
Supramolecular antibiotics: a strategy for conversion of broad-spectrum to narrow-spectrum antibiotics for Staphylococcus aureus†
 ‡b
S.
Thayumanavan
*abde
‡b
S.
Thayumanavan
*abde
Abstract
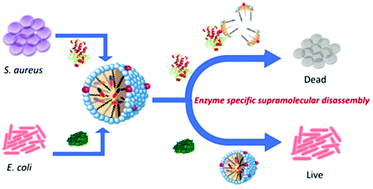
The propensity of broad-spectrum antibiotics to indiscriminately kill both pathogenic and beneficial bacteria has a profound impact on the spread of resistance across multiple bacterial species. Alternative approaches that narrow antibacterial specificity towards desired pathogenic bacterial population are of great interest. Here, we report an enzyme-responsive antibiotic-loaded nanoassembly strategy for narrow delivery of otherwise broad-spectrum antibiotics. We specifically target Staphylococcus aureus (S. aureus), an important blood pathogen that secretes PC1 β-lactamases. Our nanoassemblies selectively eradicate S. aureus grown in vitro with other bacteria, highlighting its potential capability in targeting the desired pathogenic bacterial population.
- This article is part of the themed collection: Editor’s Choice: Recent breakthroughs in nanobiotechnology
 Please wait while we load your content...
Please wait while we load your content...

