. Bolotin, S. Ahmed, R. F. Klie, J. T. Vaughey, T. T. Fister and J. Cabana, Intercalation of Magnesium into a Layered Vanadium Oxide with High Capacity, ACS Energy Lett., 2019, 4, 1528–1534 CrossRef CAS.
- G. M. Nolis, A. Adil, H. D. Yoo, L. Hu, R. D. Bayliss, S. H. Lapidus, L. Berkland, P. J. Phillips, J. W. Freeland, C. Kim, R. F. Klie and J. Cabana, Electrochemical Reduction of a Spinel-Type Manganese Oxide Cathode in Aqueous Electrolytes with Ca2+ or Zn2+, J. Phys. Chem. C, 2018, 122, 4184–4188 CrossRef.
- L. Hu, I. D. Johnson, S. Kim, G. M. Nolis, J. Freeland, H. D. Yoo, T. T. Fister, L. McCafferty, T. E. Ashton, J. A. Darr and J. Cabana, Tailoring the Electrochemical Activity of Magnesium Chromium Oxide Towards Mg Batteries Through Control of Size and Crystal Structure, Nanoscale, 2019, 11, 639–646 RSC.
- M. Liu, Z. Rong, R. Malik, P. Canepa, A. Jain, G. Ceder, K. A. Persson and M. Liu, Spinel Compounds as Multivalent Battery Cathodes: A Systematic Evaluation Based on Ab Initio Calculations, Energy Environ. Sci., 2015, 8, 964–974 RSC.
- B. J. Kwon, K. C. Lau, H. Park, Y. A. Wu, K. L. Hawthorne, H. Li, S. Kim, I. L. Bolotin, T. T. Fister, P. Zapol, R. F. Klie, J. Cabana, C. Liao, S. H. Lapidus, B. Key and J. T. Vaughey, Probing Electrochemical Mg-Ion Activity in MgCr2−xVxO4 Spinel Oxides, Chem. Mater., 2020, 32, 1162–1171 CrossRef CAS.
- X. Sun, L. Blanc, G. M. Nolis, P. Bonnick, J. Cabana and L. F. Nazar, NaV1.25Ti0.75O4: A Potential Post-Spinel Cathode Material for Mg Batteries, Chem. Mater., 2018, 30, 121–128 CrossRef CAS.
- S. H. Bo, C. P. Grey and P. G. Khalifah, Defect-Tolerant Diffusion Channels for Mg2+ Ions in Ribbon-Type Borates: Structural Insights into Potential Battery Cathodes MgVBO4 and Mgx,Fe2−xB2O5, Chem. Mater., 2015, 27, 4630–4639 CrossRef CAS.
- K. Makino, Y. Katayama, T. Miura and T. Kishi, Electrochemical Insertion of Magnesium to Mg0.5Ti2(PO4)3, J. Power Sources, 2001, 99, 66–69 CrossRef CAS.
- R. Y. Wang, C. D. Wessells, R. A. Huggins and Y. Cui, Highly Reversible Open Framework Nanoscale Electrodes for Divalent Ion Batteries, Nano Lett., 2013, 13, 5748–5752 CrossRef CAS.
- M. Wagemaker and F. M. Mulder, Properties and Promises of Nanosized Insertion Materials for Li-Ion Batteries, Acc. Chem. Res., 2013, 46, 1206–1215 CrossRef CAS.
- R. I. Gruar, C. J. Tighe and J. A. Darr, Scaling-up a Confined Jet Reactor for the Continuous Hydrothermal Manufacture of Nanomaterials, Ind. Eng. Chem. Res., 2013, 52, 5270–5281 CrossRef CAS.
- J. A. Darr, J. Zhang, N. M. Makwana and X. Weng, Continuous Hydrothermal Synthesis of Inorganic Nanoparticles; Applications and Future Directions, Chem. Rev., 2017, 117, 11125–11238 CrossRef CAS.
- I. D. Johnson, E. Blagovidova, P. A. Dingwall, D. J. L. Brett, P. R. Shearing and J. A. Darr, High Power Nb-Doped LiFePO4 Li-Ion Battery Cathodes; Pilot-Scale Synthesis and Electrochemical Properties, J. Power Sources, 2016, 326, 476–481 CrossRef CAS.
- I. D. Johnson, M. Loveridge, R. Bhagat and J. A. Darr, Mapping Structure-Composition-Property Relationships in V and Fe Doped LiMnPO4 Cathodes for Lithium-Ion Batteries, ACS Comb. Sci., 2016, 18, 665–672 CrossRef CAS.
- I. D. Johnson, M. Lübke, O. Y. Wu, N. M. Makwana, G. J. Smales, H. U. Islam, R. Y. Dedigama, R. I. Gruar, C. J. Tighe, D. O. Scanlon, F. Corà, D. J. L. Brett, P. R. Shearing and J. A. Darr, Pilot-Scale Continuous Synthesis of a Vanadium-Doped LiFePO4/C Nanocomposite High-Rate Cathodes for Lithium-Ion Batteries, J. Power Sources, 2016, 302, 410–418 CrossRef CAS.
- D. Bauer, T. E. Ashton, D. J. L. Brett, P. R. Shearing, N. Matsumi and J. A. Darr, Mixed Molybdenum and Vanadium Oxide Nanoparticles with Excellent High-Power Performance as Li-Ion Battery Negative Electrodes, Electrochim. Acta, 2019, 322, 134695 CrossRef CAS.
- I. D. Johnson, G. Nolis, K. McColl, Y. A. Wu, D. Thornton, L. Hu, H. D. Yoo, J. W. Freeland, F. Corà, J. K. Cockcroft, I. P. Parkin, R. F. Klie, J. Cabana and J. A. Darr, Probing Mg Intercalation in Tetragonal Tungsten Bronze Framework V4Nb18O55, Inorg. Chem., 2019, 59, 9783–9797 CrossRef.
- Y. Idemoto, N. Kawakami, N. Ishida and N. Kitamura, Synthesis, electrochemical properties, and changes in crystal and electronic structures during charge/discharge process of spinel-type cathode materials Mg4V5−xNixO12 (x = 0, 0.3, 0.6, 1.0) for magnesium secondary batteries, J. Power Sources, 2020, 455, 227962 CrossRef CAS.
- K. Shimokawa, T. Atsumi, M. Harada, R. E. Ward, M. Nakayama, Y. Kumagai, F. Oba, N. L. Okamoto, K. Kanamura and T. Ichitsubo, Zinc-based spinel cathode materials for magnesium rechargeable batteries: Toward the reversible spinel-rocksalt transition, J. Mater. Chem. A, 2019, 7, 12225–12235 RSC.
- W. Kaveevivitchai and A. J. Jacobson, High Capacity Rechargeable Magnesium-Ion Batteries Based on a Microporous Molybdenum-Vanadium Oxide Cathode, Chem. Mater., 2016, 28, 4593–4601 CrossRef CAS.
- M. E. Spahr, P. Novák, O. Haas and R. Nesper, J. Power Sources, 1995, 54, 346–351 CrossRef CAS.
- P. M. Marley, T. A. Abtew, K. E. Farley, G. A. Horrocks, R. V. Dennis, P. Zhang and S. Banerjee, Emptying and Filling a Tunnel Bronze, Chem. Sci., 2015, 6, 1712–1718 RSC.
- J. M. Velazquez, C. Jaye, D. A. Fischer and S. Banerjee, Near Edge X-Ray Absorption Fine Structure Spectroscopy Studies of Single-Crystalline V2O5 Nanowire Arrays, J. Phys. Chem. C, 2009, 113, 7639–7645 CrossRef CAS.
- L. R. De Jesus, G. A. Horrocks, Y. Liang, A. Parija, C. Jaye, L. Wangoh, J. Wang, D. A. Fischer, L. F. J. Piper, D. Prendergast and S. Banerjee, Mapping Polaronic States and Lithiation Gradients in Individual V2O5 Nanowires, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- M. Abbate, H. Pen, M. T. Czyzyk, F. M. F. de Groot, J. C. Fuggle, Y. J. Ma, C. T. Chen, F. Sette, A. Fujimori, Y. Ueda and K. Kosuge, Soft X-Ray Absorption Spectroscopy of Vanadium Oxides, J. Electron Spectrosc. Relat. Phenom., 1993, 62, 185–195 CrossRef CAS.
- L. R. De Jesus, J. L. Andrews, A. Parija and S. Banerjee, Defining Diffusion Pathways in Intercalation Cathode Materials: Some Lessons from V2O5 on Directing Cation Traffic, ACS Energy Lett., 2018, 3, 915–931 CrossRef CAS.
- T. Gallasch, T. Stockhoff, D. Baither and G. Schmitz, Ion Beam Sputter Deposition of V2O5 Thin Films, J. Power Sources, 2011, 196, 428–435 CrossRef CAS.
- Q. D. Truong, H. Kobayashi and I. Honma, Rapid Synthesis of MgCo2O4 and Mg2/3Ni4/3O2 Nanocrystals in Supercritical Fluid for Mg-Ion Batteries, RSC Adv., 2019, 9, 36717–36725 RSC.
- K. Shimokawa and T. Ichitsubo, Spinel–Rocksalt Transition as a Key Cathode Reaction toward High-Energy-Density Magnesium Rechargeable Batteries, in Current Opinion in Electrochemistry, Elsevier B.V., 2020, pp. 93–99 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr05060a |
| This journal is © The Royal Society of Chemistry 2020 |