
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular gold strings: aurophilicity, luminescence and structure–property correlations
Tim P.
Seifert
,
Vanitha R.
Naina
,
Thomas J.
Feuerstein
 ,
Nicolai D.
Knöfel
and
Peter W.
Roesky
*
,
Nicolai D.
Knöfel
and
Peter W.
Roesky
*
Institute of Inorganic Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstr. 15, 76131 Karlsruhe, Germany. E-mail: roesky@kit.edu
First published on 1st October 2020
Abstract
This review covers the compound class of one-dimensional gold strings. These compounds feature a formally infinite repetition of gold complexes as monomers/repeating units that are held together by aurophilic interactions, i.e. direct gold–gold contacts. Their molecular structures are primarily determined in the solid state using single crystal X-ray diffraction. The chemical composition of the employed gold complexes is diverse and furthermore plays a key role in terms of structure characteristics and the resulting properties. One of the most common features of gold strings is their photoluminescence upon UV excitation. The emission energy is often dependent on the distance of adjacent gold ions and the electronic structure of the whole string. In terms of gold strings, these parameters can be fine-tuned by external stimuli such as solvent, pH value, pressure or mechanical stress. This leads to direct structure–property correlations, not only with regard to the photophysical properties, but also electric conductivity for potential application in nanoelectronics. Concerning these correlations, gold strings, consisting of self-assembled individual complexes as building blocks, are the ideal compound class to look at, as perturbations by an inhomogeneity in the ligand sphere (such as the end of a molecule) can be neglected. Therefore, the aim of this review is to shed light on the past achievements and current developments in this area.
 Tim P. Seifert | Tim P. Seifert received his Master's Degree in Chemistry from Karlsruhe Institute of Technology in 2015. He then joined the inorganic chemistry department in the group of Professor Peter Roesky, where he obtained his PhD degree in 2018. He is currently enrolled as a postdoctoral researcher in the same group. His research interests include metallophilic interactions in a luminescence and fundamental context. |
 Vanitha R. Naina | Vanitha Reddy Naina received her Master's Degree in 2019 from the Discipline of Chemistry, Indian Institute of Technology Indore, India, during which she was a recipient of the DAAD India IIT Master Sandwich Program in 2018. She is currently pursuing her doctoral research in chemistry at Karlsruhe Institute of Technology in the research group of Prof. Dr Peter W. Roesky. Her current research work is focused on luminescent metal complexes. |
 Thomas J. Feuerstein | Thomas J. Feuerstein obtained his M.Sc. degree in Chemistry at Karlsruhe Institute of Technology (KIT) in 2015. Afterwards he joined the workgroup of Professor Peter Roesky where he obtained his PhD degree in 2019. Currently he works as a postdoctoral researcher in the same group. His research interests are photoluminescent metal compounds, especially coinage metals, and their investigation under cryogenic conditions. |
 Nicolai D. Knöfel | Nicolai D. Knöfel obtained his Master's degree in Chemistry at Karlsruhe Institute of Technology (KIT) in 2015, followed by a PhD study in the workgroup of Professor Peter W. Roesky (Inorganic Chemistry department, KIT). He received his PhD degree in 2019 with distinction and proceeded as a postdoctoral researcher in the same working group for one year. His research fields comprise metallopolymers and multiply bonded transition metal complexes. Sometimes he also writes reviews. |
 Peter W. Roesky | Peter W. Roesky obtained his diploma in 1992 from the University of Würzburg and his doctoral degree from the Technical University of Munich (with Prof. W. A. Herrmann) in 1994. He was as a postdoc with Prof. T. J. Marks at Northwestern University (1995–1996). In 1999 he completed his Habilitation at the University of Karlsruhe. As a full professor he joined the faculty of the Freie Universität Berlin in 2001. Since 2008 he has held the chair for inorganic functional materials at the University of Karlsruhe Institute of Technology. In 1999 he received a Heisenberg scholarship of the German Science Foundation, and in 2000 a Karl–Winnacker scholarship. Since 2014 he is a fellow of the Royal Society of Chemistry and since 2020 a fellow of the European Academy of Science (EurAsc). In 2019 he received a JSPS Invitational Fellowship for Research in Japan and in 2020 a Reinhart Koselleck Project of the German Science Foundation. |
Introduction
A. General considerations for molecular one-dimensional gold strings and their properties
In the age of nanotechnology, interest in materials with tailor-made properties is steadily increasing. Since physical properties are often dependent on the molecular structure, there is a general demand for precise and well-investigated materials.1 One of the crucial factors is the molecular dimensionality of the material, which, in the case of properties and applications, is impressively demonstrated by the carbon modifications graphite, graphene, carbon nanotubes and fullerenes as 3D, 2D, 1D and 0D materials, respectively.2–7 The major driving forces for research on one-dimensional substances are typically their conductive properties and potential application in nanoelectronics as molecular wires.8 Besides the development of organic 1D conductors, organometallic complexes that exhibit a chain-like structure are in the focus of interest as well.9–14 A defined linear arrangement of metal ions is not only a potential molecular wire, yet highly interesting from a fundamental and theoretical point of view. Examples range from epitaxial crystal growth,15 non-linear optical behaviour16,17 and metal-containing liquid crystals (MLC)18 to anisotropic electrical conductivity.19–25 Synthetic approaches towards such metal strings include ligand-enforced arrangement of metal ions, as seen in the undecametallic [Ni11(tentra)4Cl2](PF6)4 (tentra = tetranaphthyridyltriamine) complex26 or metallophilic interactions as design elements. In the latter, especially d8 and d10 configured ions are utilized for the synthesis of one-dimensional metal strings.27–31 In this review, focus is put on formally infinite molecular gold strings with homogeneous repeating units, because their specific properties can be assigned to defined structural features.32 In contrast, finite (oligomeric) gold strings in small molecules have ‘dead ends’ or an inhomogeneous saturation of the coordination sphere caused by the respective ligand system and thus, deviations in structure–property relationships might be induced.27,33,34 While related metal strings or metal string complexes of a series of elements have already been discussed and reviewed,25,35–48 we think a critical update on the unique chemistry of gold strings is valuable at this point.B. Aurophilicity and metallophilicity
When X-ray diffraction became more and more popular for the definite elucidation of molecular structures in the 1970s, some structural features of simple gold-containing compounds could not be explained anymore by the standard concepts of valence and chemical bonding. Namely, gold atoms (especially in their +I oxidation state) were observed to be in close proximity, even below the sum of their van der Waals radii, which suggested a strong attractive interaction between these cations.49–51 This was indeed unusual, as Coulomb repulsion should evoke the opposite effect. Furthermore, Au(I) exhibits a closed-shell ([Xe]4f145d10) electron configuration and an interaction of such systems was simply not expected. These early observations gave rise to a hitherto unknown research field. Not surprisingly, this curiosity soon became interesting from a theoretical point of view as well.52 A major breakthrough was achieved in the early 1990s, when P. Pyykkö studied varying Au–Au distances in the model complex [(AuCl(PH3))2] at the Hartree–Fock (HF) and second order Møller–Plesset (MP2) levels. While the HF curve showed a purely repulsive behaviour, attraction was observed at the MP2 level, which could be attributed to the dispersive part of electron-correlation effects.53 In addition, the interaction decreases with the Au–Au separation (R) like R−6, hence following the rules of the London dispersion force (LDF).54,55 Thus, in a simple picture, these Au–Au attractions are van der Waals interactions, but unusually strong ones due to relativistic effects. To this date, this interpretation has been supported by many studies and is generally accepted,56–61 although discussions about the concept are still on-going to this date.62,63 At first, those interactions were believed to only emerge for gold; hence the term ‘aurophilic interactions’ or ‘aurophilicity’ was established by H. Schmidbaur in 1989 to describe the phenomenon.64,65 In general, these terms are used commonly if the distance between two gold atoms falls below the limit of 3.5 Å, for which bonding can still be considered, although the respective van der Waals distance (3.32 Å)66 is shorter.67,68 Hereby, the Au–Au distance is correlated with the strength of these interactions, which can reach values of up to 50 kJ mol−1, i.e. comparable with hydrogen bonds. Based on findings by P. Schwerdtfeger, the energy can be estimated according to| EAu–Au = 1.27 × 106e−3.5d(Au–Au) | (1) |
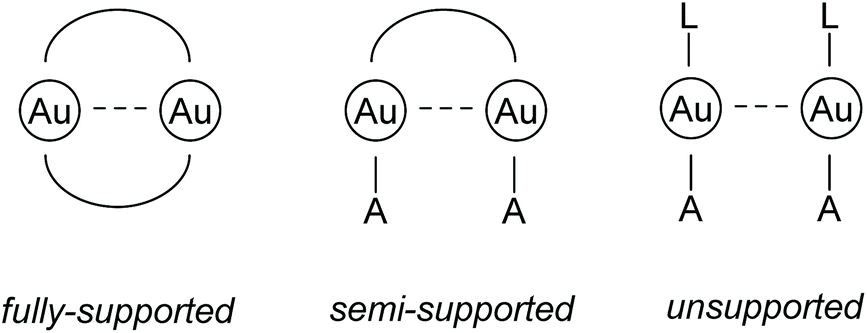 | ||
| Fig. 1 Schematic drawings of different subtypes of aurophilic interactions. L = neutral donor ligand; A = anionic ligand.68 | ||
While it is assumed that fully supported interactions are maintained in solution, the bonding situation of semi- and unsupported interactions in solution cannot be generalized and has to be evaluated in each case individually, because the solvation energy can compensate the comparably weak energy gain resulting from aurophilic interactions.67,69–73
The upcoming of aurophilicity attracted interest in similar interactions of other closed-shell ions as well. For example, R. Hoffmann and M. Jansen observed very short intermetallic distances for the lighter d10 configured homologues Cu(I) and Ag(I) in 1978 and 1987, respectively.74,75 Today, numerous elements, including open-shell ions like Au(III), Pt(II), Pd(II), Ir(I) or Ru(0), are known to interact with other heavy metal ions, expanding the concept to ‘metallophilic interactions’, even though pure aurophilic interactions are typically the strongest among them and therefore still take a special place.36,39,76–83
C. Luminescence
Among the properties of molecular 1D gold(I) chains, particular interest lies in the investigation of their photophysical characteristics.84–86 Heavy atoms like gold enhance spin–orbit coupling of the respective system, which facilitates transitions to the triplet manifold via intersystem crossing (ISC). Therefore, effective phosphorescence is regularly observed, manifested by large Stokes shifts and luminescence decay times in the lower microsecond range.87 Besides that, the emission properties can be further perturbed in the presence of aurophilic interactions. A prominent example addressing this issue was reported by Yam et al.,88 in which the luminescence of gold(I) complexes is directly affected and enhanced in the presence of an alkali metal crown ether complex, resulting in the formation of short Au–Au contacts. The perturbation induces a shift towards a lower emission energy. Therefore, it was assumed that the aurophilic interaction alters the emissive ligand to metal charge transfer (LMCT) state into a ligand to metal–metal charge transfer (LMMCT) state that goes along with a smaller HOMO–LUMO gap (Fig. 2). Additionally, besides the common LMCT emissive states, MLCT (metal to ligand charge transfer) processes for Au(I) complexes are known, which is mainly due to the oxidizing as well as reducing capability of Au(I) ions. Absorptions, which can be attributed to MLCT states, are primarily observed in complexes with π-acceptor ligands like cyanides, isocyanides and phosphines.89,90 However, these usually feature high energy absorptions and therefore, typical MLCT absorption bands are observed at comparably short wavelengths between 230 and 250 nm.91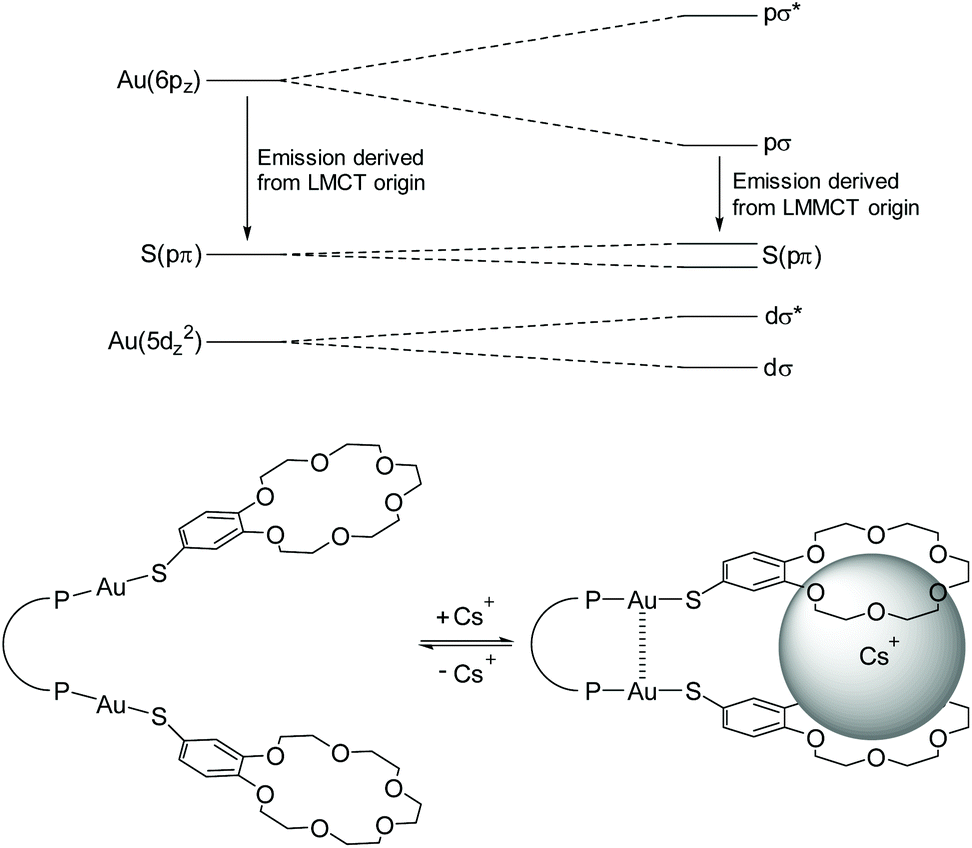 | ||
| Fig. 2 Schematic representation of orbital splittings induced by to aurophilic interactions in dinuclear gold(I) thiolate complexes. Adapted with permission.88 Copyright 2004 American Chemical Society. | ||
It has been predicted that the alteration from LMCT towards LMMCT states further enhances when three gold(I) ions interact linearly, due to a δ combination of three instead of two dx2−y2 orbitals (Fig. 3). This interesting phenomenon is particularly anticipated for ‘infinite’ 1D gold(I) chains.92 Moreover, as unsupported aurophilic interactions are the main assembly motif of 1D gold chains, a perturbation via different environmental stimuli is readily achieved in many cases and often goes along with a significant change of the structural and photophysical properties.
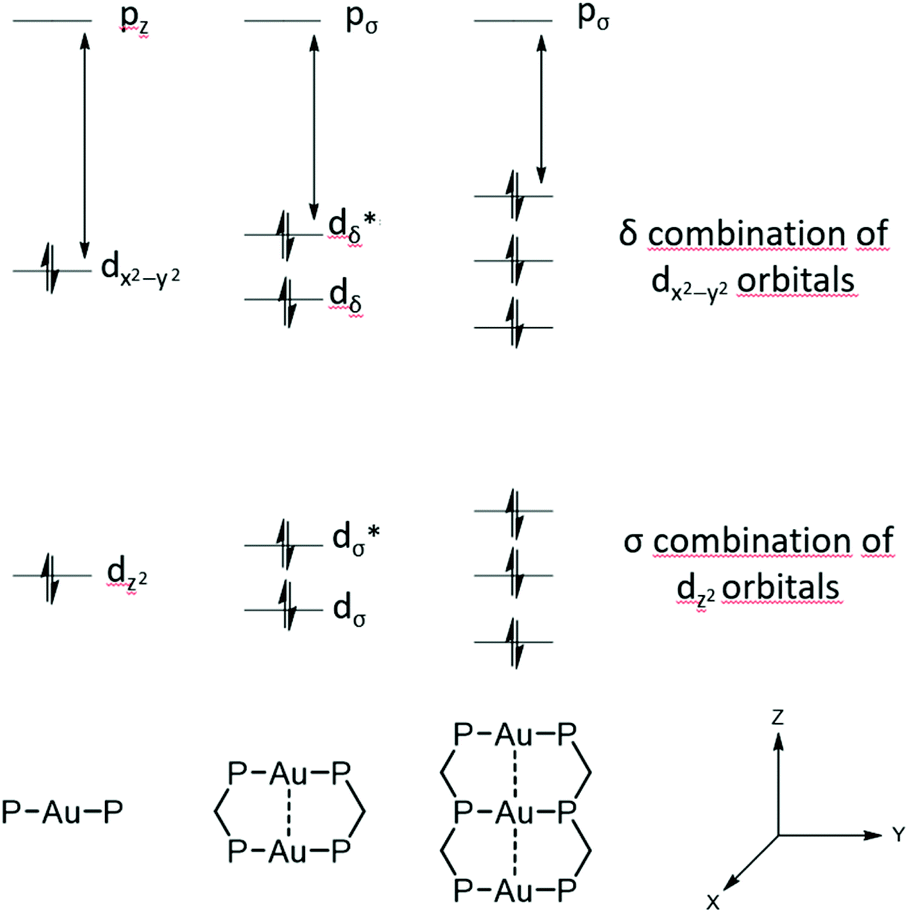 | ||
| Fig. 3 Molecular-orbital interactions for stacking of d10 AuP2 units in D2h symmetry.93 | ||
From a photophysical point of view, this represents the most important aspect of the herein presented 1D structures, as diverse slight stimuli already have an impact on the rather weak aurophilic interactions. Possible stimuli include solvent, pH value, pressure, and mechanical stress as well as temperature.84 This makes them a promising compound class towards an application as luminescent sensors and has already been remarkably demonstrated in recent reports, in which 1D gold(I) chains were found to show unique properties like solvatochromism,94 mechanochromism95 and luminescence tribochromism.96 In addition, this compound class is especially valuable to gain a better understanding of metallophilic interactions in general.
D. Classification of molecular gold strings
In the crystalline state of many gold-containing complexes, a clear tendency towards the development of aurophilic interactions is observed. However, from a synthetic point of view, it is challenging to predict whether this manifests in the formation of dimers, oligomers or ‘infinite’ (mostly one-dimensional) self-assembled polymers.97 Most importantly, the combination of steric and electronic demands of the respective ligand sphere plays a decisive role.98,99 1D gold chains are generated by an aurophilicity-induced self-assembly of molecular complexes; therefore a general distinction between ionic and neutral ‘chain links’ is reasonable, since the presence or absence of Coulomb interactions is an additional key factor. Hereby, ionic chains feature (mostly) homoleptic [A-Au-A]−, [L-Au-L]+ (A = anion; L = neutral donor) moieties or a combination of both anionic and cationic species. Due to the ionic character, these compounds often show a good solubility in polar solvents such as water. A neutral ‘chain link’ or complex, if Au(I) is taken as an example, consists of a monoanionic ligand paired with a neutral donor molecule coordinated to the central gold atom. Notably, heteroleptic neutral compounds often show a ligand exchange equilibrium in solution and an ionic homoleptic species is observed in the crystalline state. This is particularly valid for small and monodentate ligands like thiophene, pyridine, halogenides and pseudo-halogenides.100–103 Therefore, the different synthetic approaches, as well as the resulting molecular structures, are differentiated by the classification presented in Fig. 4. Within the scope of this review, the different classes are shortly introduced and selected publications, highlighting extraordinary research results and major breakthroughs, are presented in detail.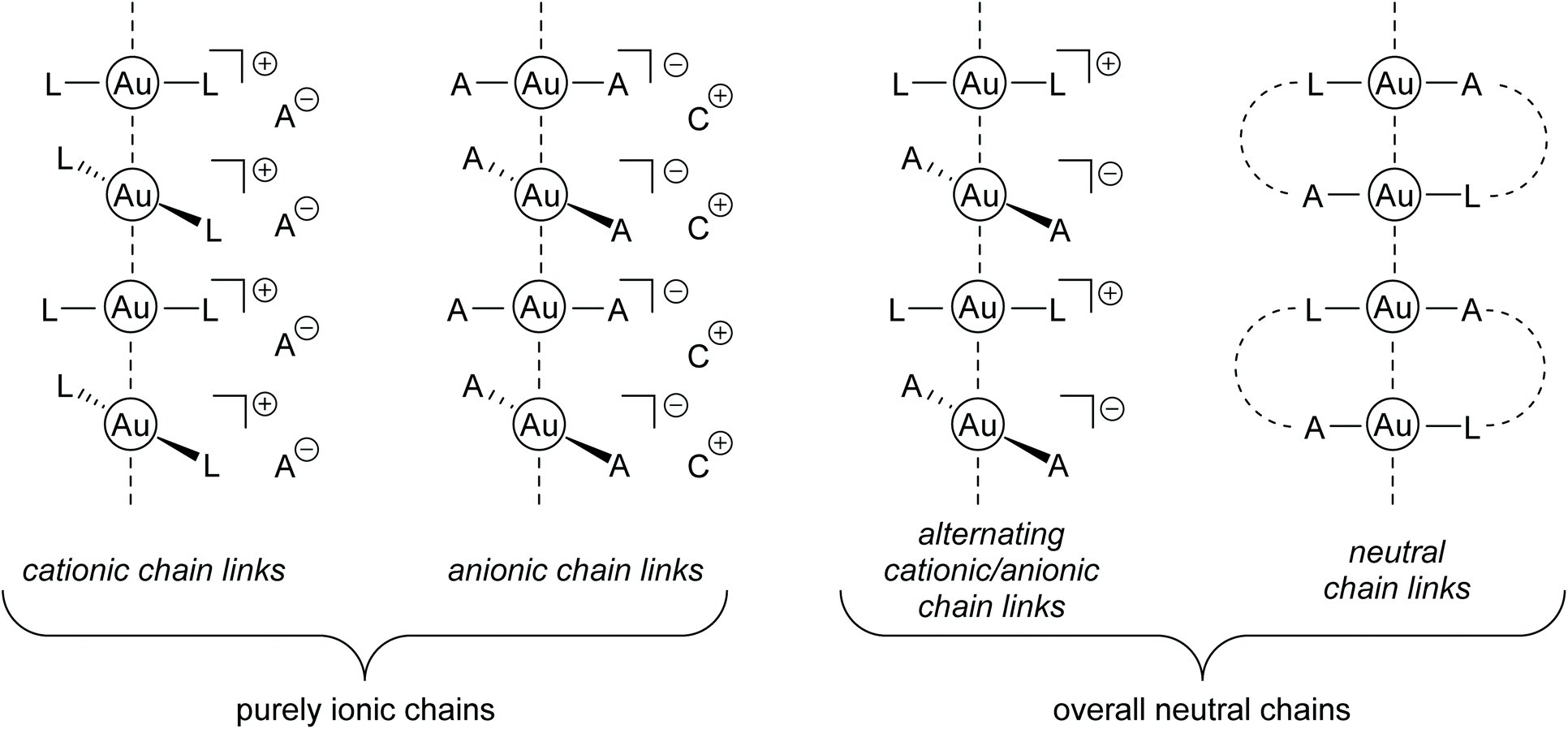 | ||
| Fig. 4 Schematic drawing of gold strings consisting of Au(I) chain links with neutral or ionic building blocks (A = anion, C = cation, L = neutral ligand). | ||
Gold strings with ionic chains
Purely cationic chain links
The few examples of gold strings, in which the respective countercations or -anions are simple organic or weakly coordinating molecules, are particularly interesting, since the aurophilic attraction overcompensates the Coulomb repulsion, resulting in a sequence of ‘infinite’ adjacent anions or cations.104–106Already in 1974, the Balch group introduced a synthetic procedure for cationic bis(carbene) gold(I) complexes by addition of isocyanides and primary amines to aqueous solutions of [AuCl4]− salts. The reaction readily yields compounds of the type [Au{C(NR2)(NR′H)}2]+.107 Following this procedure, using methylisocyanide and methylamine, a series of symmetrically substituted bis(carbene) gold(I) complexes [Au{C(NHMe)2}2]+ with varying anions (PF6− (1a), BF4− (1b), AsF6−, SbF6−) were presented in 2002 and 2008 by the same group (Scheme 1).108–110
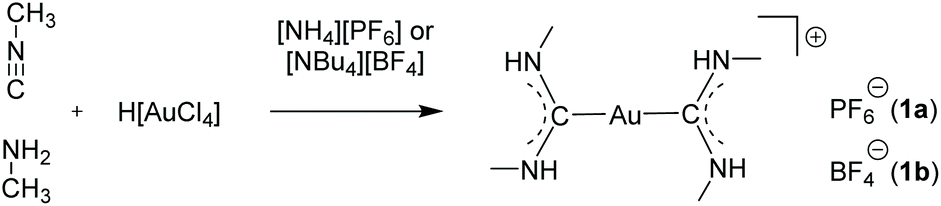 | ||
| Scheme 1 Synthesis of bis(carbene) gold complexes 1a and 1b. | ||
As observed in the crystal structure, the PF6− salt 1a consists of a linear coordinated gold(I) center with the ligands being arranged coplanarly. The cations form a linear gold chain along a crystallographic 42 axis comprising relatively short Au–Au contacts (3.1882(1) Å). Furthermore, the carbene ligands are linked to the PF6 ion via hydrogen bonds from the NH-moieties (Fig. 5, left). For 1b (anion = BF4−), the Au–Au bond lengths are elongated (dAu–Au = 3.4615(2) Å), while the angles are in a comparable range to 1a (angleC–Au–C = 178.42(12)°). In addition, the relative ligand orientation differs in a way that the outer methyl groups face one another. The same applies for the N–H groups, which allow the formation of hydrogen bonds from one cation to two different fluorine atoms of the BF4 ion (Fig. 5, right). Both 1a and 1b are colourless solids and display photoluminescence behaviour. Exemplarily, 1a shows green-blue (λmax = 460 nm) luminescence upon UV irradiation at 300 K. Upon cooling to 77 K, two emission bands are observed, whereby the maximum of the more intense emission is significantly red shifted. When dissolved in solvents like acetone, pyridine, dimethylsulfoxide (DMSO), dimethylformamide (DMF) or acetonitrile, the resulting colourless solutions are not luminescent anymore. However, upon freezing in liquid N2, the solutions become intensely luminescent again and strikingly, the emission colours differ in varying solvents (Fig. 6). This process is entirely reversible as the thawed solutions lose their photoluminescence properties. Note that the luminescence of 1b is affected in a similar fashion by dissolution in the aforementioned solvents and the freezing procedure.
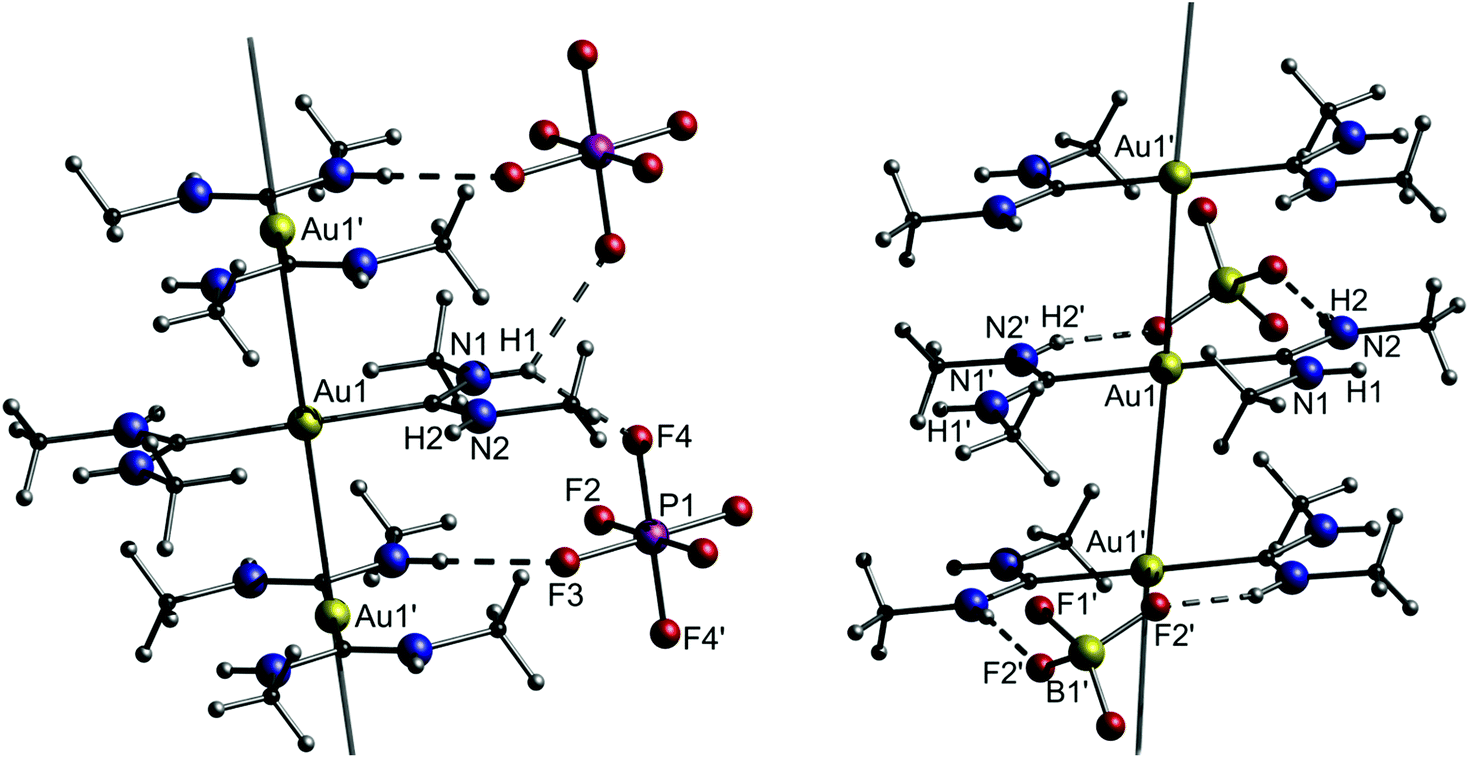 | ||
| Fig. 5 Left: A portion of the structure of crystalline [Au{C(NHMe)2}2][PF6]·0.5acetone (1a). Solvent molecules (acetone) are omitted. Right: A portion of the structure of crystalline [Au{C(NHMe)2}2][BF4] (1b).108 | ||
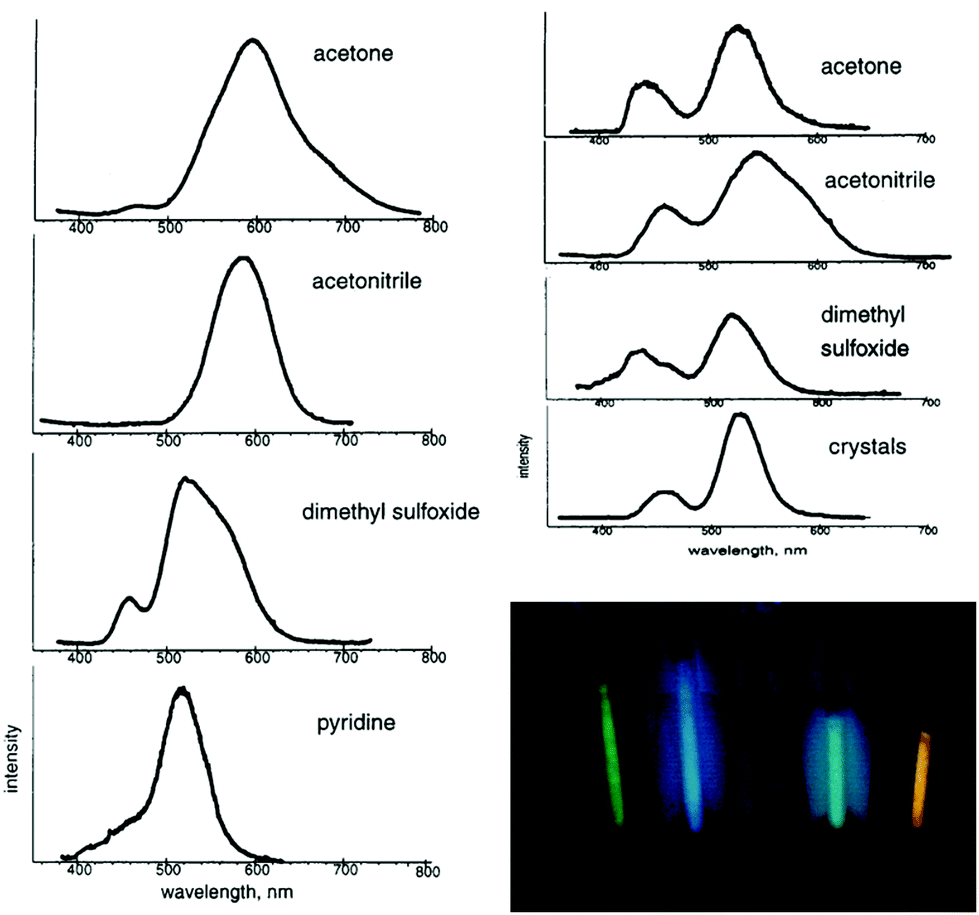 | ||
| Fig. 6 Left: Emission spectra obtained from 6 mM solutions of 1a, frozen at 77 K. Top right: Emission spectra obtained from 0.06 mM solutions of 1a frozen at 77 K and crystalline 1a at 77 K. Bottom right: Photograph of the luminescence from 6 mM solutions of 1a at 77 K. From left to right: MeCN, DMSO, DMF, pyridine, acetone. Reprinted (adapted) with permission.108 Copyright 2002 American Chemical Society. | ||
The absence of luminescence in solutions of 1a is presumably due to a breakdown of the polymeric structure into smaller units or monomers. The fact that the emission spectra of the frozen solutions strongly resemble the spectrum of polycrystalline 1a suggests similarities in the respective molecular structures. In this regard, it is reasonable that oligomeric forms of [Au{C(NHMe)2}2][PF6], generating species similar in structure to the arrangement of crystalline 1a, do form upon freezing the different solutions. The detailed molecular structures that are present in the frozen solutions could not be resolved, but it is apparent that the luminescence behaviour is affected by the number of gold ions, the Au–Au distance, the orientation of the ligands, hydrogen bonding interactions, the anion itself and the coordination of solvent molecules to the metal centers.111 A detailed understanding of these aspects might play a crucial role, e.g. in the development of sensors for different chemical environments.
Other examples for purely cationic chain links paired with weakly coordinating anions (WCA) were presented by the Krossing group in 2016.112 A series of [Au(MeCN)2][WCA] salts was prepared by direct oxidation of elemental gold in acetonitrile (MeCN) with nitrosyl salts of the type [NO]+[WCA]− ([WCA]− = [BF4]−, [GaCl4]−, [B(CF3)4]−, [Al(OC(CF3)3)4]− and [B(CF3)3CN]−). The resulting complexes [Au(MeCN)2][BF4] (2a) and [Au(MeCN)2][GaCl4] (2b) contain gold strings only composed of [Au(MeCN)2]+ cations that are interconnected by aurophilic interactions in the solid state, whereas the corresponding [B(CF3)4]− and [Al(OC(CF3)3)4]− salts form dimers and monomers, respectively (Scheme 2).
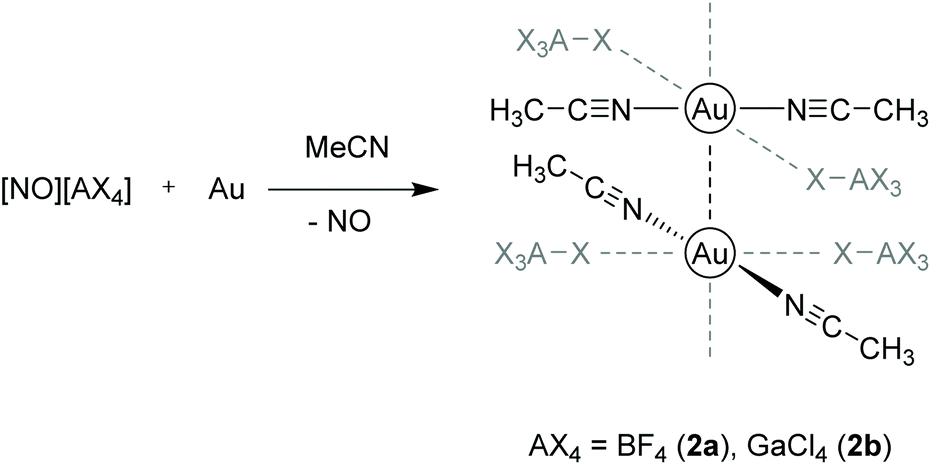 | ||
| Scheme 2 Synthesis and molecular structures of compounds 2a and 2b in the solid state.112 | ||
In both 2a and 2b, a weak interaction to the fluoride and chloride atoms of the respective anions BF4− and GaCl4− is observed in a trans ‘coordination’, although the atomic distances are outside of their van der Waals distance. In 2a, the contact lengths between adjacent gold atoms follow a repeating medium–short–short–medium–long–long pattern with the shortest contact being 3.113 Å. The respective Au–Au distances in 2b are equidistant (3.540 Å). Interestingly, Au–Au contact lengths are correlated with the coordination strength of the anions, which follows a [BF4]− > [GaCl4]− > [B(CF3)4]− > [Al(OC(CF3)3)4]− sequence.113,114 The cations thus appear in four forms: (a) as a chain with strong contacts (2a), (b) as a chain with weak contacts (2b), (c) as a dimer ([WCA]− = [B(CF3)4]−) and (d) as a monomer ([WCA]− = [Al(OC(CF3)3)4]−). This leads to the hypothesis that more electron-rich gold(I) centers feature stronger aurophilic interactions.
The assumption that the respective anion plays a decisive role regarding the molecular structure and resulting properties of cationic gold strings is further supported by a series of [(C6H11NC)2Au][XF6] salts (X = P (3a), As (3b), Sb (3c)) published by the Balch group.115–117 The hexafluoropnictogenates PF6−, AsF6− and SbF6−, although being weakly coordinating anions, alter the structure and thus the photophysical properties (including vapochromism and thermochromism) of the cationic string consisting of the two-coordinate [(C6H11NC)2Au]+. The term ‘thermochromism’ is used to describe a (typically reversible) temperature dependent change in the photophysical properties (e.g. colour or emission wavelength) of a sample. This phenomenon can be attributed to a change in the molecular or crystal structure.118,119 Both compounds 3a and 3b crystallize either as colourless, blue-emitting crystals or as yellow, green emitting crystals. Interestingly, the colourless crystals of 3a and 3b are isostructural (Fig. 7, P21/c), whereas the yellow crystals exhibit different solid state structures (Fig. 7, P212121 and P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ). The SbF6− derivative 3c is obtained as pale-yellow crystals as well, exhibiting a blue luminescence, yet its molecular structure is entirely different from that of 3a and 3b. Two different chains of cations are observed and both run parallel along the same crystallographic axis (Fig. 7). The Au–Au separations of 3a–c are summarized in Table 1.
). The SbF6− derivative 3c is obtained as pale-yellow crystals as well, exhibiting a blue luminescence, yet its molecular structure is entirely different from that of 3a and 3b. Two different chains of cations are observed and both run parallel along the same crystallographic axis (Fig. 7). The Au–Au separations of 3a–c are summarized in Table 1.
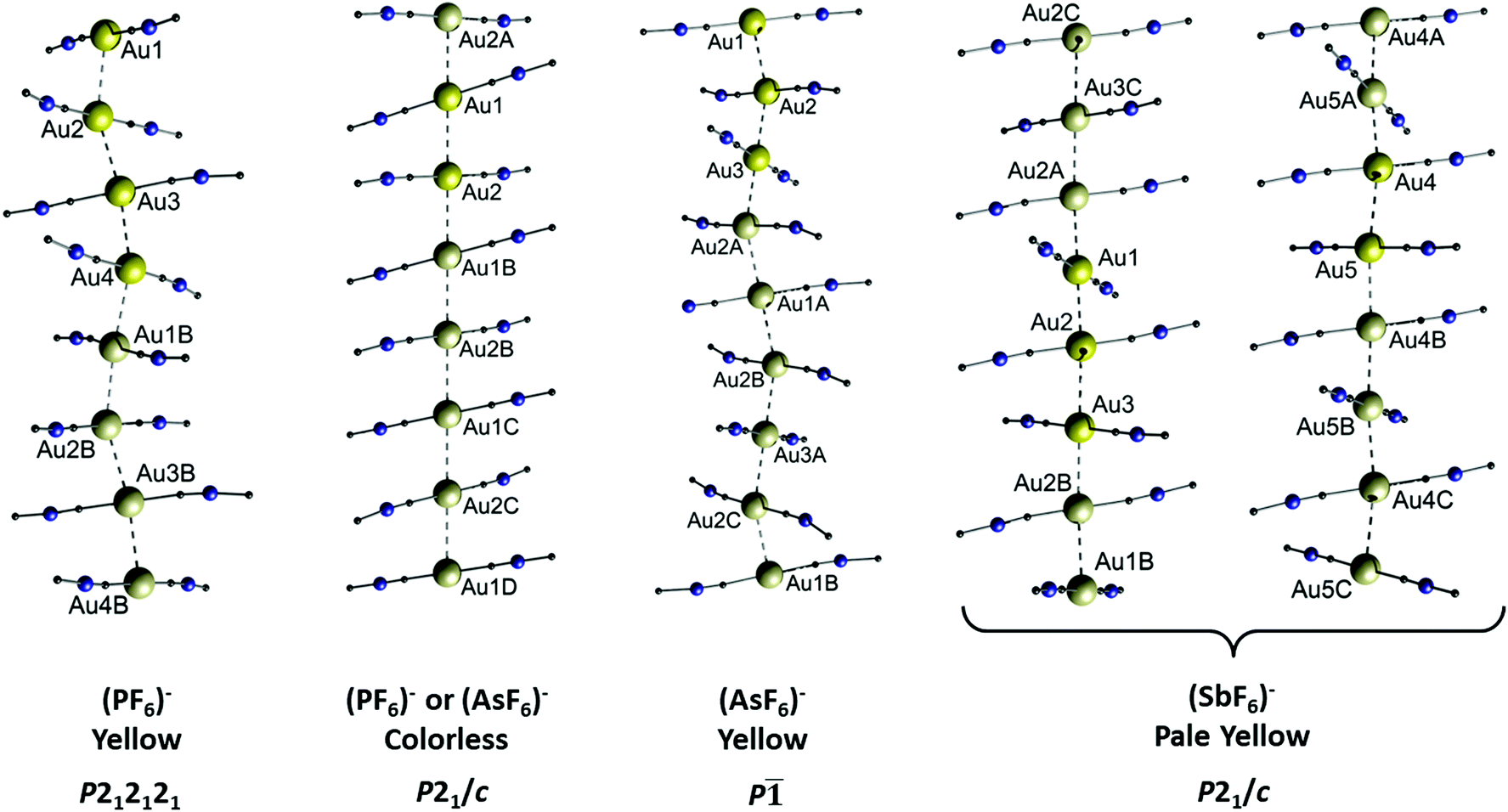 | ||
| Fig. 7 Comparison of the columnar structures of the different forms of [(C6H11NC)2Au][XF6] (X = P (3a), As (3b), Sb (3c)). For clarity, only the gold ions and the CNC portion of the ligands attached to the metals are indicated. Aurophilic interactions are shown as dashed lines. Anions are not shown. There are no solvate molecules in any of the crystal structures. Reprinted (adapted) with permission.115 Copyright 2020 American Chemical Society. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 115–117
115–117
| Sample | Colourless polymorph | Yellow polymorph |
|---|---|---|
| 3a | 3.1822(3) Å | 2.9803(6) Å |
| 2.9790(6) Å | ||
| 2.9651(6) Å | ||
| 2.9643(6) Å | ||
| 3b | 3.1983(8) Å | 3.0230(5) Å |
| 3.0097(6) Å | ||
| 3c | Chain 1: | |
| 3.0252(7) Å | ||
| 3.0858(7) Å | ||
| Chain 2: | ||
| 3.0424(9) Å | ||
| 3.0782(9) Å |
Although being intensely luminescent in the solid state, solutions of 3a–c are not emissive, which is consistent with a breakdown of the polymeric structure into monomers. However, upon exposure of the yellow, green-emitting polymorphs of 3a and 3b to vapours of selected solvents (dichloromethane, acetone, methanol or acetonitrile), a conversion to their respective colourless, blue-emitting polymorphs is observed (Fig. 8). In contrast, no change in colour or luminescence was observed in similar experiments with 3c.
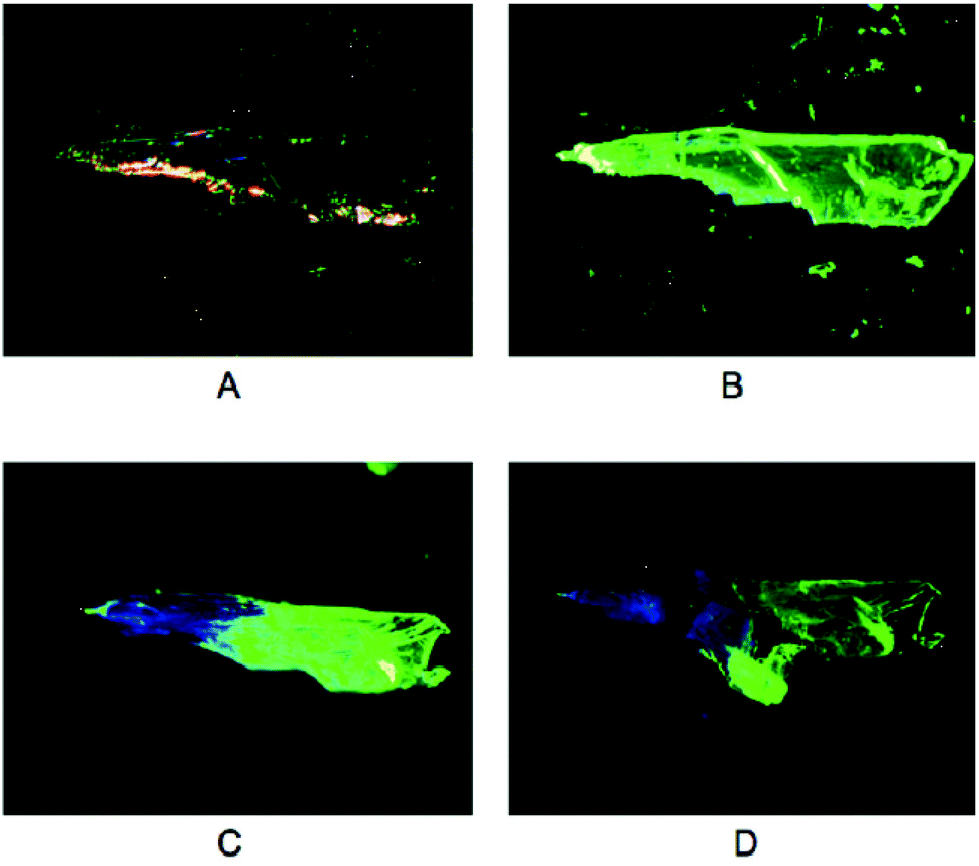 | ||
| Fig. 8 Photographs of crystalline 3b: (A) at ambient light, (B) upon UV irradiation, (C) after exposure to dichloromethane vapour upon UV irradiation, and (D) after crushing the crystal upon UV irradiation. Reprinted (adapted) with permission.115 Copyright 2020 American Chemical Society. | ||
Crystalline samples of 3a–c can be melted upon rising the temperature to a range of 113–126 °C. Independent of the polymorph type, the resulting melts of all samples are colourless and exhibit a bluish-white emission. Upon cooling, the respective yellow polymorphs of 3a and 3b are eventually formed, indicating that these are the thermodynamically more stable forms. Cooling the melt of 3c results in the reformation of crystalline 3c. However, a peculiarity is observed regarding the colourless polymorph of the AsF6− derivative 3b, which, upon rising the temperature to 100 °C, transforms into a yellow solid without melting. The emission spectrum of this solid is nearly identical to that of the respective yellow polymorph. A similar yellow solid is formed by cooling the melt of 3b, but in this case, the emission spectrum of this amorphous sample does not resemble the spectra of either the colourless or the yellow polymorph, indicating the formation of a new phase. The colourless polymorph of 3b is the only sample that shows this thermochromic behaviour. In addition, the mixed-anion salts [(C6H11NC)2Au][PF6]0.5[AsF6]0.5, [(C6H11NC)2Au] [PF6]0.5[SbF6]0.5, [(C6H11NC)2Au][AsF6]0.5[SbF6]0.5, [(C6H11NC)2Au][PF6]0.25[AsF6]0.75 and [(C6H11NC)2Au] [PF6]0.75[AsF6]0.25 were prepared by co-crystallization. Similar to 3a and 3b, colourless, blue-emitting and yellow, green-emitting polymorphs were found for all mixed-anion salts. Remarkably, all of the colourless crystals formed from a mixture of two anions show luminescence thermochromism below their melting points, similar to the colourless polymorph of 3b. In the interest of an overall view, we would like to dispense a further detailed discussion about the mixed-anion salts, yet an in-depth investigation of these compounds can be found in the original work.115 The thermochromic and melting point ranges are summarized in Table 2. A picture of the thermochromic behaviour is shown in Fig. 9.
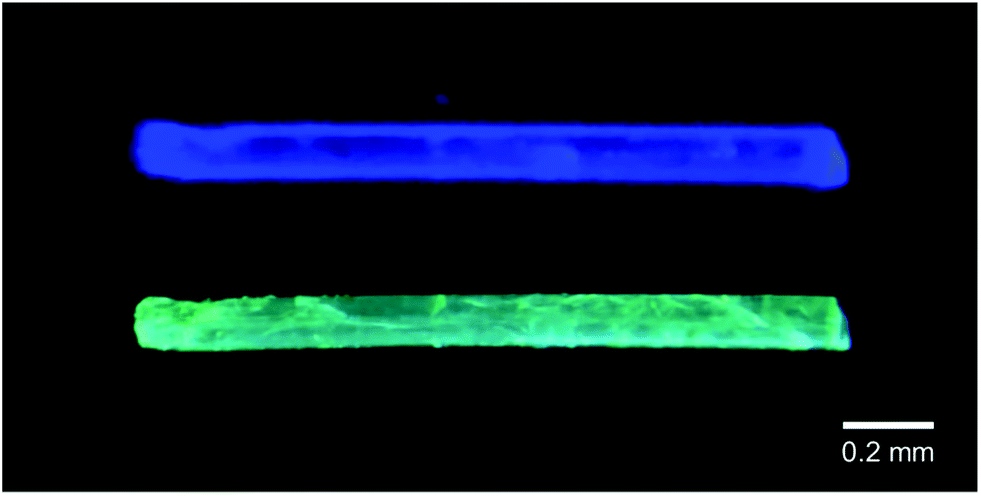 | ||
| Fig. 9 Photographs of the luminescence from a crystal of [(C6H11NC)2Au][PF6]0.5[AsF6]0.5. Top: Blue luminescence at ambient temperature. Bottom: Green luminescence after heating to 104–108 °C. Reprinted (adapted) with permission.115 Copyright 2020 American Chemical Society. | ||
| Compound | Thermochromic range [°C] | Melting point range [°C] |
|---|---|---|
| Colourless form | ||
| [(C6H11NC)2Au][PF6] | None | 115–120 |
| [(C6H11NC)2Au][AsF6] | 98–102 | 123–125 |
| [(C6H11NC)2Au][PF6]0.5[SbF6]0.5 | 87–95 | 114–116 |
| [(C6H11NC)2Au][AsF6]0.5[SbF6]0.5 | 88–92 | 113–114 |
| [(C6H11NC)2Au][PF6]0.5[AsF6]0.5 | 104–108 | 118–119 |
| [(C6H11NC)2Au][PF6]0.75[AsF6]0.25 | 105–111 | 118–121 |
| [(C6H11NC)2Au][PF6]0.25[AsF6]0.75 | 100–105 | 118–121 |
| Yellow form | ||
| [(C6H11NC)2Au][PF6] | None | 110–115 |
| [(C6H11NC)2Au][AsF6] | None | 123–126 |
| [(C6H11NC)2Au][SbF6] | None | 113–115 |
These results show the significant impact of the respective anion on the aurophilic interactions, even though being weakly coordinating and well separated from the chains of gold ions that form the 1D molecular structure. Most likely, the differences in the volume of the anions are responsible for the variations in the solid state structures and ultimately, in the thermochromic and vapochromic responses.
Purely anionic chain links
In this part, the utilization of negatively charged aurates [A-Au-A]− as building blocks for the synthesis of gold strings is presented. Among the whole compound class of gold strings, including neutral and charged chains, an anion aggregation resulting in aurophilicity-linked oligomers or polymers is only observed ‘in very special cases’, as stated by H. Schmidbaur and co-workers in 2002, referring to their own investigations.120 Consequently, studies of gold strings consisting of purely anionic chain links are rather scarce. Representative cations can be simple organic molecules like paraquat (N,N′-dimethyl-4,4′-bipyridinium or ‘Me2bipy’), as shown by the Guloy group in 1998.121 They presented the crystal structure of [Me2bipy][AuI2]2 (4), in which the diiodoaurate anions form a single 1D chain that does not interact with the staggered countercations [Me2bipy]2+ (Fig. 10). The respective Au–Au distances (3.3767(3) Å) are in the range of aurophilic interactions.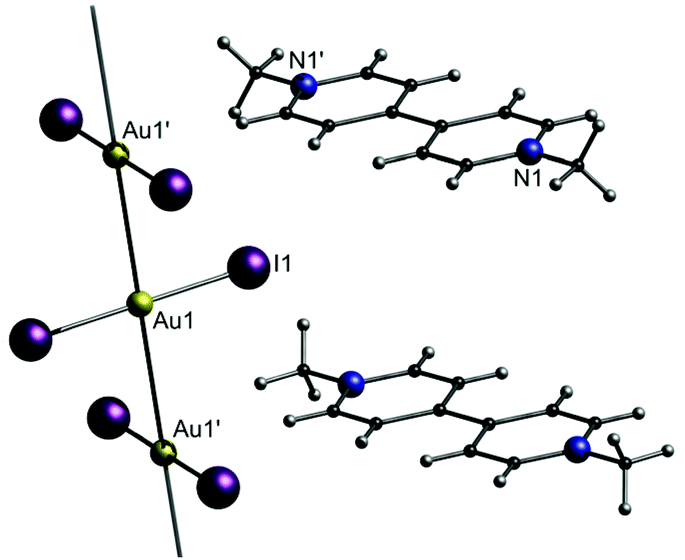 | ||
| Fig. 10 Representation of the crystal structure of 4.121 | ||
Another fascinating example for negatively charged chains is a series of bis(thiocyanato)aurates [Au(SCN)2]− with varying countercations including Na+ (5a), K+ (5b), Rb+ (5c), Cs+ (5d), NH4+ (5e), NMe4+ (5f), N(nBu)4+ (5g) and PPh4+, introduced in 2004 and 2006 by the Elder group.122,123 The phosphonium salt consists of isolated monomers, whereas the nearly isostructural alkali salts, as well as the NH4+ salt, form infinite linear gold strings with alternating long and short Au–Au distances along the chain. The NMe4+ salt forms a kinked chain of trimers and the N(nBu)4+ salt consists of isolated dimers (Fig. 11).
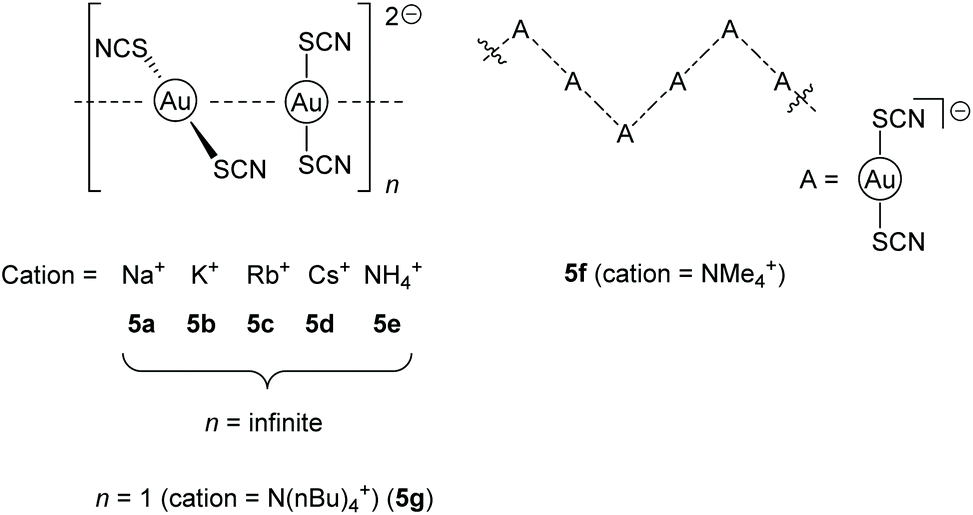 | ||
| Fig. 11 Representation of the anion-arrangement in compounds 5a–g.122,123 | ||
Polycrystalline samples of 5a–g show photoluminescence upon UV excitation at cryogenic temperature (77 K) with emission maxima ranging from 506 to 710 nm (Table 3).
122,123
| Sample | Au–Au [Å] | Exc [nm] | Em [nm] | E [cm−1] |
|---|---|---|---|---|
| 5b | 3.0064(5) | 320 | 516 | 19379 |
| 3.0430(5) | ||||
| 5g | 3.0700(8) | 360 | 539 | 18552 |
| 5c | 3.0821(4) | 320 | 506 | 19762 |
| 3.1144(5) | ||||
| 5e | 3.1409(3) | Not stated | 627 | 15949 |
| 3.1723(3) | ||||
| 5f | 3.1794(2) | 325 | 635 | 15748 |
| 3.2654(2) | ||||
| 5d | 3.2128(11) | 320 | 670 | 14925 |
| 3.2399(11) | ||||
| 5a | Unknown | 320 | 710 | 14084 |
In the crystal structures of the gold string compounds 5b–5f, alternating short and long Au–Au distances can be observed. Thus, their molecular structures can be described as a polymer of dimers (or trimers in the case of 5f). Interestingly, when the emission energy (cm−1) is plotted against the reciprocal of the short Au–Au bond lengths (Å−1), a strong correlation can be observed (Fig. 12), which is in accordance with theoretical predictions.124
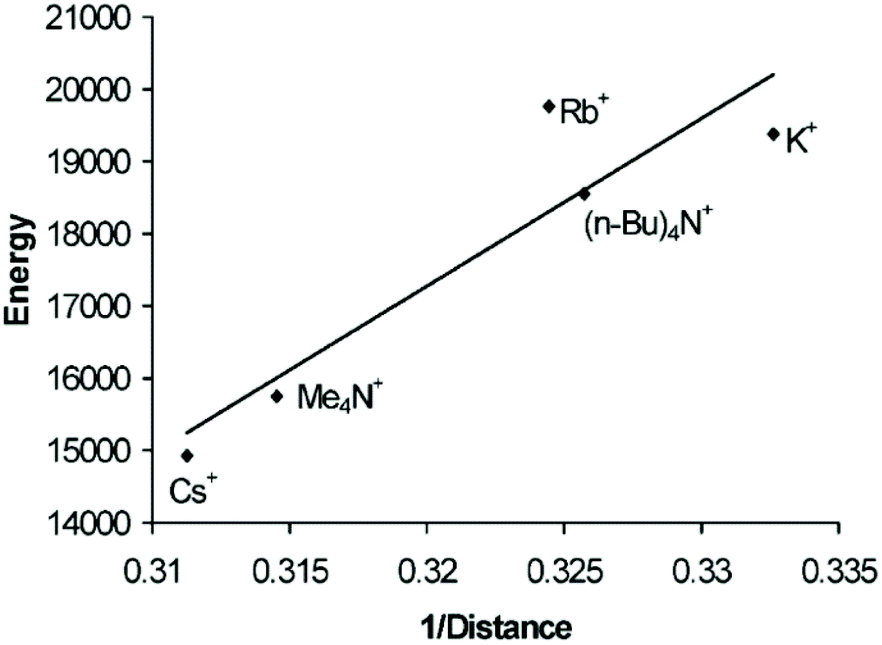 | ||
| Fig. 12 Energy [cm−1] versus 1/d [Å−1] of the short Au–Au interaction of [Au(SCN)2]nn−. Reprinted (adapted) with permission.122 Copyright 2004 American Chemical Society. | ||
The overall length of the gold chains appears not to be a crucial factor in this case, as the dimeric complex 5g and the assembly of trimers in 5f fall in line with the data for the ‘infinite’ chains. Instead, it is very likely that not only the supramolecular string, but also single gold–gold pairs or ‘Au3’ trimers with equidistant gold–gold contacts serve as the source of the emission.
A different picture emerges in ionic liquids containing [Au(SCN)2]− moieties, when differently substituted imidazolium cations instead of ‘inorganic’ cations are used, as presented by Aoyagi and co-workers in 2015.125 In this case, ionic liquids of the type [Cnmim][Au(SCN)2] (mim = methylimidazolium) are formed (Scheme 3).
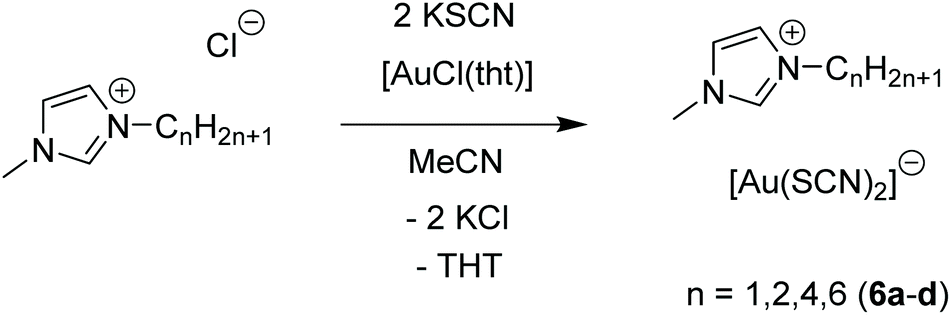 | ||
| Scheme 3 Synthesis of 6a and metal-containing ionic liquids (M-IL) 6b–d.125 | ||
The imidazolium salts 6b–d are ionic liquids at ambient temperatures with very low glass transition temperatures around 200 K. An exception is compound 6a, which forms a solid at room temperature. Compound 6a consists of [Au(SCN)2]nn− gold strings with Au–Au distances of 3.1773(3) Å, as observed in the corresponding crystal structure (Fig. 13). Interestingly, using photocrystallography, it was possible to resolve the structural changes induced upon UV light irradiation. Besides a change in the unit cell parameters, an excimeric contraction of the (Au–Au)* distance is observed as well. A list of changes regarding bond lengths is summarized in Table 4.
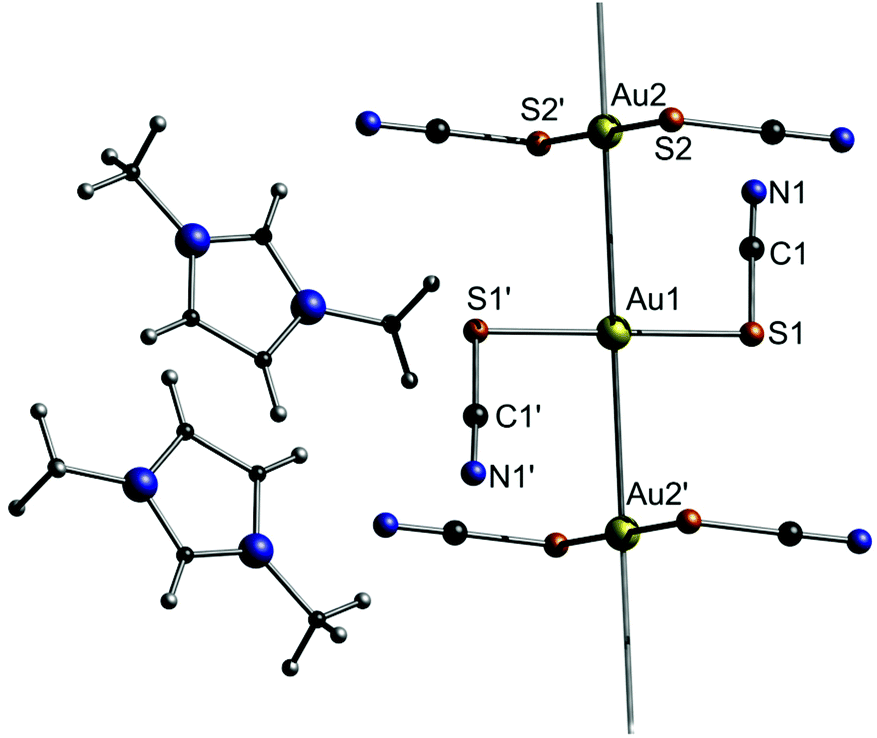 | ||
| Fig. 13 Excerpt of the crystal structure of 6a.125 | ||
| Bond | Lamp off [Å] | Lamp on [Å] | Δoff–on [Å] |
|---|---|---|---|
| Au1–S1 | 2.304(3) | 2.319(5) | −0.015(6) |
| Au2–S2 | 2.294(2) | 2.300(5) | −0.006(5) |
| S2–C2 | 1.680(10) | 1.66(2) | 0.02(2) |
| S1–C1 | 1.668(10) | 1.65(2) | 0.018(10) |
| Au1–Au2 | 3.1773(3) | 3.1659(3) | 0.0114(4) |
Furthermore, for the M-ILs 6b–d, time-resolved laser fluorescence spectroscopy (TRLFS) was performed. The spectra of the crude products are shown in Fig. 14. After a delay time of 100 μs to the excitation pulse, the peaks between 519 and 526 nm disappear, indicating fluorescence from the S1 state, and all envelopes of the spectra undergo a blue-shift. The data indicate a colour change in a single luminescence decay event. The existence of multiple different aggregates of [Au(SCN)2]nn−, possessing individual spectral features and luminescence lifetimes, is therefore suggested. For example, since the band gap of the T1 phosphorescent state to the S0 state is reduced by the number of metal ions involved, the peak in the deconvolution curves (green) at around 548 nm (Fig. 14, inset (a)) is attributed to a small aggregate, presumably a dimer [Au(SCN)2]22−, since the monomer (with no aurophilic interaction) shows no luminescence behaviour.
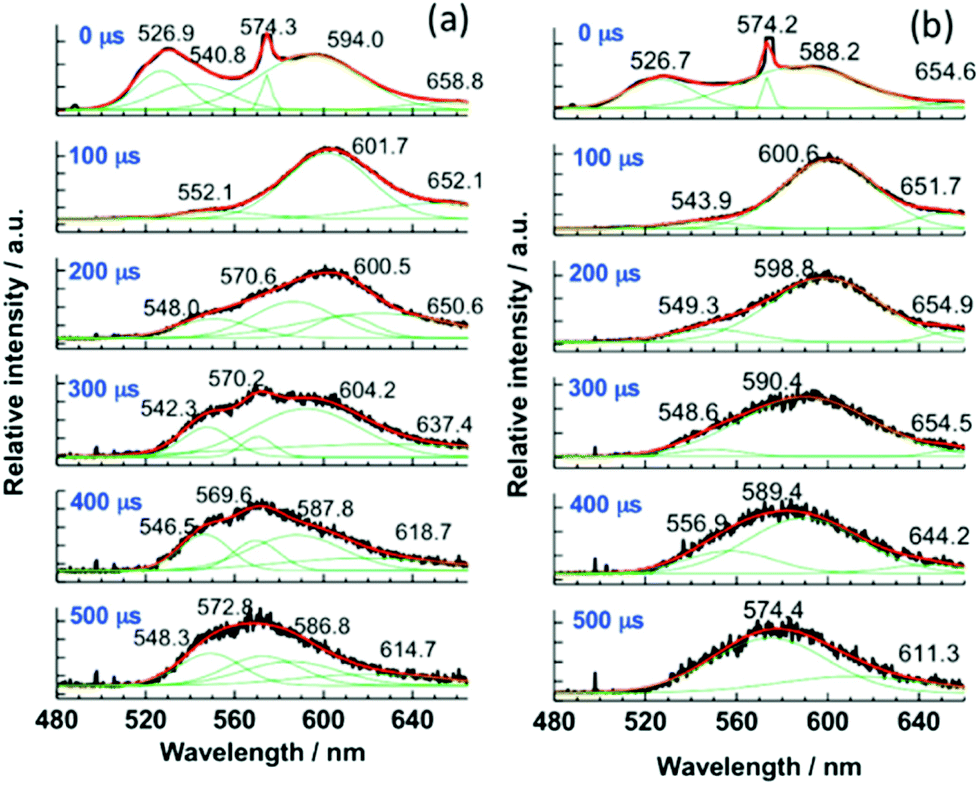 | ||
| Fig. 14 Time-resolved luminescence spectra of 6b (a), 6c (b) and their deconvolution curves (green) at 77 K (λex = 355 nm). Reprinted (adapted) with permission.125 Copyright 2015 American Chemical Society. | ||
The dicyanoaurate anion [Au(CN)2]− is a highly stable ion and is therefore used or observed in many different processes, such as cyanide leaching,126 gold electroplating127 or in protein crystallography as a phasing agent.128 A number of crystallographically investigated compounds containing [Au(CN)2]− are described in the literature. However, the aggregation of multiple [Au(CN)2]− units seems disfavoured in terms of Coulomb repulsion. On the other hand, the low steric demand, the simple linear structure and not least the hydrogen bond acceptor ability of the cyanides facilitate a self-aggregation process.129,130 In this context, the Attar and Balch groups investigated the formation and luminescence of dicyanoaurate strings comprising common ammonium counter cations.131 Among the presented piperidinium [C5H10NH2][Au(CN)2] (7a), pyrrolidinium [C4H8NH2][Au(CN)2] (7b), 1,1-diphenylhydrazinium [Ph2NNH3][Au(CN)2] (7c) and tetrapropylammonium [(C3H7)4N][Au(CN)2] (7d) salts, only the latter does not form a polymeric string in the solid state, presumably due to a lack of N–H groups suitable for hydrogen bonding to the cyanide moieties of the anions. Exemplarily, the crystal structure of 7c is shown in Fig. 15.
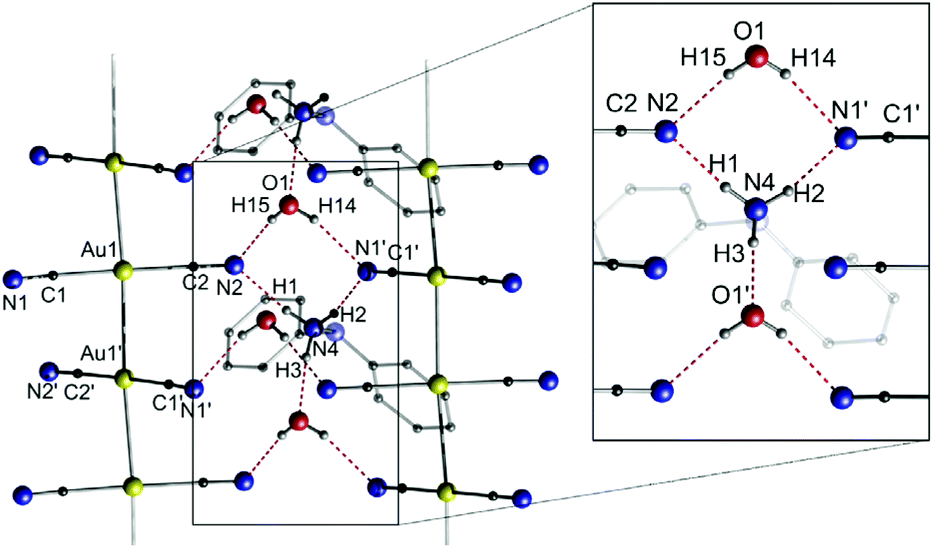 | ||
| Fig. 15 Excerpt of the crystal structure of 7c·H2O.131 | ||
In compound 7c, the aurates form an infinite gold string with equidistant Au–Au separations of 3.0866(4) Å. It is noteworthy that hydrogen bonds between the cyanides and the 1,1-diphenylhydrazinium cations and additional water molecules stabilize and support the formation of the string compound. The crystal structures of the analogues 7a and 7b are comparable to that of 7c, although hydrogen bonds are exclusively formed between the cyanide and the ammonium moieties.
In general, the Au–Au distances of 7a–c are remarkably short and lie in a narrow range of 3.0795(4)–3.0969(3) Å, whereas the shortest metal–metal distance in the tetrapropylammonium derivative [(C3H7)4N][Au(CN)2] (7d) is 7.526(1) Å and therefore far outside the aurophilic range. In consequence, crystalline 7a–c show intense blue luminescence, even at ambient temperature. The small range of the emission maxima (436 nm (7a), 451 nm (7b) and 400 nm (7c)) reflects the fact that their molecular structures in the solid state and their respective Au–Au separations are similar. In contrast, the charge separated salt [(C3H7)4N][Au(CN)2] without aurophilic contacts is non-luminescent at ambient and cryogenic (77 K) temperatures.
Purely anionic chains often include heterometallic compounds in which the chains are cross-linked by additional metal ions. By using dicyanoaurates [Au(CN)2]− as anions, similar to compounds 7a–c, but employing a metal complex as a [cation]+, (hetero)bimetallic coordination polymers are obtained.132 In this compound class, the bidentate character of the cyanide ligand is the driving force for the formation of supramolecular architectures. Representative compounds consist of [M(L)n]x[Au(CN)2]y (M = Cu, Mn, Zn, Ni, Co, Cd, Au; L = multidentate N-donor ligand).133–148 The general connectivity is illustrated in Scheme 4.
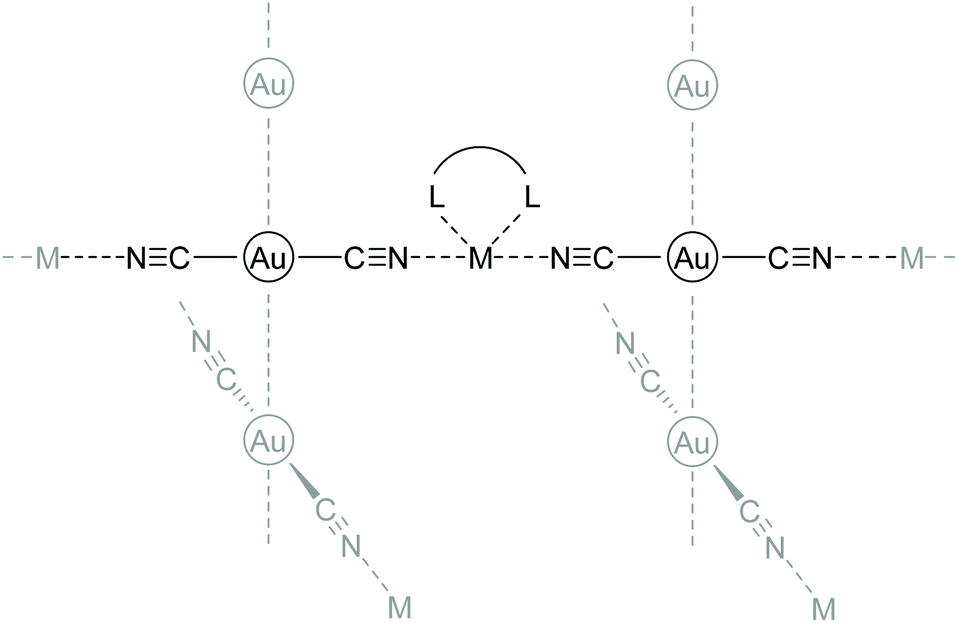 | ||
| Scheme 4 General connectivity of heterometallic compounds with [Au(CN)2]− as chain links. | ||
In many cases, the respective cationic [M(L)n] block consists of a divalent transition metal paired with a neutral multidentate ligand like phenanthroline, bipyridine or TMEDA. Additionally, +2 charged metal complex cations [M(L)n]2+, which consist of metal ions in higher oxidation states and negatively charged ligands, were employed for the construction of bimetallic coordination polymers, which tend to grow in two or three dimensions. The properties of the compounds, e.g. magnetism142 or biological activity,141 are mainly derived from the cationic [M(L)n]2+ building block. As an example, the first uranium-containing [Au(CN)2]− coordination polymer [UO2(bipy)(MeO)(MeOH)]2[(μ-Au(CN)2)(Au(CN)2)] (8) was presented by Leznoff and co-workers in 2017 (Fig. 16).149
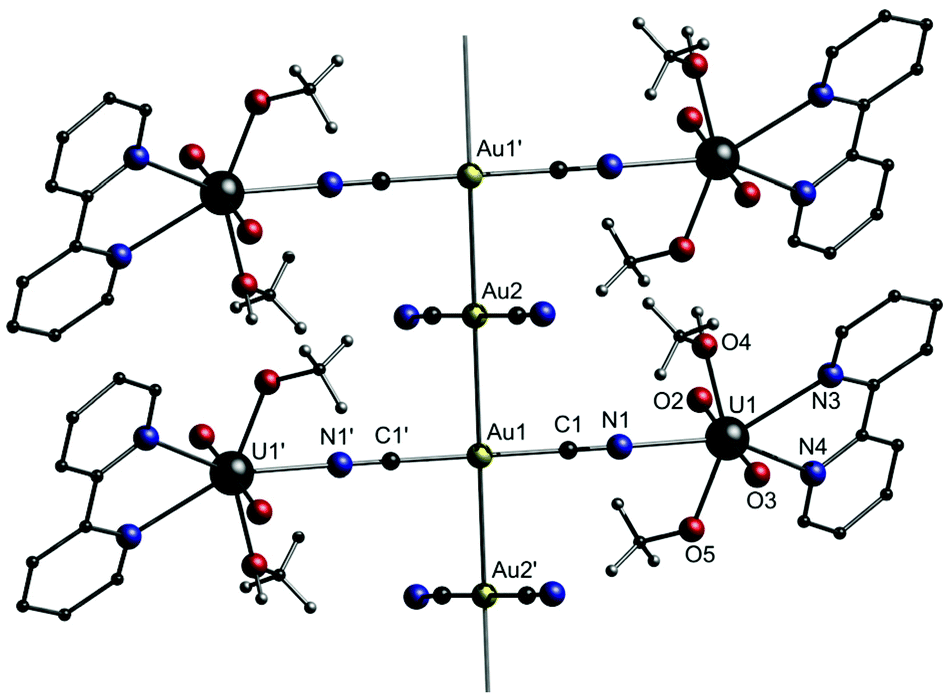 | ||
| Fig. 16 Excerpt of the crystal structure of 8.149 | ||
Compound 8 was synthesized by the reaction of [NBu4][Au(CN)2]·0.5H2O, UO2(NO3)2·6H2O and 2,2′-bipyridine in methanol. The molecular structure in the solid state consists of a one-dimensional gold chain structure of trinuclear [UO2(bipy)(MeO)(MeOH)]2[Au(CN)2] units linked by aurophilic contacts of 3.0528(5) Å to adjacent, unbound [Au(CN)2]− units. The dicyanoaurate anions are staggered, with a C–Au–Au–C torsion angle of 75.1(6)°. The different stretching frequencies of the cyanide ligands can be observed in the IR spectrum (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) CN = 2169 and 2133 cm−1) as well as in the corresponding Raman spectrum (CN = 2187 and 2148 cm−1). The blue-shifted bands can be assigned to the uranyl-bound [Au(CN)2]− units, whereas the resonance at lower energy is assigned to the free [Au(CN)2]− units.
CN = 2169 and 2133 cm−1) as well as in the corresponding Raman spectrum (CN = 2187 and 2148 cm−1). The blue-shifted bands can be assigned to the uranyl-bound [Au(CN)2]− units, whereas the resonance at lower energy is assigned to the free [Au(CN)2]− units.
Gold strings with overall neutral chains
Alternating cationic and anionic chain links
In this part, gold chains comprising gold double salts are presented. The term ‘double salt’ is used to describe ionic species in which both cations and anions contain metal ions, although initially, it was primarily used in solid-state chemistry to refer to (M)(M′)(X)n species such as KAl(SO4)2·12H2O.35 In case both ions are gold complexes, their aurophilic attraction is moreover supported by attractive Coulomb interactions.In 2018, Lu, Chen and co-workers presented a library of 35 different combinations of [Au(NHC)2][MX2] (NHC = N-heterocyclic carbene; M = Au or Cu; X = halide, cyanide or arylacetylide) complex salts (Fig. 17), outlining the large diversity of the double-salt approach.15,150,151 Although not every compound was analysed by single crystal X-ray diffraction, it is assumed that all double salts feature infinite metal strings in the solid state. A representative crystal structure (C3–A8) is shown in Fig. 17. The intermetallic distances of the double salts, of which crystal structures were determined, are in the range of 3.182–3.384 Å (Au–Au) and 3.373–3.398 Å (Au–Cu). The prepared complex salts are emissive in the solid state upon UV irradiation with emission maxima ranging from 448 nm (C1–A1) to 785 nm (C3–A14), emission lifetimes ranging from 0.1 μs (C3–A13) to 7.1 μs (C1–A2) and quantum yields ranging from 10% (C3–A10) to 99% (C1–A5 and C2–A5). A picture of selected double salts upon UV-irradiation and representative emission spectra are shown in Fig. 18.
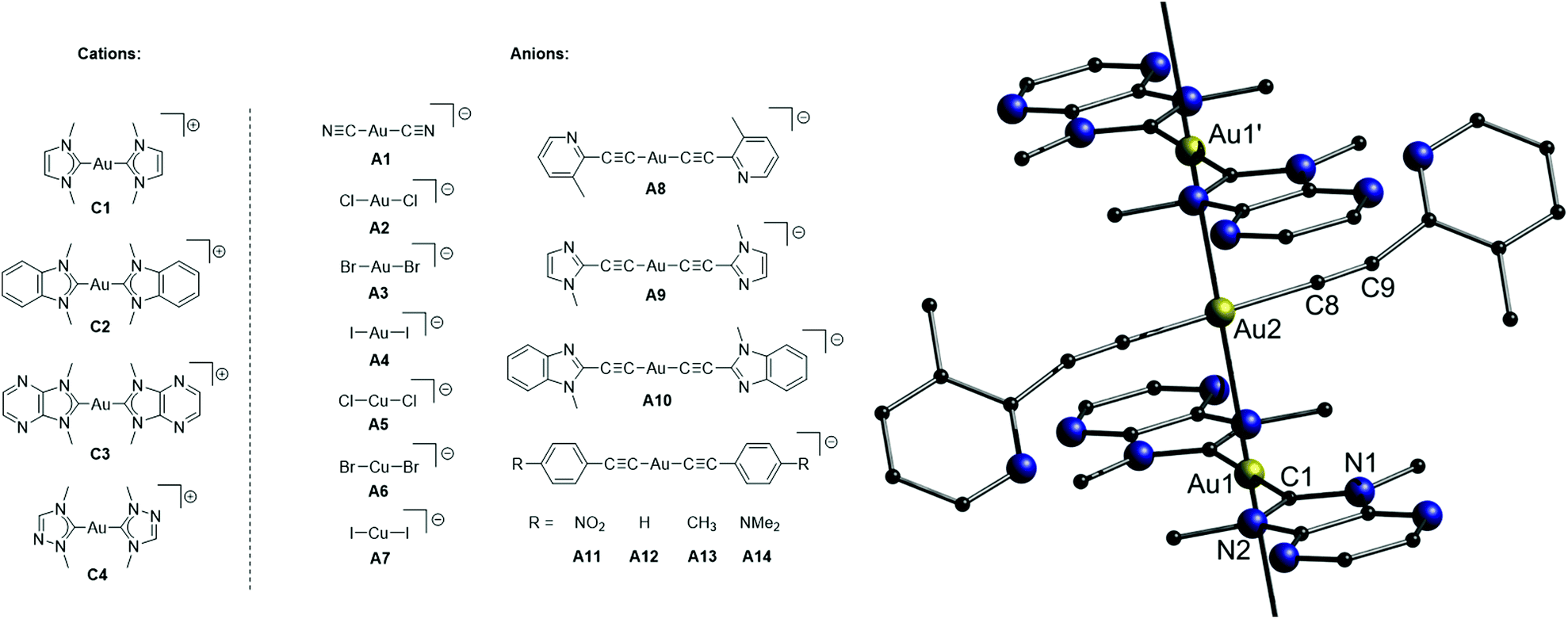 | ||
| Fig. 17 Left: Chemical structures of four cationic (C1–C4) and fourteen anionic (A1–A14) precursors. Right: Representation of the crystal structure of the double salt C3–A8.15 | ||
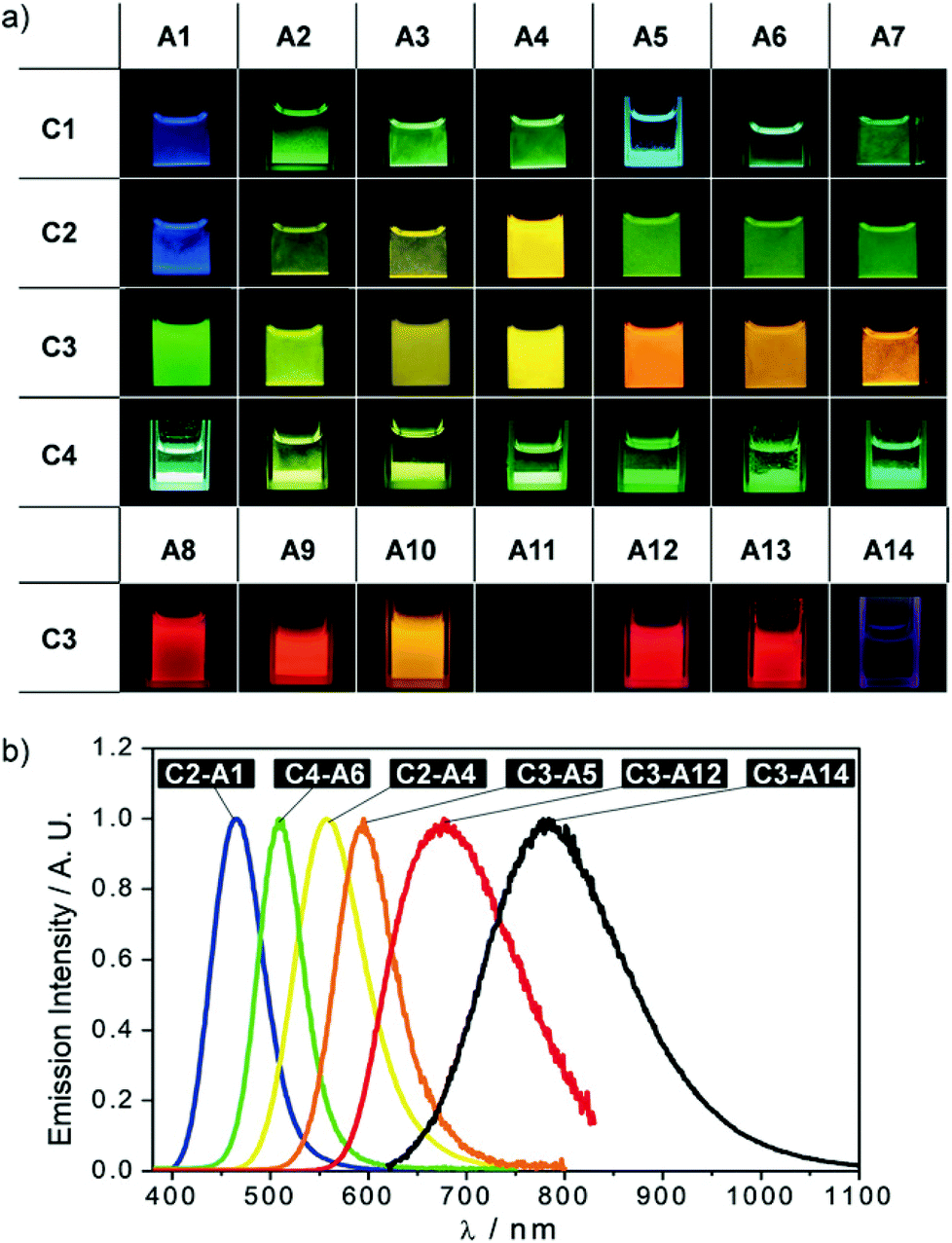 | ||
| Fig. 18 (a) The images of the 35 complex salts dispersed in water or acetonitrile observed under a 365 UV lamp; (b) normalized solid-state emission spectra of selected complex salts showing the multi-colour luminescence. Adapted with permission from John Wiley & Sons, copyright 2018.15 | ||
The following structure–property relationships can be derived from in-depth analysis of the photophysical properties: (1) Maintaining the anion, the emission energy of the resulting double salt red-shifts with more electron withdrawing NHCs on the cations. (2) Maintaining the cation, the emission energy of the resulting double salt red-shifts with more electron donating ligands on the anion series of [AuX2]−, yet it remains constant for the respective [CuX2]− salts. (3) The double salts of the form [Au(NHC)2][CuCl2] generate the highest quantum yields.
Interestingly, the combinatorial pool of double salts allows for multi-colour emission by combining two phosphors with different emission colour. Therefore, co-crystallization of C2, A1 and A4 (molar ratio 2:1:1) results in the formation of the triple salt C2–A1/A4 (Fig. 19).
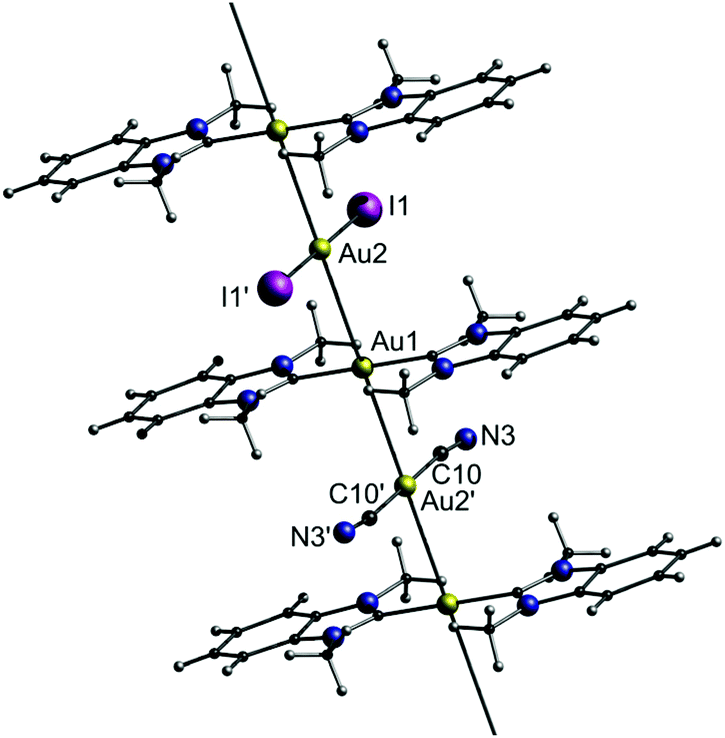 | ||
| Fig. 19 Representation of the crystal structure of the triple salt C2–A1/A4.15 | ||
The triple salt C2–A1/A4 indeed shows dual emission at 586 and 652 nm. The latter is significantly red-shifted compared to the corresponding parent complex salts C2–A1 (469 nm) and C2–A4 (570 nm), which can be explained by a simultaneous, yet not equal raise in energy of the HOMO and LUMO. This is due to a stronger σ-donating and weaker π-accepting capability of the iodide ligand in comparison with that of cyanide. The emission colour of C2–A1/A4 can further be fine-tuned by varying the molar ratios of A1 and A4 upon co-crystallization (Fig. 20). By screening the combinatorial pool, Lu, Chen and co-workers impressively demonstrated that metallophilic interactions, in the form of 1D chains, provide an easy access to new phosphorescent materials with high quantum yields and versatile colour tunability.
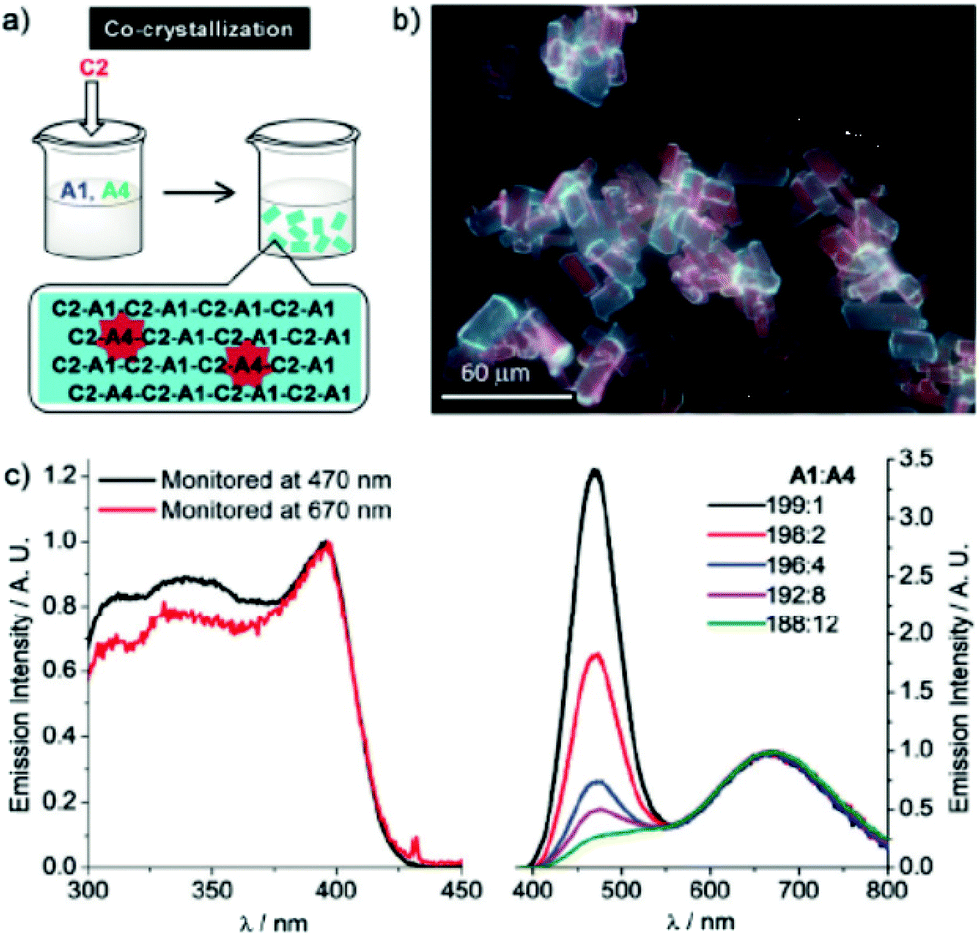 | ||
| Fig. 20 (a) A sketch for the co-crystallization process; (b) fluorescence micrographs of C2–A1/A4 microcrystals prepared via co-crystallization; (c) excitation and emission spectra of C2–A1/A4 microcrystals with a variety of A1:A4 ratios. Adapted with permission from John Wiley & Sons, copyright 2018.15 | ||
Besides the combination of the typical monovalent cationic and anionic gold complexes152,153 that were highlighted above, this concept can be readily expanded to mixed-valence gold(I/III) complexes,154,155 as shown e.g. by Heck and co-workers.156 A reaction of AuCl, AuCl3 and cyclohexylcarbonitrile (NC-Cy) yielded the complex [Au(NC-Cy)2][AuCl4] (9).
The crystal structure consists of linear gold strings with alternating [Au(NC-Cy)2]+ and [AuCl4]− units (Fig. 21). The distances between the Au(I) and Au(III) centers are equidistant (polymorph A: 3.2382(4) Å; polymorph B: 3.2499(2) Å) and furthermore display the shortest unsupported Au(I)–Au(III) interactions that have been observed to date.
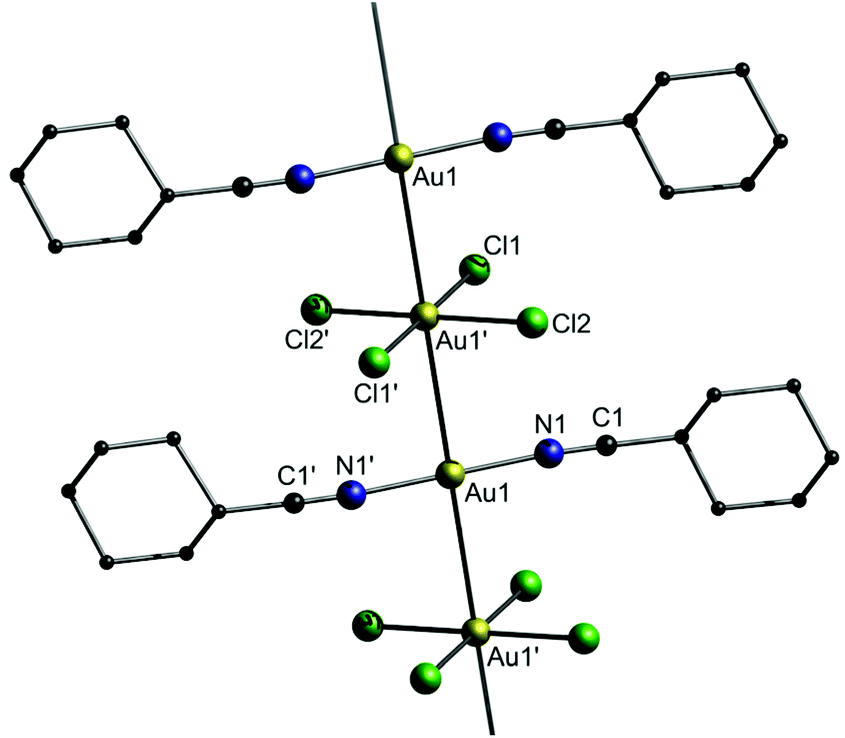 | ||
| Fig. 21 Representation of the crystal structure of 9.156 | ||
Neutral chain links
As shown in Fig. 4, gold strings with neutral chain links consist of neutral complexes, which form polymeric gold strings through unsupported aurophilic interactions.157–171 As an example, Ecken et al. studied the effect of anions on the solid state structure of (o-xylyl isocyanide)gold(I) (Cl (10a), Br (10b), I (10c), CN (10d)).172 Their study revealed that the Au–Au distances decrease in the order I− > Br− > Cl−. For CN−, which is a soft base as well, the Au–Au distances are similar to those of I−. However, the trend differs from that of [AuX(PMe2Ph)]2 dimers and theoretical predictions, for which Au–Au distances decrease following a Cl− > Br− > I− sequence.173–175 For 10a, the presence of Cl− anions results in the formation of dimers. In contrast, 10b forms an asymmetric unit comprising two Au atoms linked by aurophilic interactions and setting off the third one aside, resulting in a slightly kinked chain (Fig. 22). Individual molecules of 10c are aligned in chains through aurophilic interactions. 10d also forms a structure similar to 10b, but unlike 10c, all three Au atoms in the asymmetric unit are involved in aurophilic interactions and thereby, a 2D structure is obtained. The intra- and interaggregate Au–Au distances are summarized in Table 5.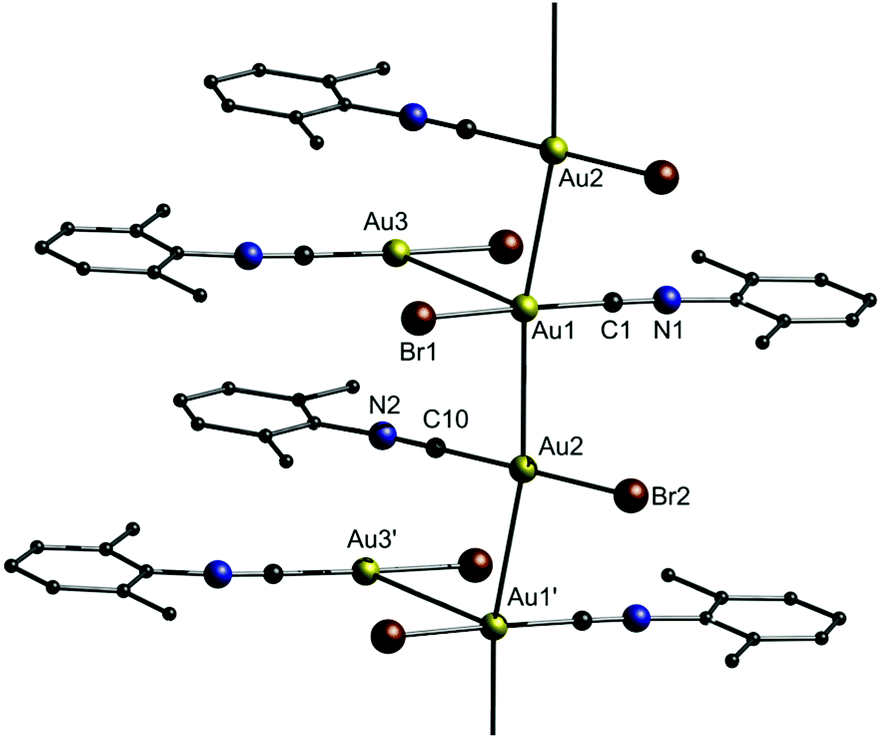 | ||
| Fig. 22 Representation of the crystal structure of 10b.172 | ||
| Sample | Intra Au–Au [Å] | Inter Au–Au [Å] | Solid-state structure |
|---|---|---|---|
| (o-xylylNC)AuCl (10a) | 3.3570(11) | 4.0225(12) | Dimers |
| (o-xylylNC)AuBr (10b) | 3.3480(5) | 3.7071(10) | Slightly kinked chain |
| (o-xylylNC)AuI (10c) | 3.4602(3) | 3.4602(3) | Slightly kinked chain |
| (o-xylylNC)AuCN (10d) | 3.4220(6) | 3.4615(6) | 2D structure |
| 3.1706(4) |
In accordance, Scheer and co-workers studied Au(I) complexes containing phosphanyl and arsanyl borane ligands and concluded that softer co-ligands coordinated to Au(I) tend to form extended polymeric structures due to multiple Au–Au interactions when compared to hard Lewis bases.176 In fact, gold in its +I oxidation state is a very soft Lewis acid and therefore has a pronounced affinity towards soft bases such as sulfur or selenium,177–180 which is in accordance with Pearson's principle of hard and soft acids and bases (HSAB).181–184 In this context, many gold string compounds are supported by bidentate dithiophosphate,185–189 dithiocarboxylate,190,191 diselenophosph(in)ate192,193 or related ligand systems.194–199 The Ch-X-Ch donor sites (Ch = chalcogenide, X = C, P) exhibit a low steric demand, which allows for the formation of gold strings comprising intermolecular connected homoleptic digold complexes (Fig. 23).
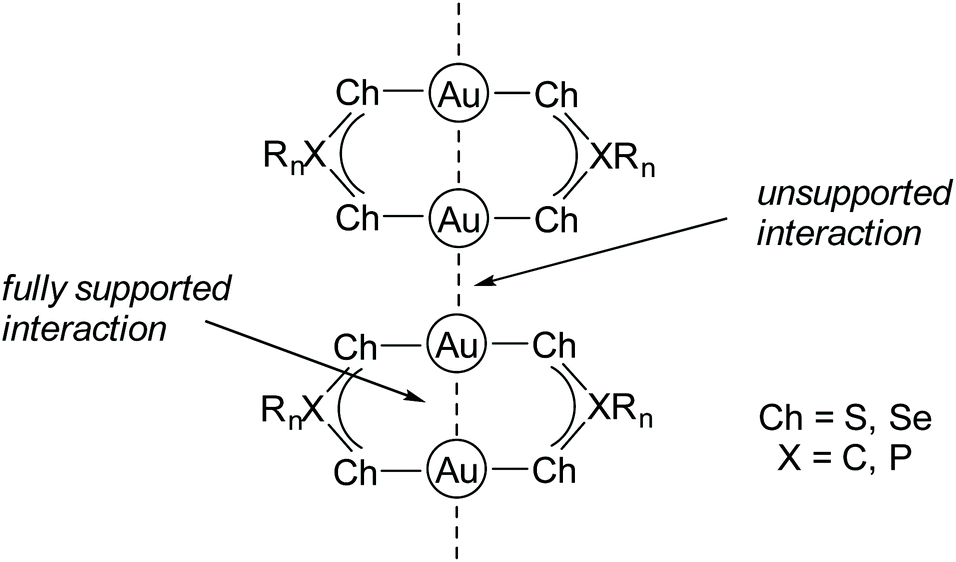 | ||
| Fig. 23 General connectivity of gold strings comprising Ch-X-Ch ligand systems (Ch = chalcogenide, X = C, P). | ||
In 2014, Katrusiak and co-workers studied the behaviour of a dithiocarbamate-ligated one-dimensional gold string towards the application of pressure.200 The two known polymorphs of [Au(Et2DTC)]2 (11) (DTC = dithiocarbamate) differ in their respective crystal structures as shown in Fig. 24. The α-phase consists of repeating identical units of [Au(Et2DTC)]2, whereas the β-phase exhibits an interval two times longer. For both, the intramolecular Au–Au bond lengths are about 2.78 Å, while the respective unsupported intermolecular Au–Au distance is about 0.2 Å longer.
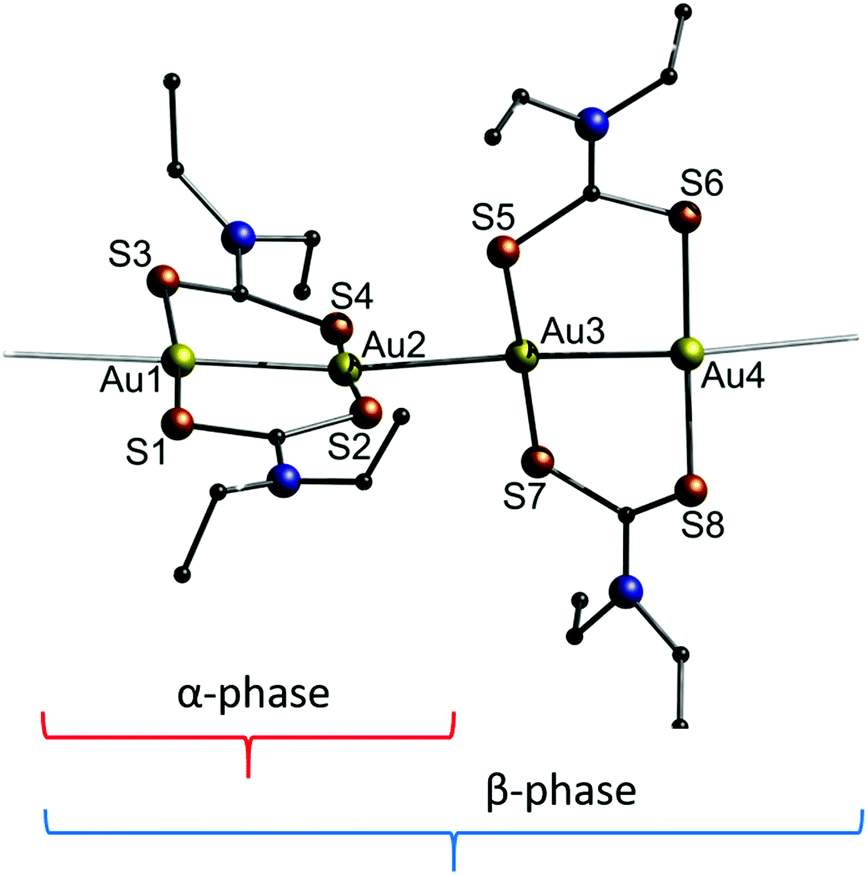 | ||
| Fig. 24 Representation of the crystal structure of 11. The brackets indicate the repeating unit of the two different polymorphs.200 | ||
The selective formation of either the α- or the β-phase can be controlled by varying the crystallization conditions or by the application of pressure. It is observed that above 50 MPa, the tetragonal crystals of the α-phase transform to an orthorhombic β-phase. Interestingly, the Au–Au distances can be further compressed by increasing the pressure. Thereby, the intermolecular Au–Au bonds are shortened at an average rate of −0.1006 Å per applied GPa. The average compressibility of the fully supported intramolecular Au–Au bonds, measured between 0.1 MPa and 1.0 GPa, is −0.0435 Å GPa−1 (Fig. 25). Thus, the fully supported bond is more than two times harder (i.e. deforms twice less). At 0.1 GPa, the supported Au–Au bonds are similar in length to that in metallic gold, whereas a pressure of 1.0 GPa diminishes the contact length to 2.72 Å, which therefore is about 0.05 Å shorter in comparison with that for the metal.
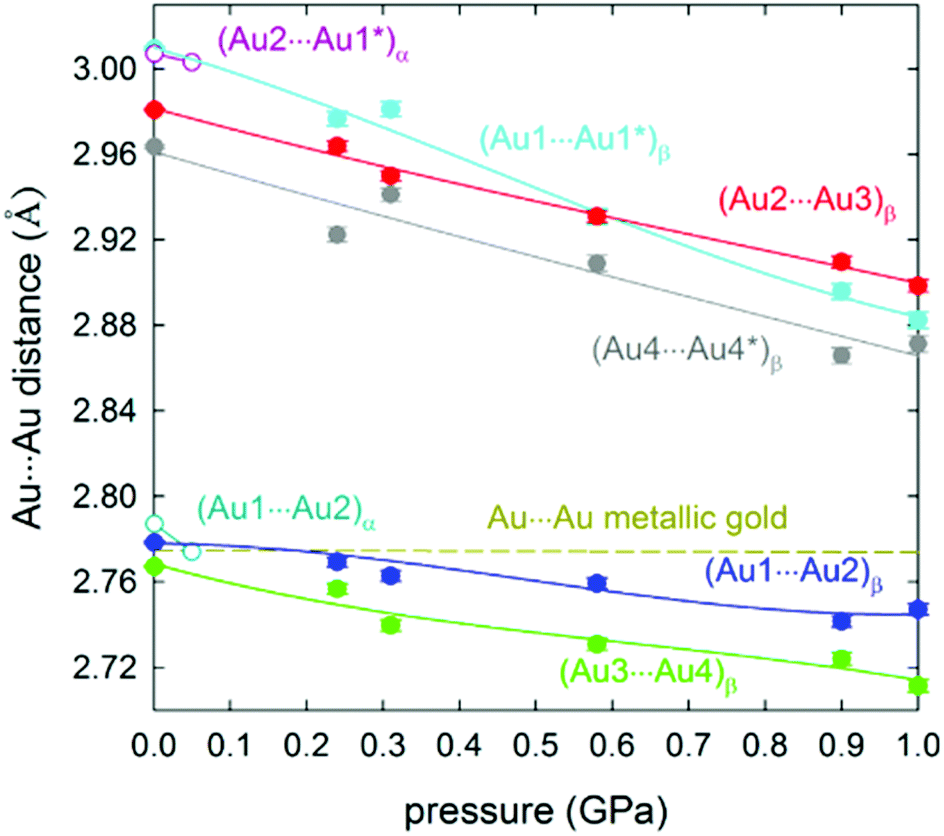 | ||
| Fig. 25 Compression of the Au–Au distances in phases α (open circles) and β (full symbols) of 11. The pressure dependence of the Au–Au distance (hardly changing to 1.0 GPa) in metallic gold is shown as the dashed line. Intramolecular and intermolecular bonds are shown. Reprinted with permission.200 Further permissions to this material should be directed to the ACS. | ||
Another example for dithiocarbamate ligated gold strings involves the report of solvochromic luminescence regarding a dinuclear gold(I)-(aza-[18]crown-6)dithiocarbamate (12), as presented by Chao and co-workers.201 The study investigates the effect of different solvates on the Au–Au distance of 12 and its resulting luminescence behaviour. Initially, the title compound was synthesized in the presence of MeCN to obtain 12·2MeCN (Fig. 26).
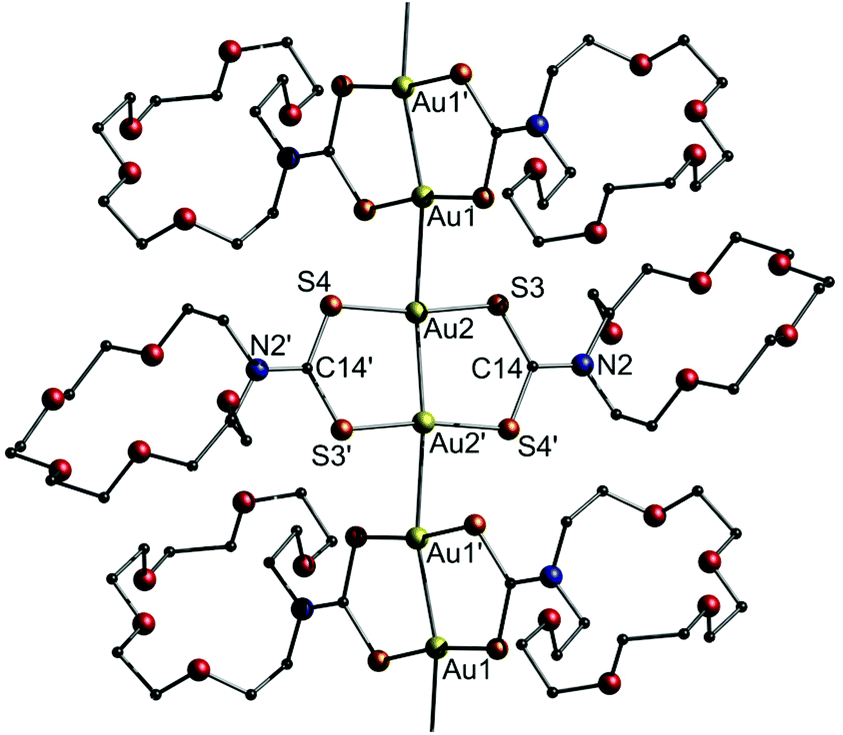 | ||
| Fig. 26 Representation of the crystal structure of 12·2(MeCN). Solvent molecules are omitted.201 | ||
Immersing the crystals of 12·2MeCN in a tert-butylbenzene or m-xylene solution yielded 12·tert-butyl-benzene·H2O and 12·0.5m-xylene respectively through single-crystal-to-single-crystal (SCSC) phase transformation. Hereby, 12 forms 1D chains irrespective of the presence of the aforementioned solvates. The Au–Au distances in 12, when crystallized with different solvates and their corresponding emission wavelengths, are summarized in Table 6. From the values, a direct correlation between the Au–Au distance and the emission wavelength is observed. The study was extended to other aromatic solvates with electron donating groups and it was observed that the emission energies vary in the following order: tert-butylbenzene (546 nm) > dry (553 nm) > benzene (566 nm) > p-xylene (568 nm) > m-xylene (583 nm) > o-xylene, anisole, toluene (595 nm) > MeCN (602 nm). It is suggested that even though the electron-donating substituents in the solvates do not interact with the complexes, they influence stacking in the crystal lattice, affecting i.e. the Au–Au distance. As a result, the complexes exhibit distinct emission wavelengths as shown in Fig. 27. In a similar study, Ito and co-workers presented the formation of crystals comprising gold isocyanides as a versatile host system with guest-dependent luminescence.202
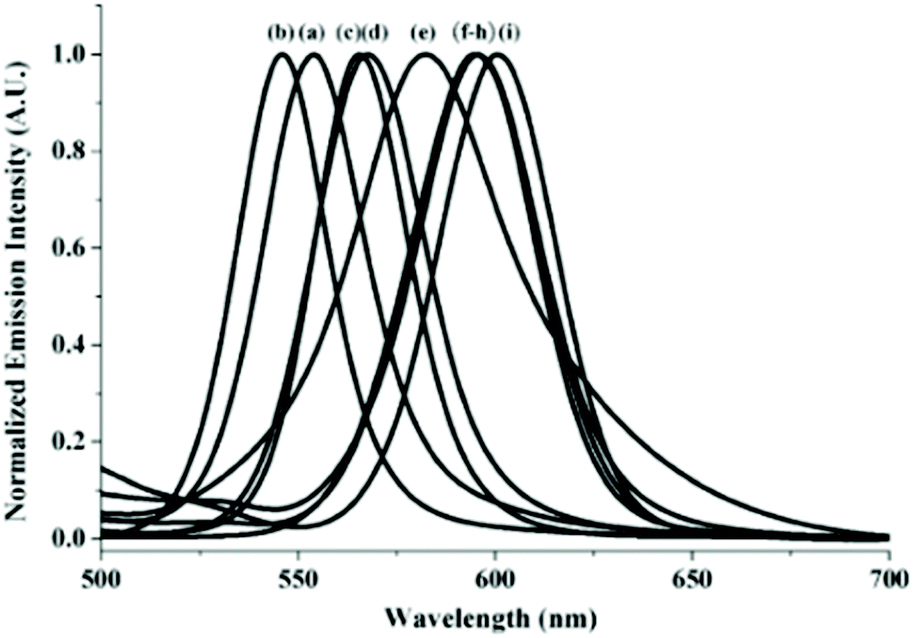 | ||
| Fig. 27 Solvochromic luminescence (λexc = 380 nm) of (a) dry samples of 12·2MeCN and upon grinding with (b) tert-butylbenzene, (c) benzene, (d) p-xylene, (e) m-xylene, (f) o-xylene, (g) anisole, (h) toluene, and (i) MeCN. Adapted with permission from John Wiley & Sons, copyright 2014.201 | ||
| Sample | Intra Au–Au [Å] | Inter Au–Au [Å] | Em [nm] |
|---|---|---|---|
| 12·2MeCN | 2.7186(3) | 2.8355(3) | 602 |
| 2.7249(3) | |||
| 12·0.5m-xylene | 2.755(2) | 2.890(2)–2.902(2) | 583 |
| 2.764(2) | |||
| 12·tBu-benzene·H2O | 2.7713(4) | 2.9420(5) | 546 |
| 12 (dry sample) | Unknown | Unknown | 553 |
In 2016, Ito and co-workers reported the synthesis of phenyl(phenylisocyanide)gold(I) (13a) and phenyl(3,5-dimethylphenylisocyanide)gold(I) (13b), which exhibit mechano triggered SCSC phase transformations and possess interesting optical and electronic properties.23,203–205 Complexes 13a and 13b were obtained as blue- and green-emitting single crystals respectively. However, a rapid phase transition was observed when the crystals were mechanically triggered. The phase transition of 13a and 13b propagated to the entire crystal to yield the daughter phases 13aSCSC and 13bSCSC. The values determining the photoluminescence properties of the corresponding crystals are summarized in Table 7 and their respective PL spectra are displayed in Fig. 28. The PL spectra indicate that the phase transition induces a red- and blue-shift of the emission bands of 13a and 13b, respectively.
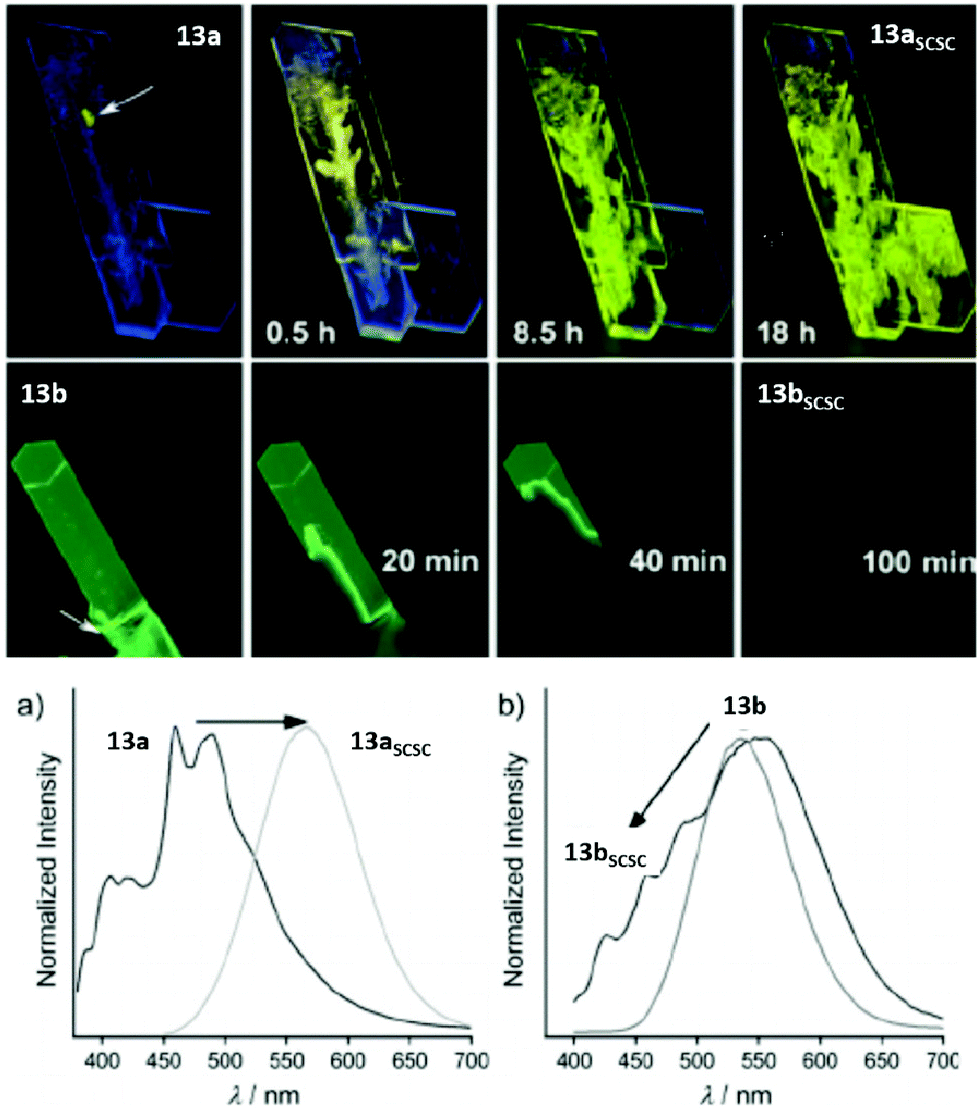 | ||
| Fig. 28 Top: Mechano-triggered SCSC phase transition of 13a to 13aSCSC and 13b to 13bSCSC over time. Bottom: PL spectra of crystals 13a and 13b in comparison with their daughter phases obtained upon slight mechanical stimulation. Excitation wavelength: 365 nm. Adapted with permission from John Wiley & Sons, copyright 2016.23 | ||
| Sample | Em [nm] | Φ em [%] | τ av [μs] |
|---|---|---|---|
| 13a | 460 | 4 | 108 |
| 490 | |||
| 13aSCSC | 566 | 16 | 5.33 |
| 13b | 540 | 84 | 0.97 |
| 13bSCSC | 535 | 6 | 13.8 |
The changes in the emission properties were rationalized by the formation of different crystal structures (shown in Fig. 29).23 The crystal structure of 13a comprises dimeric units (formed by CH/π interactions) which are further stacked into 1D columns through additional CH/π interactions. The shortest Au–Au distance of 5.733 Å indicates the absence of aurophilic interactions. However, on mechanical stimulation, the monomeric units are twisted and aurophilic contacts of 3.177 Å are formed within the dimeric units of 13aSCSC, which is in accordance with the red-shift of the emission band of 13a. The crystal structure of 13b consists of hexamers held together by aurophilic interactions, which are in the range of 3.274–3.112 Å. The hexamers are stacked into 1D columns supported by π–π interactions. Mechanical stimulation of the green-emitting species 13b leads to the formation of the blue-emitting daughter phase 13bSCSC (the Au–Au distance is 4.784 Å) by disconnecting the aurophilic interactions. Additionally, weak CH/π interactions are formed in the daughter phase.
 | ||
| Fig. 29 Crystal structures of (a) 13a (b) 13aSCSC (c) 13b and (d) 13bSCSC. Dotted lines and solid lines indicate CH/π interactions and aurophilic interactions, respectively.23 | ||
Furthermore, intergrowth (IG) crystals comprising multiple crystal domains of different polymorphs were prepared by mechanical stimulation. Quantitatively, SEM images show the difference between the IG crystals and their respective daughter phases in terms of charging ability which is demonstrated in Fig. 30. Moreover, the conductive properties of the samples were analysed quantitatively. The evaluation of conductive properties typically requires an electrode–sample contact, which is not very effective for 13a and 13b, because such a contact would trigger SCSC phase transition. For this reason, flash-photolysis time-resolved microwave conductivity (FP-TRMC) measurements were performed. Although this method only gives insight into short-range (approx. 10 nm) conductivity, it is contactless and therefore suitable to increase the knowledge about the electronic properties of the polymorphs.
 | ||
| Fig. 30 Photoluminescence microscopy images of (a) 13aIG and (c) 13bIG upon excitation at 365 nm and sputtering-free SEM images of (b) 13aIG and (d) 13bIG. Schematic representations of charging in (e) 13aIG and (f) 13bIG, when observed by SEM. Adapted with permission from John Wiley & Sons, copyright 2018.23 | ||
The maximum transient conductivity of species 13a was determined to be φ∑μ = 1.3 × 10−5 cm2 V−1 s−1, while the conductivity of species 13aSCSC was significantly higher at φ∑μ = 1.3 × 10−4 cm2 V−1 s−1. This increase of conductivity is most likely attributed to the formation of aurophilic interactions, as observed in the respective crystal structures (Fig. 29). For 13b and 13bSCSC, maximum transient conductivities of 3.2 × 10−5 and 2.8 × 10−6 cm2 V−1 s−1 were determined. The fact that the conductivity of 13bSCSC is one order of magnitude lower than that for 13b supports the assumption that the difference in conductivity of the samples is directly attributable to the presence or absence of aurophilic interactions in each species.
Another example focussing on the correlation between a change in the crystal structures and the photophysical properties due to external stimulation was presented by Eisenberg and co-workers in their report on luminescence tribochromism.206 It describes the phenomenon that luminescence of suitable solid samples can be turned on by gentle grinding. Their work focussed on Au(I) thiouracilate complexes which were synthesized by reacting [Au2Cl2(μ-dppm)] and AgCO2CF3 with 2-thiouracil (TU) and 6-methyl-2-thiouracil (Me-TU) to give [Au2(μ-TU)(μ-dppm)]CF3COO (14a) and [Au2(μ-Me-TU)(μ-dppm)]CF3COO (14b), respectively (Scheme 5).
 | ||
| Scheme 5 Synthesis of 14a and 14b.206 | ||
The obtained complex 14a is a monocationic dinuclear Au(I) complex, which forms a helical structure in the solid state. A head to tail arrangement of the TU moieties is observed and an intramolecular Au–Au distance of 2.8797(4) Å and an intermolecular Au–Au distance of 3.3321(5) Å were determined. The structure of 14b is similar to that of 14a, except that the helix is discontinuous for head to head arrangement with an inter Au–Au distance of 4.344 Å and for the head to tail arrangement, the average Au–Au distance is 3.236–3.354 Å. 14a and 14b are either weakly luminescent or non-emissive. However, on gently grinding the solids, a bright cyan coloured luminescence was observed at room temperature. Recrystallization of the emissive forms in the presence of CF3COOH again results in the formation of 14a or 14b. However, the emissive forms (15a and 15b) can be selectively prepared by stirring CH2Cl2/MeOH solutions of 14a or 14b over Na2CO3 followed by filtration. The changes seen in the presence of the base/acid might be due to the deprotonation/protonation of the pyrimidine N–H moiety of thiouracil. The PL spectra of both emissive (15b) and non-emissive (14b) forms are displayed in Fig. 31.
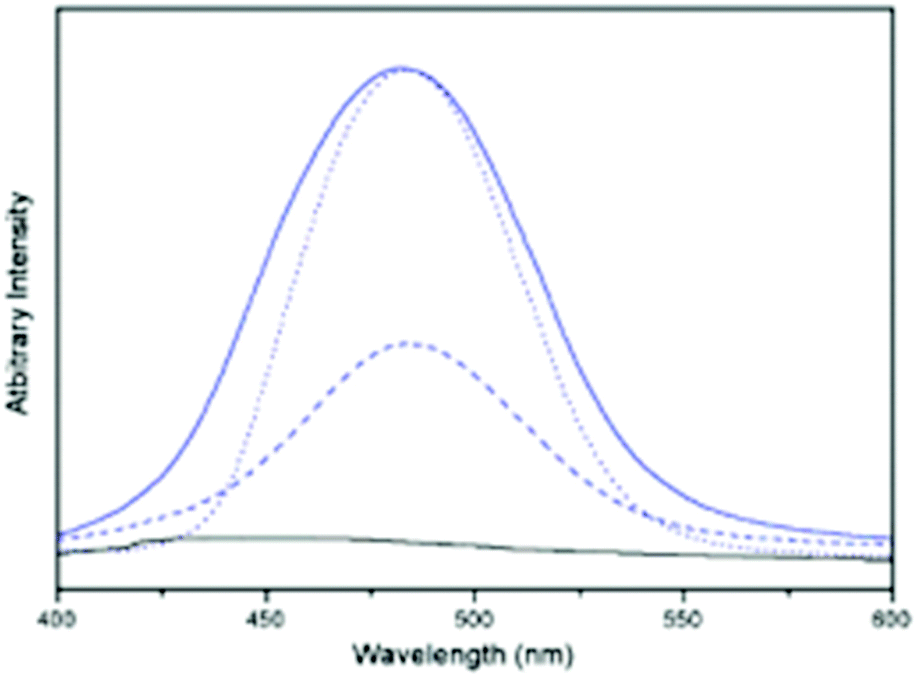 | ||
| Fig. 31 Emission spectra of 14b (black line) and ground crystals of 14b (blue line), CH2Cl2 solution of 14b (---) and crystals of 15b (⋯) (prepared by reaction with Na2CO3) at room temperature (at λex = 375 nm). Reprinted (adapted) with permission.206 Copyright 2003 American Chemical Society. | ||
The emissive compound 15b has a similar structure to 14a and 14b but different packing arrangements, and comprises strong intermolecular aurophilic interactions between the dimers with a distance of 2.9235(4) Å (Fig. 32). Hence, it can be concluded that the delicate balance between changes in the Au–Au interactions and acidity of the sample results in this fascinating luminescence tribochromism.
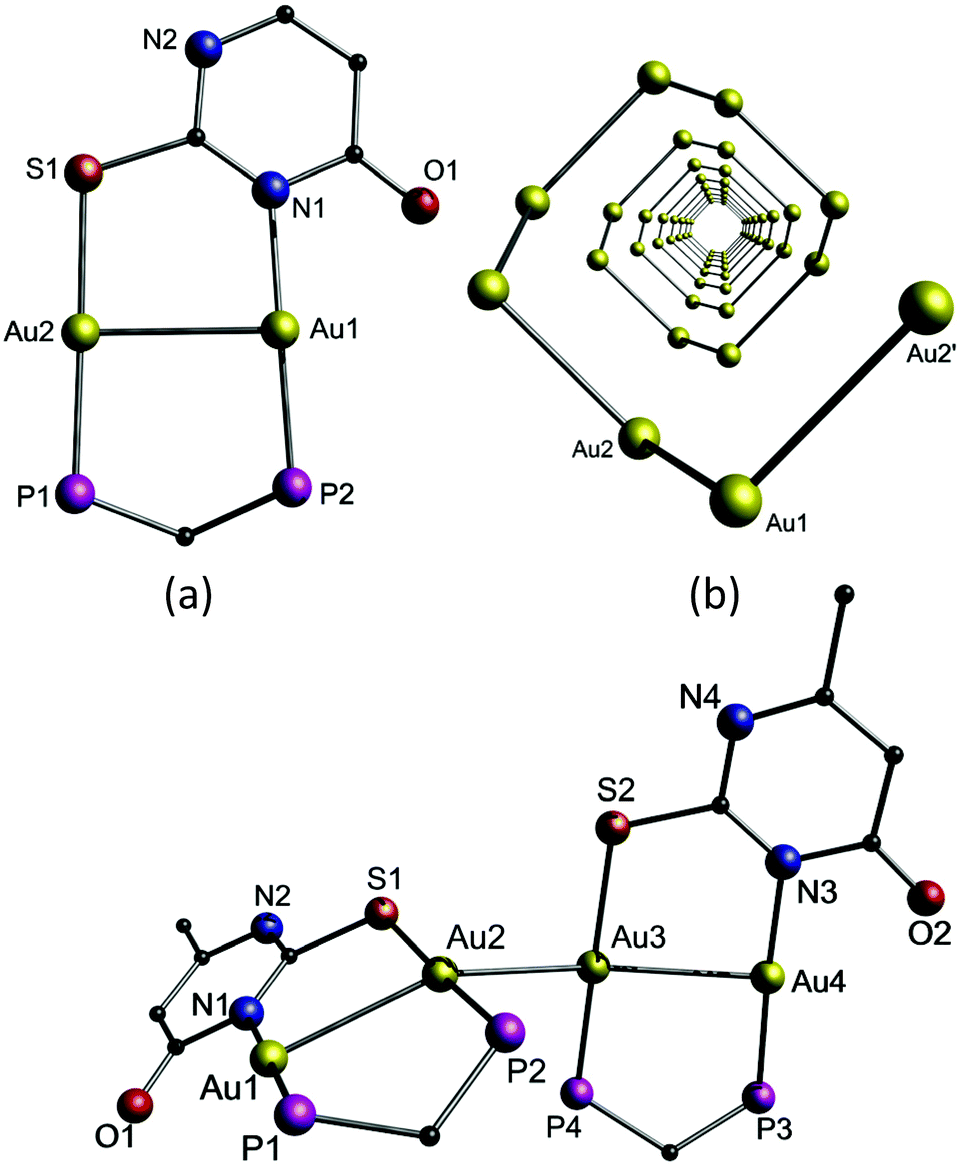 | ||
| Fig. 32 Top: (a) View of cationic 14a with phenyl rings omitted; (b) helical arrangement of the gold ions in 14a with ligands omitted for clarity. Bottom: Perspective view of 15b with phenyl rings omitted.206 | ||
Among the compound class of neutral gold strings, cyclic trinuclear gold complexes represent a special case as they exhibit unique electronic properties.207 Their properties are majorly impacted by intermolecular interactions in the solid state that involve aurophilic contacts between the trimers.208 Balch and co-workers have reported on the synthesis and intermolecular interactions in the polymorphs of [Au3(MeN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) COMe)3] (16a), [Au3(n-PentNCOMe)3] (16b) and [Au3(i-PrNCOMe)3] (16c) in 2005 (Fig. 34).209 The trinuclear gold complexes of the form [Au3(RNCOR′)3] stack in four different arrangements, dependent on the bulkiness of the ligand substituents (R, R′), which retard or enhance intertrimer interactions, as reported by Omary and co-workers (Fig. 33). The stacking arrangements mainly differ regarding the nature of relative disposition of trimers with respect to each other.24
COMe)3] (16a), [Au3(n-PentNCOMe)3] (16b) and [Au3(i-PrNCOMe)3] (16c) in 2005 (Fig. 34).209 The trinuclear gold complexes of the form [Au3(RNCOR′)3] stack in four different arrangements, dependent on the bulkiness of the ligand substituents (R, R′), which retard or enhance intertrimer interactions, as reported by Omary and co-workers (Fig. 33). The stacking arrangements mainly differ regarding the nature of relative disposition of trimers with respect to each other.24
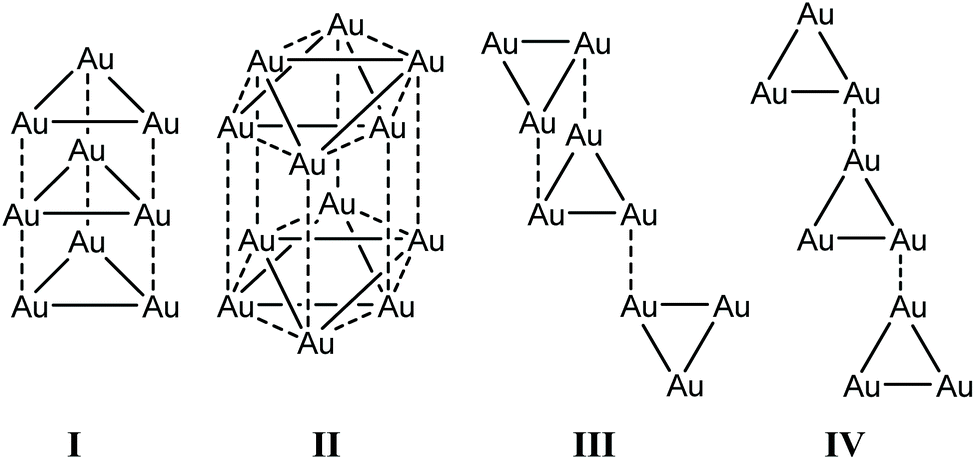 | ||
| Fig. 33 Different stacking arrangements of trimers, in which the dashed lines and solid lines indicate inter and intramolecular Au–Au interactions. I, II, III and IV represent eclipsed, disorder staggered, chair and staircase stacking arrangements.24 | ||
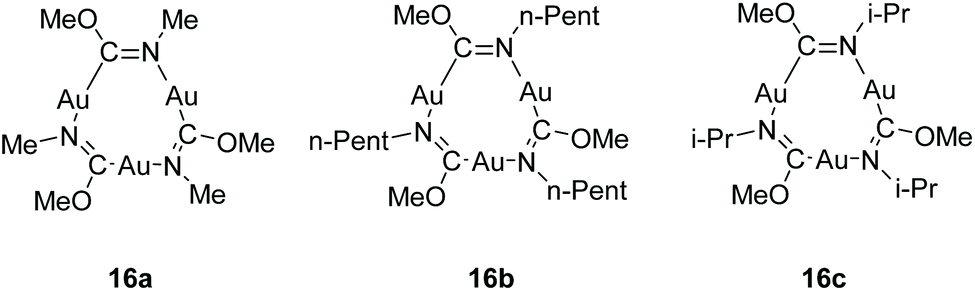 | ||
| Fig. 34 Chemical structure of trinuclear Au(I) carbeniate complexes.209 | ||
16a forms three polymorphs, namely hexagonal, monoclinic and triclinic. The hexagonal polymorph forms the stack arrangements I and II, while the triclinic and monoclinic polymorphs are present in the structural motifs III and IV, respectively. The Au–Au distances of the different polymorphs are summarized in Table 8. The hexagonal polymorphs of 16a, in which extended, linear chains of gold ions are present, show some remarkable photophysical properties. Upon irradiation with near-UV light, a dual emission, consisting of a short-lived (t ≈ 1 ms) emission at 446 nm and a broad long-lived (triexponential, t ≈ 1.4, 4.4 and 31 s) emission at 552 nm, is observed. The yellow emission at 552 nm is readily detected by the human eye even seconds after the irradiation has stopped. Interestingly, if a suitable solvent (chloroform, dichloromethane, toluene, methanol, hexane, water) is dropped onto previously irradiated and still glowing crystals of 16a, a bright burst of yellow light is observed. No overall chemical transformation appears to be involved in this solvoluminescence process. This phenomenon can be reproduced until the sample is entirely dissolved. For comparison, the sterically more demanding derivative [Au3(C6H5CH2NCOMe)3], which consists of separated individual molecules in the solid state, does not show long-lived luminescence behaviour. Consequently, the extended supramolecular aggregation of 16a is believed to be important for the energy storage within the solid and may be involved in the solvoluminescence process. In this context, the recombination of a charge or electron separation may be responsible for the emission that occurs by exposure to the solvent.94
209
| Polymorph of 16a | Intra Au–Au [Å] | Inter Au–Au [Å] | Em [nm] |
|---|---|---|---|
| Hexagonal I | 3.308 | 3.346 | 450 |
| Hexagonal II | 3.280 | 3.384 | 520 |
| Triclinic III | 3.339 | 3.528 | 444 |
| Monoclinic IV | 3.323 | 3.653 | 431 |
Moreover, the unique electronic characteristics of hexagonal 16a are further demonstrated by the successive oxidation with molecular bromine or iodine, yielding in [Au3Xn(MeNCOMe)3] (X = Br, I), where n is 2, 4 or 6.210 A study of the electrochemical oxidation of this compound was presented as well.211 In a 0.1 M N(nBu)4ClO4 dichloromethane solution as the supporting electrolyte, 16a was oxidized at +650 mV, accompanied by the formation of fine, needle-like crystals on the electrode. Not suitable for single crystal XRD measurements, their composition was determined by bulk electrolysis and quartz crystal microbalance techniques to be [Au3(MeNCOMe)3][ClO4]0.34, being the first partially oxidized extended linear chain gold complex. It is assumed that the molecular structure of [Au3(MeNCOMe)3][ClO4]0.34 resembles the hexagonal polymorph of 16a. Chronoamperometric studies confirmed this new phase to be conductive, which offers further insight into the solvoluminescence, energy storage and electron mobility along chains of gold complexes.
The n-pentyl substituted compound 16b crystallizes in an orthorhombic (16b-1) and a triclinic polymorph (16b-2). 16b-1 exhibits a stair case arrangement, in which the intermolecular Au–Au distance between the trimers is 3.618 Å, whereas the intramolecular Au–Au distances are 3.315(3), 3.260(2) and 3.332(2) Å. 16b-2 comprises four independent trimers in the asymmetric unit linked by aurophilic interactions (Au–Au: 3.4614(6), 3.3458(6), 3.6126(6) Å). The shortest Au–Au distance between the asymmetric units is 3.8125(6) Å. Among both polymorphs only 16b-2 is luminescent at room temperature with an emission band at 654 nm (excitation at 326 nm), resulting in a large Stokes shift. This indicates the possible occurrence of a metal-centered excimeric emission due to the contraction of the ground-state aurophilic association.24 The structure of 16c comprises only one trinuclear unit in the asymmetric unit. However, the shortest Au–Au distance in the trimers is 6.417 Å, which is far outside the aurophilic range. Moreover, 16c is not luminescent at ambient temperature, most likely due to the absence of aurophilic interactions. Furthermore, it is suggested that steric effects of the side groups (R, R′) influence the formation of Au–Au interactions and thereby the photophysical properties.
Omary and co-workers have also reported their research on cyclotrimeric gold(I) carbeniates, [Au3(RNCOR′)3] (17a: R = Me, R′ = nBu; 17b: R = nBu, R′ = Me; 17c: R = R′ = nBu and 17d: R = cyclo-pentyl, R′ = Me).24 The crystal structure of [Au3(MeNCOnBu)3] (17a) is exemplarily shown in Fig. 35.
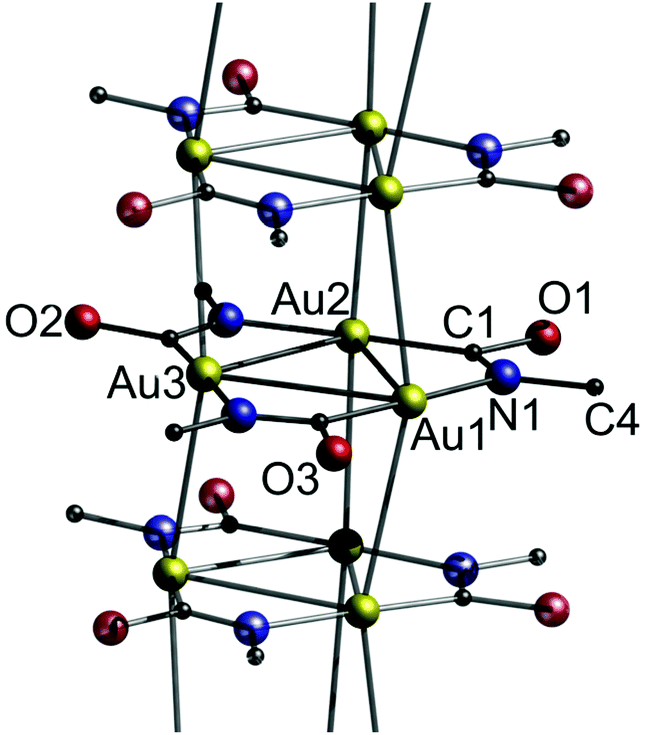 | ||
| Fig. 35 Excerpt of the crystal structure of 17a with n-butyl groups omitted for clarity.24 | ||
Their study revealed that a substitution of R or R′ leads to an alteration in solid-state stacking of the trinuclear units and thereby affecting opto-electronic and electronic properties. Bulky R and R′ groups tend to lie above or below the plane of the trinuclear units, which makes stacking of trinuclear Au units into 1D polymeric chains difficult. Molecular simulations of [Au3(HNCOH)3] reveal that infinitely extended chains of eclipsed trinuclear units with head to tail arrangement tend to have smaller Stokes shifts and lower band gaps and reorganization energies (λ), which are ideal characteristics required for the design of semiconducting materials for molecular electronic devices.
Charge transport properties of organic semiconductors in principle depend on the coupling between the geometric and electronic structures of the respective molecules.212 The degree of chain orders in polymers and solid-state stacking in crystals majorly impact the charge transport ability in the organic molecules.213,214 According to Marcus theory, the electron transfer rate (ket) can be expressed as
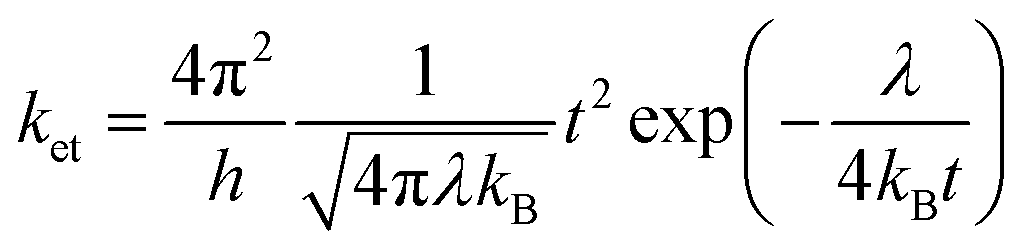 | (2) |
COMe)3]) facilitates strong electronic coupling and results in significant t values.218 The λ value varies with the change in R and R′ groups as reported by Omary and co-workers (Table 9).
| Sample | λ [eV] |
|---|---|
| [Au3(NHCOH)3] |
0.448 |
| [Au3(MeNCOMe)3] |
0.664 |
| [Au3(MeNCOnBu)3] |
0.696 |
The molecular simulations predict that, if extended chains of [Au3(RNCOR′)3] with non-bulky groups can be synthesized, they can probably act as ideal semiconductor materials for fabricating molecular electronic devices.
Conclusions and perspective
The compound class of molecular gold strings is presented in this review. Their unique characteristic is their periodicity, which makes them valuable from synthetic and fundamental points of view. The physical properties of the strings are derived from the distance between adjacent gold ions and the electronic nature of the supporting ligands. The exposure to different external stimuli such as temperature, solvent interaction or mechanical stress alters the Au–Au distance, which goes along with a change in the physical properties. This is primarily demonstrated in the respective photoluminescence characteristics, as the emission energies typically decrease with larger metallic distances. Furthermore, the overall length of the gold strings is an important factor for the electronic structure, as observed e.g. by TRLFS measurements. Studying these structure–property correlations can help researchers to understand the nature of the aurophilicity phenomenon and to further develop potential applications. In this regard, the most interesting features certainly are their luminescence behaviour and electronic properties. In the case of luminescence, the development and application of smart sensor materials, which e.g. are already known for lanthanide-based compounds219,220 or molecular switches, which to date are majorly based on organic compounds,221 are the next logical steps. In addition, by controlling the Au–Au distance of molecular gold strings and utilizing ligands with tailor-made electronic properties, emitters with predictable and tuneable emission wavelengths might be developed and used e.g. for OLED materials,222 as it is already the case for copper and iridium complexes.223–227 Another interesting aspect of molecular gold strings is their energy storage and conductive behaviour, as shown by the Balch, Omary and Ito groups,23,24,211 making them promising candidates for molecular wires. The synthetic approaches towards molecular metal-containing wires and comprehensively understanding their electric characteristics have been in the focus of materials science for decades.9,11,228 As a consequence, addressing these research opportunities is only possible by a multidisciplinary approach involving experimental and theoretical scientists.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This review was written during the SARS-CoV-2 pandemic. We would like to thank all frontline workers and people who keep our society running.The DFG funded Research Training Group (RTG) 2039 (Molecular architecture for fluorescence cell imaging) is acknowledged for financial support.
Notes and references
- Z. Allahyari and A. R. Oganov, npj Comput. Mater., 2020, 6, 55 CrossRef.
- Y. Kopelevich and P. Esquinazi, Adv. Mater., 2007, 19, 4559–4563 CrossRef CAS.
- B. Partoens and F. M. Peeters, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 075404 CrossRef.
- I. Szleifer and R. Yerushalmi-Rozen, Polymer, 2005, 46, 7803–7818 CrossRef CAS.
- N. Todorova, A. J. Makarucha, N. D. M. Hine, A. A. Mostofi and I. Yarovsky, PLoS Comput. Biol., 2013, 9, e1003360 CrossRef.
- T. Nakanishi, W. Schmitt, T. Michinobu, D. G. Kurth and K. Ariga, Chem. Commun., 2005, 5982–5984 RSC.
- R. Nadiv, G. Shachar, S. Peretz-Damari, M. Varenik, I. Levy, M. Buzaglo, E. Ruse and O. Regev, Carbon, 2018, 126, 410–418 CrossRef CAS.
- J. N. Tiwari, R. N. Tiwari and K. S. Kim, Prog. Mater. Sci., 2012, 57, 724–803 CrossRef CAS.
- K. Jang, I. G. Jung, H. J. Nam, D.-Y. Jung and S. U. Son, J. Am. Chem. Soc., 2009, 131, 12046–12047 CrossRef CAS.
- R. Hayoun, D. K. Zhong, A. L. Rheingold and L. H. Doerrer, Inorg. Chem., 2006, 45, 6120–6122 CrossRef CAS.
- I.-W. P. Chen, M.-D. Fu, W.-H. Tseng, J.-Y. Yu, S.-H. Wu, C.-J. Ku, C.-h. Chen and S.-M. Peng, Angew. Chem., Int. Ed., 2006, 45, 5814–5818 CrossRef CAS.
- C.-C. Chiu, M.-C. Cheng, S.-H. Lin, C.-W. Yan, G.-H. Lee, M.-C. Chang, T.-S. Lin and S.-M. Peng, Dalton Trans., 2020, 49, 6635–6643 RSC.
- J. M. Tour, Acc. Chem. Res., 2000, 33, 791–804 CrossRef CAS.
- I. P.-C. Liu, C.-h. Chen and S.-M. Peng, Bull. Jpn Soc. Coord. Chem., 2012, 59, 3–10 CrossRef.
- Q. Liu, M. Xie, X. Chang, S. Cao, C. Zou, W.-F. Fu, C.-M. Che, Y. Chen and W. Lu, Angew. Chem., Int. Ed., 2018, 57, 6279–6283 CrossRef CAS.
- I. R. Whittall, M. G. Humphrey and D. C. R. Hockless, Aust. J. Chem., 1997, 50, 991–998 CrossRef CAS.
- J. Vicente, M. T. Chicote, M. D. Abrisqueta, P. González-Herrero and R. Guerrero, Gold Bull., 1998, 31, 83–87 CrossRef CAS.
- S. J. Hsu, K. M. Hsu, M. K. Leong and I. J. B. Lin, Dalton Trans., 2008, 1924–1931 RSC.
- S. Myllynen and M. Wasberg, Electrochem. Commun., 2009, 11, 1453–1456 CrossRef CAS.
- M. Mitsumi, H. Ueda, K. Furukawa, Y. Ozawa, K. Toriumi and M. Kurmoo, J. Am. Chem. Soc., 2008, 130, 14102–14104 CrossRef CAS.
- A. Guijarro, O. Castillo, A. Calzolari, P. J. S. Miguel, C. J. Gómez-García, R. di Felice and F. Zamora, Inorg. Chem., 2008, 47, 9736–9738 CrossRef CAS.
- M. Mitsumi, H. Goto, S. Umebayashi, Y. Ozawa, M. Kobayashi, T. Yokoyama, H. Tanaka, S.-i. Kuroda and K. Toriumi, Angew. Chem., Int. Ed., 2005, 44, 4164–4168 CrossRef CAS.
- T. Seki, K. Sakurada, M. Muromoto, S. Seki and H. Ito, Chem. – Eur. J., 2016, 22, 1968–1978 CrossRef CAS.
- R. N. McDougald, B. Chilukuri, H. Jia, M. R. Perez, H. Rabaâ, X. Wang, V. N. Nesterov, T. R. Cundari, B. E. Gnade and M. A. Omary, Inorg. Chem., 2014, 53, 7485–7499 CrossRef CAS.
- V. P. Georgiev, P. J. Mohan, D. DeBrincat and J. E. McGrady, Coord. Chem. Rev., 2013, 257, 290–298 CrossRef CAS.
- R. H. Ismayilov, W.-Z. Wang, G.-H. Lee, C.-Y. Yeh, S.-A. Hua, Y. Song, M.-M. Rohmer, M. Bénard and S.-M. Peng, Angew. Chem., Int. Ed., 2011, 50, 2045–2048 CrossRef CAS.
- S. Bestgen, M. T. Gamer, S. Lebedkin, M. M. Kappes and P. W. Roesky, Chem. – Eur. J., 2015, 21, 601–614 CrossRef CAS.
- C. Kaub, S. Lebedkin, S. Bestgen, R. Köppe, M. M. Kappes and P. W. Roesky, Chem. Commun., 2017, 53, 9578–9581 RSC.
- S. Schäfer, M. T. Gamer, S. Lebedkin, F. Weigend, M. M. Kappes and P. W. Roesky, Chem. – Eur. J., 2017, 23, 12198–12209 CrossRef.
- T. P. Seifert, N. D. Knoefel, T. J. Feuerstein, K. Reiter, S. Lebedkin, M. T. Gamer, A. C. Boukis, F. Weigend, M. M. Kappes and P. W. Roesky, Chem. – Eur. J., 2019, 25, 3799–3808 CrossRef CAS.
- N. D. Knöfel, C. Schoo, T. P. Seifert and P. W. Roesky, Dalton Trans., 2020, 49, 1513–1521 RSC.
- G. S. M. Tong, S. C. F. Kui, H.-Y. Chao, N. Zhu and C.-M. Che, Chem. – Eur. J., 2009, 15, 10777–10789 CrossRef CAS.
- D. Li, C.-M. Che, S.-M. Peng, S.-T. Liu, Z.-Y. Zhou and T. C. W. Mak, J. Chem. Soc., Dalton Trans., 1993, 189–194 RSC.
- T. Tanase, R. Otaki, T. Nishida, H. Takenaka, Y. Takemura, B. Kure, T. Nakajima, Y. Kitagawa and T. Tsubomura, Chem. – Eur. J., 2014, 20, 1577–1596 CrossRef CAS.
- L. H. Doerrer, Dalton Trans., 2010, 39, 3543–3553 RSC.
- A. Aliprandi, D. Genovese, M. Mauro and L. D. Cola, Chem. Lett., 2015, 44, 1152–1169 CrossRef CAS.
- H. B. Gray, S. Záliš and A. Vlček, Coord. Chem. Rev., 2017, 345, 297–317 CrossRef CAS.
- M. J. Katz, K. Sakai and D. B. Leznoff, Chem. Soc. Rev., 2008, 37, 1884–1895 RSC.
- S. Sculfort and P. Braunstein, Chem. Soc. Rev., 2011, 40, 2741–2760 RSC.
- M. Stollenz, Chem. – Eur. J., 2019, 25, 4274–4298 CrossRef CAS.
- S.-A. Hua, M.-C. Cheng, C.-h. Chen and S.-M. Peng, Eur. J. Inorg. Chem., 2015, 2015, 2510–2523 CrossRef CAS.
- J. K. Bera and K. R. Dunbar, Angew. Chem., Int. Ed., 2002, 41, 4453–4457 CrossRef CAS.
- B. Lippert, Coord. Chem. Rev., 1999, 182, 263–295 CrossRef.
- J. A. Chipman and J. F. Berry, Chem. Rev., 2020, 120, 2409–2447 CrossRef CAS.
- G. Aromí, Comments Inorg. Chem., 2011, 32, 163–194 CrossRef.
- R. D. Riley, D. A. Dickie, M. A. Land, R. A. Kemp, C. L. B. Macdonald, U. Werner-Zwanziger, K. N. Robertson and J. A. C. Clyburne, Chem. – Eur. J., 2020, 26, 7711–7719 CrossRef CAS.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS.
- V. W.-W. Yam, K. M.-C. Wong and N. Zhu, J. Am. Chem. Soc., 2002, 124, 6506–6507 CrossRef CAS.
- V. G. Andrianov, Y. T. Struchkov and E. R. Rossinskaja, J. Chem. Soc., Chem. Commun., 1973, 338–339 RSC.
- P. G. Jones, Gold Bull., 1981, 14, 102–118 CrossRef CAS.
- M. Melnínk and R. V. Parish, Coord. Chem. Rev., 1986, 70, 157–257 CrossRef.
- J. J. Rehr, E. Zaremba and W. Kohn, Phys. Rev. B: Condens. Matter Mater. Phys., 1975, 12, 2062–2066 CrossRef CAS.
- P. Pyykkö and Y. Zhao, Angew. Chem., Int. Ed. Engl., 1991, 30, 604–605 CrossRef.
- P. Pyykkö and F. Mendizabal, Chem. – Eur. J., 1997, 3, 1458–1465 CrossRef.
- N. Runeberg, M. Schütz and H.-J. Werner, J. Chem. Phys., 1999, 110, 7210–7215 CrossRef CAS.
- P. Pyykkö, Angew. Chem., Int. Ed., 2004, 43, 4412–4456 CrossRef.
- P. Schwerdtfeger, A. E. Bruce and M. R. M. Bruce, J. Am. Chem. Soc., 1998, 120, 6587–6597 CrossRef CAS.
- P. Pyykkö, Chem. Soc. Rev., 2008, 37, 1967–1997 RSC.
- P. Pyykkö, Inorg. Chim. Acta, 2005, 358, 4113–4130 CrossRef.
- G. J. Hutchings, M. Brust and H. Schmidbaur, Chem. Soc. Rev., 2008, 37, 1759–1765 RSC.
- H. Schmidbaur, Gold Bull., 2000, 33, 3–10 CrossRef CAS.
- M. B. Brands, J. Nitsch and C. F. Guerra, Inorg. Chem., 2018, 57, 2603–2608 CrossRef CAS.
- E. Andris, P. C. Andrikopoulos, J. Schulz, J. Turek, A. Růžička, J. Roithová and L. Rulíšek, J. Am. Chem. Soc., 2018, 140, 2316–2325 CrossRef CAS.
- H. Schmidbaur, Gold Bull., 1990, 23, 11–21 CrossRef CAS.
- F. Scherbaum, A. Grohmann, G. Müller and H. Schmidbaur, Angew. Chem., Int. Ed. Engl., 1989, 28, 463–465 CrossRef.
- S. Alvarez, Dalton Trans., 2013, 42, 8617–8636 RSC.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931–1951 RSC.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- M. A. Rawashdeh-Omary, M. A. Omary and H. H. Patterson, J. Am. Chem. Soc., 2000, 122, 10371–10380 CrossRef CAS.
- J. C. Y. Lin, S. S. Tang, C. S. Vasam, W. C. You, T. W. Ho, C. H. Huang, B. J. Sun, C. Y. Huang, C. S. Lee, W. S. Hwang, A. H. H. Chang and I. J. B. Lin, Inorg. Chem., 2008, 47, 2543–2551 CrossRef CAS.
- F. Balzano, A. Cuzzola, P. Diversi, F. Ghiotto and G. Uccello-Barretta, Eur. J. Inorg. Chem., 2007, 2007, 5556–5562 CrossRef.
- Z. Lei, J.-Y. Zhang, Z.-J. Guan and Q.-M. Wang, Chem. Commun., 2017, 53, 10902–10905 RSC.
- H. de la Riva, A. Pintado-Alba, M. Nieuwenhuyzen, C. Hardacre and M. C. Lagunas, Chem. Commun., 2005, 4970–4972 RSC.
- P. K. Mehrotra and R. Hoffmann, Inorg. Chem., 1978, 17, 2187–2189 CrossRef CAS.
- M. Jansen, Angew. Chem., Int. Ed. Engl., 1987, 26, 1098–1110 CrossRef.
- S. Sculfort, P. Croizat, A. Messaoudi, M. Bénard, M.-M. Rohmer, R. Welter and P. Braunstein, Angew. Chem., Int. Ed., 2009, 48, 9663–9667 CrossRef CAS.
- E. J. Fernández, P. G. Jones, A. Laguna, J. M. López-de-Luzuriaga, M. Monge, J. Pérez and M. E. Olmos, Inorg. Chem., 2002, 41, 1056–1063 CrossRef.
- E. M. Gussenhoven, M. M. Olmstead, J. C. Fettinger and A. L. Balch, Inorg. Chem., 2008, 47, 4570–4578 CrossRef CAS.
- L. H. Doerrer, Comments Inorg. Chem., 2008, 29, 93–127 CrossRef CAS.
- M. Kim, T. J. Taylor and F. P. Gabbaï, J. Am. Chem. Soc., 2008, 130, 6332–6333 CrossRef CAS.
- N. Masciocchi, M. Moret, P. Cairati, F. Ragaini and A. Sironi, J. Chem. Soc., Dalton Trans., 1993, 471–475 RSC.
- H. Schmidbaur, S. Cronje, B. Djordjevic and O. Schuster, Chem. Phys., 2005, 311, 151–161 CrossRef CAS.
- P. Pyykkö, Annu. Rev. Phys. Chem., 2012, 63, 45–64 CrossRef.
- X. He and V. W.-W. Yam, Coord. Chem. Rev., 2011, 255, 2111–2123 CrossRef CAS.
- V. W.-W. Yam and E. C.-C. Cheng, Chem. Soc. Rev., 2008, 37, 1806–1813 RSC.
- V. W.-W. Yam and K. K.-W. Lo, Chem. Soc. Rev., 1999, 28, 323–334 RSC.
- J. C. Koziar and D. O. Cowan, Acc. Chem. Res., 1978, 11, 334–341 CrossRef CAS.
- C.-K. Li, X.-X. Lu, K. M.-C. Wong, C.-L. Chan, N. Zhu and V. W.-W. Yam, Inorg. Chem., 2004, 43, 7421–7430 CrossRef CAS.
- S. K. Chastain and W. R. Mason, Inorg. Chem., 1982, 21, 3717–3721 CrossRef CAS.
- M. M. Savas and W. R. Mason, Inorg. Chem., 1987, 26, 301–307 CrossRef CAS.
- A. Vogler and H. Kunkely, Coord. Chem. Rev., 2001, 219–221, 489–507 CrossRef CAS.
- V. W.-W. Yam and E. C.-C. Cheng, in Photochemistry and Photophysics of Coordination Compounds II, ed. V. Balzani and S. Campagna, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, pp. 269–309 Search PubMed.
- V. W.-W. Yam, T.-F. Lai and C.-M. Che, J. Chem. Soc., Dalton Trans., 1990, 3747–3752 RSC.
- J. C. Vickery, M. M. Olmstead, E. Y. Fung and A. L. Balch, Angew. Chem., Int. Ed. Engl., 1997, 36, 1179–1181 CrossRef CAS.
- M. Jin, T. Seki and H. Ito, J. Am. Chem. Soc., 2017, 139, 7452–7455 CrossRef CAS.
- A. L. Balch, Angew. Chem., Int. Ed., 2009, 48, 2641–2644 CrossRef CAS.
- K. M. Anderson, A. E. Goeta and J. W. Steed, Inorg. Chem., 2007, 46, 6444–6451 CrossRef CAS.
- H. Schmidbaur and H. G. Raubenheimer, Angew. Chem., Int. Ed., 2020, 59, 14748–14771 CrossRef CAS.
- A. Wuttke, M. Feldt and R. A. Mata, J. Phys. Chem. A, 2018, 122, 6918–6925 CrossRef CAS.
- S. Ahrland, B. Noren and A. Oskarsson, Inorg. Chem., 1985, 24, 1330–1333 CrossRef CAS.
- M. Strey, C. Döring and P. G. Jones, Z. Naturforsch., B: J. Chem. Sci., 2018, 73, 125 CAS.
- S. Ahrland, K. Dreisch, B. Norén and Å. Oskarsson, Mater. Chem. Phys., 1993, 35, 281–289 CrossRef CAS.
- W. Conzelmann, W. Hiller, J. Strähle and G. M. Sheldrick, Z. Anorg. Allg. Chem., 1984, 512, 169–176 CrossRef CAS.
- J. Vicente, M.-T. Chicote, M.-D. Abrisqueta, R. Guerrero and P. G. Jones, Angew. Chem., Int. Ed. Engl., 1997, 36, 1203–1205 CrossRef CAS.
- S.-L. Zheng, C. L. Nygren, M. Messerschmidt and P. Coppens, Chem. Commun., 2006, 3711–3713 RSC.
- M. Saitoh, A. L. Balch, J. Yuasa, K. Tada, M. Onoda, T. Nakashima and T. Kawai, Langmuir, 2011, 27, 10947–10952 CrossRef CAS.
- J. E. Parks and A. L. Balch, J. Organomet. Chem., 1974, 71, 453–463 CrossRef CAS.
- R. L. White-Morris, M. M. Olmstead, F. Jiang, D. S. Tinti and A. L. Balch, J. Am. Chem. Soc., 2002, 124, 2327–2336 CrossRef CAS.
- D. Rios, D. M. Pham, J. C. Fettinger, M. M. Olmstead and A. L. Balch, Inorg. Chem., 2008, 47, 3442–3451 CrossRef CAS.
- D. Rios, M. M. Olmstead and A. L. Balch, Dalton Trans., 2008, 4157–4164 RSC.
- M. Saitoh, A. L. Balch, J. Yuasa and T. Kawai, Inorg. Chem., 2010, 49, 7129–7134 CrossRef CAS.
- T. A. Engesser, C. Friedmann, A. Martens, D. Kratzert, P. J. Malinowski and I. Krossing, Chem. – Eur. J., 2016, 22, 15085–15094 CrossRef CAS.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS.
- T. A. Engesser, M. R. Lichtenthaler, M. Schleep and I. Krossing, Chem. Soc. Rev., 2016, 45, 789–899 RSC.
- L. M. C. Luong, M. A. Malwitz, V. Moshayedi, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2020, 142, 5689–5701 CrossRef CAS.
- R. L. White-Morris, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2003, 125, 1033–1040 CrossRef CAS.
- M. A. Malwitz, S. H. Lim, R. L. White-Morris, D. M. Pham, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2012, 134, 10885–10893 CrossRef CAS.
- T. H. Kim, Y. W. Shin, J. H. Jung, J. S. Kim and J. Kim, Angew. Chem., Int. Ed., 2008, 47, 685–688 CrossRef CAS.
- R. Li, F.-F. Xu, Z.-L. Gong and Y.-W. Zhong, Inorg. Chem. Front., 2020 10.1039/D0QI00779J.
- R. Y. Liau, H. Ehlich, A. Schier and H. Schmidbaur, Z. Naturforsch., B: J. Chem. Sci., 2002, 57b, 1085–1089 Search PubMed.
- Z. Tang, A. P. Litvinchuk, H.-G. Lee and A. M. Guloy, Inorg. Chem., 1998, 37, 4752–4753 CrossRef CAS.
- N. L. Coker, J. A. Krause Bauer and R. C. Elder, J. Am. Chem. Soc., 2004, 126, 12–13 CrossRef CAS.
- N. L. Coker, C. E. Bedel, J. A. Krause and R. C. Elder, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m319–m321 CrossRef CAS.
- Z. Assefa, B. G. McBurnett, R. J. Staples, J. P. Fackler, B. Assmann, K. Angermaier and H. Schmidbaur, Inorg. Chem., 1995, 34, 75–83 CrossRef CAS.
- N. Aoyagi, Y. Shinha, A. Ikeda-Ohno, Y. Haga, K. Shimojo, N. R. Brooks, A. Izuoka, H. Naganawa, T. Kimura and K. Binnemans, Cryst. Growth Des., 2015, 15, 1422–1429 CrossRef CAS.
- M. I. Jeffrey and P. L. Breuer, Miner. Eng., 2000, 13, 1097–1106 CrossRef CAS.
- R. Y. Wan and J. D. Miller, JOM, 1986, 38, 35–40 CrossRef CAS.
- T. J. Boggon and L. Shapiro, Structure, 2000, 8, R143–R149 CrossRef CAS.
- D. M. Pham, D. Rios, M. M. Olmstead and A. L. Balch, Inorg. Chim. Acta, 2005, 358, 4261–4269 CrossRef CAS.
- A. D. Nicholas, R. M. Bullard, R. D. Pike and H. H. Patterson, Eur. J. Inorg. Chem., 2019, 2019, 956–962 CrossRef CAS.
- M. Stender, M. M. Olmstead, A. L. Balch, D. Rios and S. Attar, Dalton Trans., 2003, 4282–4287 RSC.
- D. B. Leznoff and J. Lefebvre, Gold Bull., 2005, 38, 47–54 CrossRef CAS.
- W. Han, L. Yi, Z.-Q. Liu, W. Gu, S.-P. Yan, P. Cheng, D.-Z. Liao and Z.-H. Jiang, Eur. J. Inorg. Chem., 2004, 2004, 2130–2136 CrossRef.
- J. R. Thompson, J. S. Ovens, V. E. Williams and D. B. Leznoff, Chem. – Eur. J., 2013, 19, 16572–16578 CrossRef CAS.
- J. Qu, W. Gu and X. Liu, J. Coord. Chem., 2008, 61, 618–626 CrossRef CAS.
- F. Baril-Robert, X. Li, M. J. Katz, A. R. Geisheimer, D. B. Leznoff and H. Patterson, Inorg. Chem., 2011, 50, 231–237 CrossRef CAS.
- J. S. Ovens and D. B. Leznoff, Dalton Trans., 2011, 40, 4140–4146 RSC.
- D. B. Leznoff, B.-Y. Xue, B. O. Patrick, V. Sanchez and R. C. Thompson, Chem. Commun., 2001, 259–260 RSC.
- D. B. Leznoff, B.-Y. Xue, R. J. Batchelor, F. W. B. Einstein and B. O. Patrick, Inorg. Chem., 2001, 40, 6026–6034 CrossRef CAS.
- H. Zhang, J. Cai, X.-L. Feng, T. Li, X.-Y. Li and L.-N. Ji, Inorg. Chem. Commun., 2002, 5, 637–641 CrossRef CAS.
- A. Karadağ, A. Aydın, S. Dede, Ş. Tekin, Y. Yanar, B. H. Çadırcı, M. S. Soylu and Ö. Andaç, New J. Chem., 2015, 39, 8136–8152 RSC.
- J. Suárez-Varela, H. Sakiyama, J. Cano and E. Colacio, Dalton Trans., 2007, 249–256 RSC.
- M.-C. Brandys and R. J. Puddephatt, Chem. Commun., 2001, 1280–1281 RSC.
- G.-F. Xu, Z.-Q. Liu, H.-B. Zhou, Y. Guo and D.-Z. Liao, Aust. J. Chem., 2006, 59, 640–646 CrossRef CAS.
- Y. Guo, Z.-Q. Liu, B. Zhao, Y.-H. Feng, G.-F. Xu, S.-P. Yan, P. Cheng, Q.-L. Wang and D.-Z. Liao, CrystEngComm, 2009, 11, 61–66 RSC.
- J. C. Ahern, R. J. Roberts, P. Follansbee, J. McLaughlin, D. B. Leznoff and H. H. Patterson, Inorg. Chem., 2014, 53, 7571–7579 CrossRef CAS.
- R. J. Roberts, J. C. Ahern, H. H. Patterson and D. B. Leznoff, Eur. J. Inorg. Chem., 2016, 2016, 2082–2087 CrossRef CAS.
- R. B. Arthur, A. D. Nicholas, R. J. Roberts, Z. Assefa, D. B. Leznoff and H. H. Patterson, Gold Bull., 2018, 51, 1–10 CrossRef CAS.
- M. L. Brown, J. S. Ovens and D. B. Leznoff, Dalton Trans., 2017, 46, 7169–7180 RSC.
- Q. Liu, M. Xie, X. Chang, Q. Gao, Y. Chen and W. Lu, Chem. Commun., 2018, 54, 12844–12847 RSC.
- Y. Chen, G. Cheng, K. Li, D. P. Shelar, W. Lu and C.-M. Che, Chem. Sci., 2014, 5, 1348–1353 RSC.
- W. C. Kaska, H. A. Mayer, M. R. J. Elsegood, P. N. Horton, M. B. Hursthouse, C. Redshaw and S. M. Humphrey, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, m563–m565 CrossRef CAS.
- G. A. Koutsantonis, G. I. Jenkins, P. A. Schauer, B. Szczepaniak, B. W. Skelton, C. Tan and A. H. White, Organometallics, 2009, 28, 2195–2205 CrossRef CAS.
- A. Bauer, W. Schneider and H. Schmidbaur, Inorg. Chem., 1997, 36, 2225–2226 CrossRef CAS.
- H. Ehlich, A. Schier and H. Schmidbaur, Z. Naturforsch., B: J. Chem. Sci., 2002, 57, 890 CAS.
- M. Böge and J. Heck, Chem. – Eur. J., 2016, 22, 6787–6792 CrossRef.
- N. Mirzadeh, D. W. Drumm, J. Wagler, S. P. Russo and S. Bhargava, Dalton Trans., 2013, 42, 12883–12890 RSC.
- J. D. E. T. Wilton-Ely, H. Ehlich, A. Schier and H. Schmidbaur, Helv. Chim. Acta, 2001, 84, 3216–3232 CrossRef CAS.
- F. Mohr, E. Cerrada and M. Laguna, Organometallics, 2006, 25, 644–648 CrossRef CAS.
- M.-E. Núñez Gaytán, S. Bernès, E. Rodríguez de San Miguel and J. de Gyves, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, m414–m417 CrossRef.
- U. Siemeling, D. Rother, C. Bruhn, H. Fink, T. Weidner, F. Träger, A. Rothenberger, D. Fenske, A. Priebe, J. Maurer and R. Winter, J. Am. Chem. Soc., 2005, 127, 1102–1103 CrossRef CAS.
- D. Parker, P. S. Roy, G. Ferguson and M. M. Hunt, Inorg. Chim. Acta, 1989, 155, 227–230 CrossRef CAS.
- M. Arita, K. Naka, Y. Morisaki and Y. Chujo, Heteroat. Chem., 2012, 23, 16–26 CrossRef CAS.
- M. O. Awaleh, F. Baril-Robert, C. Reber, A. Badia and F. Brisse, Inorg. Chem., 2008, 47, 2964–2974 CrossRef CAS.
- S. Ahrland, B. Aurivillius, K. Dreisch, B. Norén and Å. Oskarsson, Acta Chem. Scand., 1992, 46, 262–265 CrossRef CAS.
- A. J. Moro, B. Rome, E. Aguiló, J. Arcau, R. Puttreddy, K. Rissanen, J. C. Lima and L. Rodríguez, Org. Biomol. Chem., 2015, 13, 2026–2033 RSC.
- S. S. Y. Chui, M. F. Y. Ng and C.-M. Che, Chem. – Eur. J., 2005, 11, 1739–1749 CrossRef CAS.
- X. Lu, M. S. Yavuz, H.-Y. Tuan, B. A. Korgel and Y. Xia, J. Am. Chem. Soc., 2008, 130, 8900–8901 CrossRef CAS.
- H. Imoto, S. Nishiyama, T. Yumura, S. Watase, K. Matsukawa and K. Naka, Dalton Trans., 2017, 46, 8077–8082 RSC.
- R. L. White-Morris, M. Stender, D. S. Tinti, A. L. Balch, D. Rios and S. Attar, Inorg. Chem., 2003, 42, 3237–3244 CrossRef CAS.
- R. L. White-Morris, M. M. Olmstead, A. L. Balch, O. Elbjeirami and M. A. Omary, Inorg. Chem., 2003, 42, 6741–6748 CrossRef CAS.
- H. Ecken, M. M. Olmstead, B. C. Noll, S. Attar, B. Schlyer and A. L. Balch, J. Chem. Soc., Dalton Trans., 1998, 3715–3720 RSC.
- D. V. Toronto, B. Weissbart, D. S. Tinti and A. L. Balch, Inorg. Chem., 1996, 35, 2484–2489 CrossRef CAS.
- B. Weissbart, D. V. Toronto, A. L. Balch and D. S. Tinti, Inorg. Chem., 1996, 35, 2490–2496 CrossRef CAS.
- P. Pyykkö, J. Li and N. Runeberg, Chem. Phys. Lett., 1994, 218, 133–138 CrossRef.
- J. Braese, A. Schinabeck, M. Bodensteiner, H. Yersin, A. Y. Timoshkin and M. Scheer, Chem. – Eur. J., 2018, 24, 10073–10077 CrossRef CAS.
- S. Nath, S. K. Ghosh, S. Kundu, S. Praharaj, S. Panigrahi and T. Pal, J. Nanopart. Res., 2006, 8, 111–116 CrossRef CAS.
- F. Guyon, A. Hameau, A. Khatyr, M. Knorr, H. Amrouche, D. Fortin, P. D. Harvey, C. Strohmann, A. L. Ndiaye, V. Huch, M. Veith and N. Avarvari, Inorg. Chem., 2008, 47, 7483–7492 CrossRef CAS.
- P. J. Sadler, J. Rheumatol., Suppl., 1982, 8, 71–78 CAS.
- C. Lavenn, N. Guillou, M. Monge, D. Podbevšek, G. Ledoux, A. Fateeva and A. Demessence, Chem. Commun., 2016, 52, 9063–9066 RSC.
- R. G. Pearson, J. Chem. Educ., 1968, 45, 581 CrossRef CAS.
- R. G. Pearson, J. Chem. Educ., 1968, 45, 643 CrossRef CAS.
- D. Datta, Inorg. Chem., 1992, 31, 2797–2800 CrossRef CAS.
- R. G. Pearson, Inorg. Chim. Acta, 1995, 240, 93–98 CrossRef CAS.
- T. A. Rodina, E. V. Korneeva, O. N. Antzutkin and A. V. Ivanov, Spectrochim. Acta, Part A, 2015, 149, 881–888 CrossRef CAS.
- Y.-A. Lee, J. E. McGarrah, R. J. Lachicotte and R. Eisenberg, J. Am. Chem. Soc., 2002, 124, 10662–10663 CrossRef CAS.
- W. E. van Zyl, J. M. López-de-Luzuriaga, A. A. Mohamed, R. J. Staples and J. P. Fackler, Inorg. Chem., 2002, 41, 4579–4589 CrossRef CAS.
- E. V. Korneeva, T. A. Rodina, A. V. Ivanov, A. V. Gerasimenko and A. C. Larsson, Russ. J. Coord. Chem., 2014, 40, 748–756 CrossRef CAS.
- S. L. Lawton, W. J. Rohrbaugh and G. T. Kokotailo, Inorg. Chem., 1972, 11, 2227–2233 CrossRef CAS.
- J. Grote, B. Neumann, H.-G. Stammler and N. W. Mitzel, Dalton Trans., 2018, 47, 4701–4706 RSC.
- M. L. Gallego, A. Guijarro, O. Castillo, T. Parella, R. Mas-Balleste and F. Zamora, CrystEngComm, 2010, 12, 2332–2334 RSC.
- H.-J. You, C.-S. Fang, J.-L. Lin, S.-S. Sun and C. W. Liu, Inorg. Chem., 2010, 49, 7641–7643 CrossRef CAS.
- C. Latouche, Y.-C. Lee, J.-H. Liao, E. Furet, J.-Y. Saillard, C. W. Liu and A. Boucekkine, Inorg. Chem., 2012, 51, 11851–11859 CrossRef CAS.
- R. J. Roberts, D. Le and D. B. Leznoff, Chem. Commun., 2015, 51, 14299–14302 RSC.
- M.-R. Azani, O. Castillo, M. L. Gallego, T. Parella, G. Aullón, O. Crespo, A. Laguna, S. Alvarez, R. Mas-Ballesté and F. Zamora, Chem. – Eur. J., 2012, 18, 9965–9976 CrossRef CAS.
- S. Naeem, S. A. Serapian, A. Toscani, A. J. P. White, G. Hogarth and J. D. E. T. Wilton-Ely, Inorg. Chem., 2014, 53, 2404–2416 CrossRef CAS.
- S. S. Tang, C.-P. Chang, I. J. B. Lin, L.-S. Liou and J.-C. Wang, Inorg. Chem., 1997, 36, 2294–2300 CrossRef CAS.
- P. Bishop, P. Marsh, A. K. Brisdon, B. J. Brisdon and M. F. Mahon, J. Chem. Soc., Dalton Trans., 1998, 675–682 RSC.
- A. C. Lane, C. L. Barnes, M. V. Vollmer and J. R. Walensky, Inorganics, 2014, 2, 540–551 CrossRef CAS.
- D. Paliwoda, P. Wawrzyniak and A. Katrusiak, J. Phys. Chem. Lett., 2014, 5, 2182–2188 CrossRef CAS.
- B.-C. Tzeng and A. Chao, Chem. – Eur. J., 2015, 21, 2083–2089 CrossRef CAS.
- T. Seki, K. Ida, H. Sato, S. Aono, S. Sakaki and H. Ito, Chem. – Eur. J., 2020, 26, 735–744 CrossRef CAS.
- H. Ito, M. Muromoto, S. Kurenuma, S. Ishizaka, N. Kitamura, H. Sato and T. Seki, Nat. Commun., 2013, 4, 2009 CrossRef.
- T. Seki, K. Sakurada and H. Ito, Angew. Chem., Int. Ed., 2013, 52, 12828–12832 CrossRef CAS.
- T. Seki, Y. Takamatsu and H. Ito, J. Am. Chem. Soc., 2016, 138, 6252–6260 CrossRef CAS.
- Y.-A. Lee and R. Eisenberg, J. Am. Chem. Soc., 2003, 125, 7778–7779 CrossRef CAS.
- A. Hayashi, M. M. Olmstead, S. Attar and A. L. Balch, J. Am. Chem. Soc., 2002, 124, 5791–5795 CrossRef CAS.
- L. D. Earl, J. K. Nagle and M. O. Wolf, Inorg. Chem., 2014, 53, 7106–7117 CrossRef CAS.
- R. L. White-Morris, M. M. Olmstead, S. Attar and A. L. Balch, Inorg. Chem., 2005, 44, 5021–5029 CrossRef CAS.
- J. C. Vickery and A. L. Balch, Inorg. Chem., 1997, 36, 5978–5983 CrossRef CAS.
- K. Winkler, M. Wysocka-Żołopa, K. Rećko, L. Dobrzyński, J. C. Vickery and A. L. Balch, Inorg. Chem., 2009, 48, 1551–1558 CrossRef CAS.
- J.-L. Brédas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971–5004 CrossRef.
- J.-L. Brédas, J. Cornil, D. Beljonne, D. A. dos Santos and Z. Shuai, Acc. Chem. Res., 1999, 32, 267–276 CrossRef.
- S. T. Bromley, M. Mas-Torrent, P. Hadley and C. Rovira, J. Am. Chem. Soc., 2004, 126, 6544–6545 CrossRef CAS.
- M. C. R. Delgado, K. R. Pigg, D. A. da Silva Filho, N. E. Gruhn, Y. Sakamoto, T. Suzuki, R. M. Osuna, J. Casado, V. Hernández, J. T. L. Navarrete, N. G. Martinelli, J. Cornil, R. S. Sánchez-Carrera, V. Coropceanu and J.-L. Brédas, J. Am. Chem. Soc., 2009, 131, 1502–1512 CrossRef CAS.
- C.-H. Li, C.-H. Huang and M.-Y. Kuo, Phys. Chem. Chem. Phys., 2011, 13, 11148–11155 RSC.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS.
- L. Zhu, V. Coropceanu, Y. Yi, B. Chilukuri, T. R. Cundari and J.-L. Brédas, J. Phys. Chem. Lett., 2013, 4, 2186–2189 CrossRef CAS.
- D. Yao, Y. Wang, P. Li, K. Müller-Buschbaum and H. Li, Opt. Mater., 2019, 96, 109371 CrossRef CAS.
- T. Wehner, J. Heck, C. Feldmann and K. Müller-Buschbaum, Chem. – Eur. J., 2019, 25, 16630–16638 CrossRef CAS.
- M. Natali and S. Giordani, Chem. Soc. Rev., 2012, 41, 4010–4029 RSC.
- M.-C. Tang, A. K.-W. Chan, M.-Y. Chan and V. W.-W. Yam, in Photoluminescent Materials and Electroluminescent Devices, ed. N. Armaroli and H. J. Bolink, Springer International Publishing, Cham, 2017, pp. 67–109 Search PubMed.
- C. Bizzarri, E. Spuling, D. M. Knoll, D. Volz and S. Bräse, Coord. Chem. Rev., 2018, 373, 49–82 CrossRef CAS.
- Z. Liu, J. Qiu, F. Wei, J. Wang, X. Liu, M. G. Helander, S. Rodney, Z. Wang, Z. Bian, Z. Lu, M. E. Thompson and C. Huang, Chem. Mater., 2014, 26, 2368–2373 CrossRef CAS.
- L. P. Ravaro, K. P. S. Zanoni and A. S. S. de Camargo, Energy Rep., 2020, 6, 37–45 CrossRef.
- I. Omae, Coord. Chem. Rev., 2016, 310, 154–169 CrossRef CAS.
- R. Hamze, J. L. Peltier, D. Sylvinson, M. Jung, J. Cardenas, R. Haiges, M. Soleilhavoup, R. Jazzar, P. I. Djurovich, G. Bertrand and M. E. Thompson, Science, 2019, 363, 601 CrossRef CAS.
- D. M. Adams, L. Brus, C. E. D. Chidsey, S. Creager, C. Creutz, C. R. Kagan, P. V. Kamat, M. Lieberman, S. Lindsay, R. A. Marcus, R. M. Metzger, M. E. Michel-Beyerle, J. R. Miller, M. D. Newton, D. R. Rolison, O. Sankey, K. S. Schanze, J. Yardley and X. Zhu, J. Phys. Chem. B, 2003, 107, 6668–6697 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |