
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThrombo-tag, an in vivo formed nanotracer for the detection of thrombi in mice by fast pre-targeted molecular imaging†
José M.
Adrover‡
 a,
Juan
Pellico‡
bc,
Irene
Fernández-Barahona
de,
Sandra
Martín-Salamanca
a,
Jesús
Ruiz-Cabello
cefg,
Andrés
Hidalgo
*a and
Fernando
Herranz
*cd
a,
Juan
Pellico‡
bc,
Irene
Fernández-Barahona
de,
Sandra
Martín-Salamanca
a,
Jesús
Ruiz-Cabello
cefg,
Andrés
Hidalgo
*a and
Fernando
Herranz
*cd
aArea of Cell and Developmental Biology, Fundación Centro Nacional de Investigaciones Cardiovasculares Carlos III (CNIC), 28029 Madrid, Spain. E-mail: ahidalgo@cnic.es
bSchool of Biomedical Engineering & Imaging Sciences, King's College London, St. Thomas’ Hospital, London, SE1 7EH, UK
cCIBER de Enfermedades Respiratorias (CIBERES), 28029 Madrid, Spain
dNanoMedMol, Instituto de Química Médica, Consejo Superior de Investigaciones Científicas (IQM-CSIC), 28006 Madrid, Spain. E-mail: fherranz@iqm.csic.es
eFacultad de Farmacia, Universidad Complutense de Madrid, 28040 Madrid, Spain
fCenter for Cooperative Research in Biomaterials (CIC biomaGUNE), Basque Research and Technology Alliance (BRTA), 2014 Donostia-San Sebastián, Spain
gIKERBASQUE, Basque Foundation for Science, Bilbao, Spain
First published on 8th October 2020
Abstract
Radioisotope-labelled nanoparticles permit novel applications in molecular imaging, while recent developments in imaging have enabled direct visualization of biological processes. While this holds true for pathological processes that are stable in time, such as cancer, imaging approaches are limited for phenomena that take place in the range of minutes, such as thrombotic events. Here, we take advantage of bioorthogonal chemistry to demonstrate the concept of nanoparticle-based fast pre-targeted imaging. Using a newly designed nanoparticle that targets platelets we show the applicability of this approach developing thrombo-tag, an in vivo produced nanoparticle that labels thrombi. We show that thrombo-tag allows specific labelling of platelets that accumulate in the injured pulmonary vasculature, or that aggregate in brains of mice suffering thrombotic processes. The fast kinetics and high specificity features of thrombo-tag may critically expand the application of molecular imaging to the most prevalent and debilitating diseases in the clinics.
Introduction
On average, every 40 seconds a new case of stroke arises in the US alone, making thrombo-inflammatory disease (which includes ischemic heart disease and stroke) the single most common underlying cause of death worldwide.1 Stroke is a devastating disease with both physical and long-term disability consequences that illustrate the dramatic consequences of thrombotic events. Furthermore, current COVID-19 pandemic points towards multiorganic thrombi formation as one of the main reasons for its potentially devastating consequences.2 Obtaining information on the size and nature of individual patients’ thrombi is a long-sought goal that would be desirable for clinicians to correctly set thrombolytic drug dosage and to predict efficacy of a treatment.3Thrombus detection in human patients by computed tomography (CT) or magnetic resonance imaging (MRI) is currently performed indirectly, by visualizing the lack of blood flow instead of the thrombus itself.4 This translates into two main bedside limitations: the inability to detect scattered or non-occlusive thrombi, and the impossibility of directly assessing the size of the thrombus. This is of importance in many clinical settings including stroke or deep vein thrombosis.5 Thus, developing new modalities for direct assessment of thrombi in tissues is an urgent biomedical need. Molecular imaging can be one of these new modalities.6,7 The remote detection and quantification of a biological process at the cellular and molecular levels is a powerful diagnostic tool that dependent on a chemical probe providing the signal and a targeted vector to the chosen biomarker. We have previously reported the use of 68Ga core-doped iron oxide nanoparticles ([68Ga]Ga-IONP) as a versatile tool for molecular imaging.8 These [68Ga]Ga-IONP consist of a small iron oxide core, about 3 nm in size, doped with 68Ga isotopes in the crystal structure and coated by a thick organic layer of citrate molecules. They can be used as a new positron emission tomography (PET) platform and also for multimodal PET/MRI. In this sense the small iron oxide core has proven to be an excellent “chelator” for 68Ga isotopes in terms of radiolabeling efficiency and stability. We have used this nanotracer for tumor detection,8in vivo neutrophil labeling,9 and for improved in vivo MRI signal.10 Bioorthogonal chemistry, on the other hand, allows for the rapid and selective in vivo conjugation of two compounds that have been previously modified with the corresponding complementary groups. This unparalleled selectivity, even in the presence of the complex mixture of the biological milieu (e.g. plasma), has boosted the use of this type of chemistry. Since the pioneering work by Bertozzi et al.11 this type of chemistry has been used for the in vivo modification of cells,12 DNA or RNA13 and antibodies.14 Among the different types of bioorthogonal reactions,15 tetrazine ligation is the most advantageous for imaging purposes due to its fast kinetics and the diversity of commercially available building blocks incorporating the tetrazine (TZ) and the trans-cyclooctene (TCO) moieties. Bioorthogonal chemistry has further allowed for the introduction of pretargeted imaging (PreI). This consists of a two-step process where the biomolecule of interest is previously modified with one of the chemical groups participating in the bioorthogonal reaction. In PreI this biomolecule is typically an antibody. This modified antibody is then injected for its complete biodistribution and, usually after 24–48 hours, the imaging probe is injected. This imaging probe has been previously modified with the complementary group to that grafted into the antibody. The use of PreI has been mainly centered in cancer diagnosis, starting with the work by Robillard et al.16 where it was clearly demonstrated the advantages of a delay between the biomolecule and imaging probe injections for tumor detection. Recently we have shown how PreI can also allow for unparalleled detection of atherosclerosis.17 This approach has two key advantages for nuclear imaging techniques:18 firstly, it allows using short-lived radioisotopes with biomolecules that require long biodistribution times, thereby reducing the dose the patient receives compared to the use of long-lived radiosiotopes. Secondly, the excellent selectivity of the reaction reduces off-target accumulation of the probe by improving signal-to-noise ratios. However, if we consider clinical scenarios of pathologies in which rapid diagnosis is needed, traditional PreI is not an optimal choice since it normally takes more than 24 hours for the full process to be completed. This timescale is not an issue for cancer detection, the pathology for which PreI was originally designed.16,19 However, fast detection is critical to be clinically useful in the context of acute thrombotic events (e.g., stroke patients), in which it must be implemented for much shorter timescales.
Here, we introduce fast pretargeted imaging (f-PreI), where the two chemical components of the bioorthogonal reaction are rapidly mixed and co-injected such that the accumulation of the biomolecule and the bioorthogonal reaction occur simultaneously in vivo. We demonstrate that f-PreI can be used for fast detection of biological processes without the need of a long delay between the two injections. We take advantage of f-PreI to develop thrombo-tag, an in vivo generated probe for non-invasive detection of thrombus formation by PET in only minutes, and validates its pre-clinical applicability in murine models of ischemia/reperfusion and acute lung injury. Fig. 1 shows the experimental design followed for rapid non-invasive detection of thrombi in vivo. The key aspects in PreI are the biomolecule, the imaging probe and the delay between the two injections. The biomolecule chosen to label thrombi was an anti-CD41 antibody, an IgG targeting integrin alpha chain IIb on the platelet membrane of platelets that accumulates in thrombi. The anti-CD41 antibody was prepared for the tetrazine ligation by covalent immobilization of the TCO group (using just 200 μg of the antibody), targeting the free amines of the protein and controlling the stoichiometry of the reaction to minimize the amount of TCO bound to the antibody (Fig. 1a). We then prepared the nanotracer as previously described,17 by covalent binding of the TZ moiety on the surface of the 68Ga core-doped iron oxide nanoparticles (Fig. 1b).
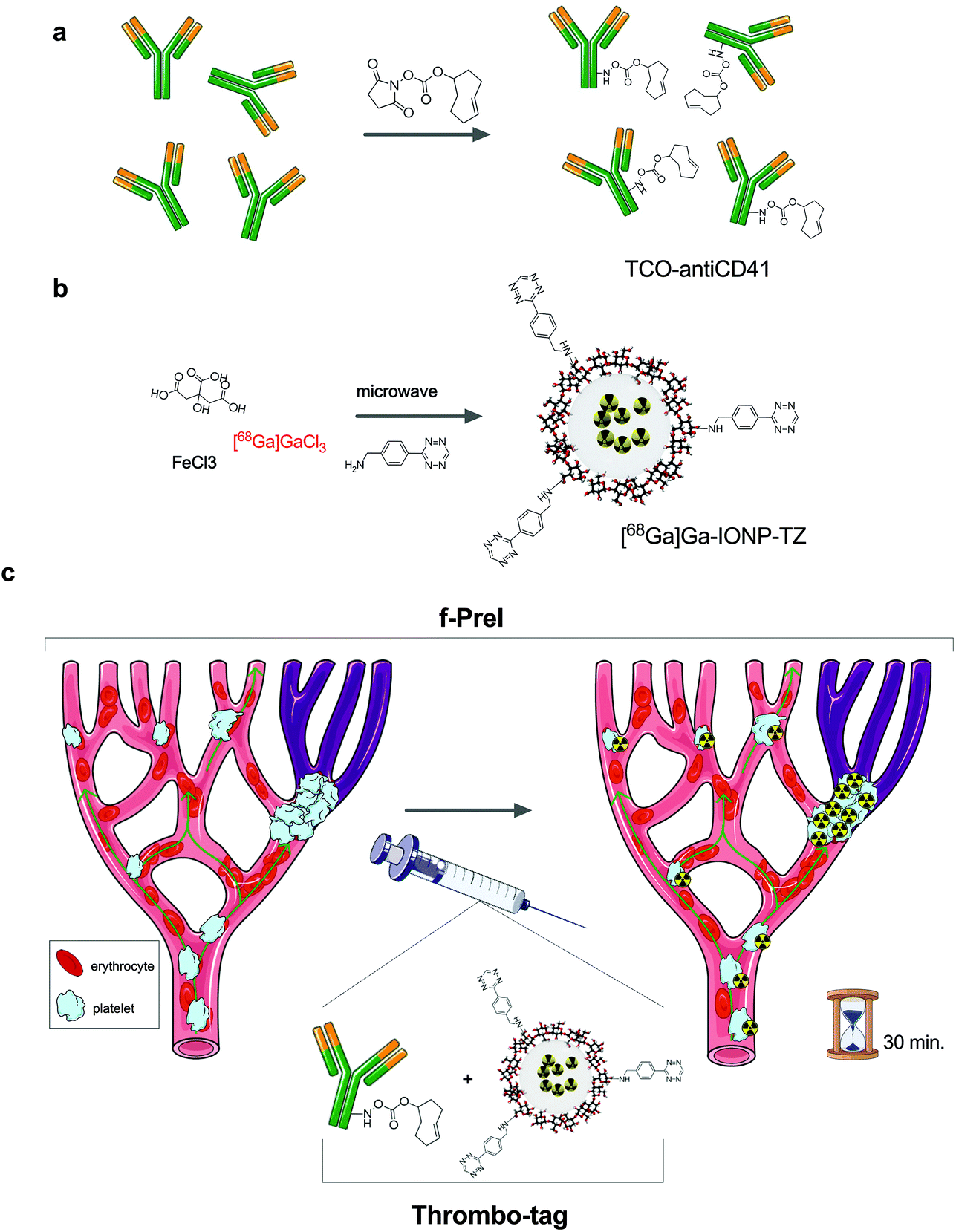 | ||
| Fig. 1 Experimental design for the in vivo detection of thrombi by fast pretargeted imaging; (a) modification of antiCD41 with transcyclooctene (TCO) groups; (b) synthesis of nanoparticles modified with tetrazine (TZ), [68Ga]Ga-IONP-TZ; (c) in vivo use of thrombo-tag for the detection of platelets thrombi. | ||
Results and discussion
Contrary to previous pretargeted experiments, time was a critical variable here. Due to the need for rapid imaging, we prepared in advance the thrombo-tag building blocks so that subsequent administration would allow both, TCO-antiCD41 and [68Ga]Ga-IONP-TZ, to react in vivo after i.v. injection. In this sense, thrombo-tag is a unique imaging probe that only exists in vivo, once the components have been co-injected (Fig. 1c) with images dynamically obtained up to 60 minutes after injection. We synthetized tetrazine-functionalized iron oxide nanoparticles doped with 68Ga as previously described.17 Briefly, we mixed precursors (FeCl3 and 68GaCl3) with sodium citrate, as a coating, in a microwave vial with water. Hydrazine hydrate was added just before the microwave protocol began. After that, we purified the [68Ga]Ga-IONP nanoparticles by gel chromatography. The citrate-coated iron oxide nanoparticles doped in the core with 68Ga ([68Ga]Ga-IONP) were then functionalized with the tetrazine groups by forming amides with free carboxylic groups on the surface, which made the [68Ga]Ga-IONP functionalized with tetrazine ([68Ga]Ga-IONP-TZ, Fig. 2a). The STEM images for [68Ga]Ga-IONP-TZ (Fig. 2b) show small spherical nanoparticles with a core size of 2.9 ± 0.9 nm that were well dispersed. | ||
| Fig. 2 (a) [68Ga]Ga-IONP-TZ structure; (b) scanning transmission electron microscopy images for [68Ga]Ga-IONP-TZ, scale bars are 10 nm (left) and 20 nm (right); (c) Z-average hydrodynamic size values for [68Ga]Ga-IONP and [68Ga]Ga-IONP-TZ measured by dynamic light scattering (N = 3); (d) zeta potential values for [68Ga]Ga-IONP and [68Ga]Ga-IONP-TZ; (e) hydrodynamic size profiles for [68Ga]Ga-IONP, [68Ga]Ga-IONP-TZ and [68Ga]Ga-IONP-antiCD41. | ||
The Z-average (the intensity-weighted mean hydrodynamic size of the collection of particles in the ensemble) was measured by dynamic light scattering (DLS) showing an average size of 7.6 ± 0.3 nm for [68Ga]Ga-IONP and 18.3 ± 0.7 nm for [68Ga]Ga-IONP-TZ (Fig. 2c). This small increase in size was expected due to the hydrophobic nature of the tetrazine groups.
We quantified with a TCO-dye the amount of TZ in the sample that reacts with the [68Ga]Ga-IONP-TZ. The conjugation conditions rendered 675 nmol of tetrazine per mmol Fe, maintaining the hydrophilic character of the nanoparticle surface but with enough tetrazine groups to undergo the bioorthogonal reaction.17 Zeta potential measurements further confirmed the surface modification due to the incorporation of tetrazine with a change in the value from −36 mV to −14 mV, due to the formation of amides with many of the free carboxylic groups (Fig. 2d).
To demonstrate the feasibility of the bioorthogonal reaction in vitro, we studied the coupling between TCO-antiCD41 and the [68Ga]Ga-IONP-TZ. We controlled the reaction by fluorescent labeling of TCO-antiCD41 with aliquots of the reaction that are analyzed at different timepoints. After the reaction was completed, we measured the hydrodynamic size (Fig. 2e). We found that after binding of the anti-CD41 antibody there was a large increase in the hydrodynamic size of the nanoparticle, some in vitro cross-linking was observed as expected; the typical size of an IgG is 14.5 nm × 8.5 nm × 4.0 nm, an important aspect that further justified our strategy, as explained below.
To confirm that the anti-CD41 antibody was bound to the surface of the [68Ga]Ga-IONP, we purified the mixture by gel chromatography and measured the fluorescent signal. Since we labeled the anti-CD41 antibody with Alexa647 dye, any fluorescent signal from [68Ga]Ga-IONP was a confirmation of the presence of the antibody on the surface. Fig. 3a shows the normalized fluorescent signal for [68Ga]Ga-IONP-TZ, the dye-labeled antibody and [68Ga]Ga-IONP after reaction with the antibody. There was a fluorescent signal in the nanoparticle that corresponds to the fluorescent signal initially presented in the antibody, with no signal in the initial nanoparticles, confirming the successful bioorthogonal reaction. We studied the kinetics of the reaction between the TCO-anti-CD41 and [68Ga]Ga-IONP-TZ (Fig. 3b) to form the thrombo-tag. The fluorescence signal shows a rapid and reproducible reaction, with 75% of the reaction completed after 10 minutes.
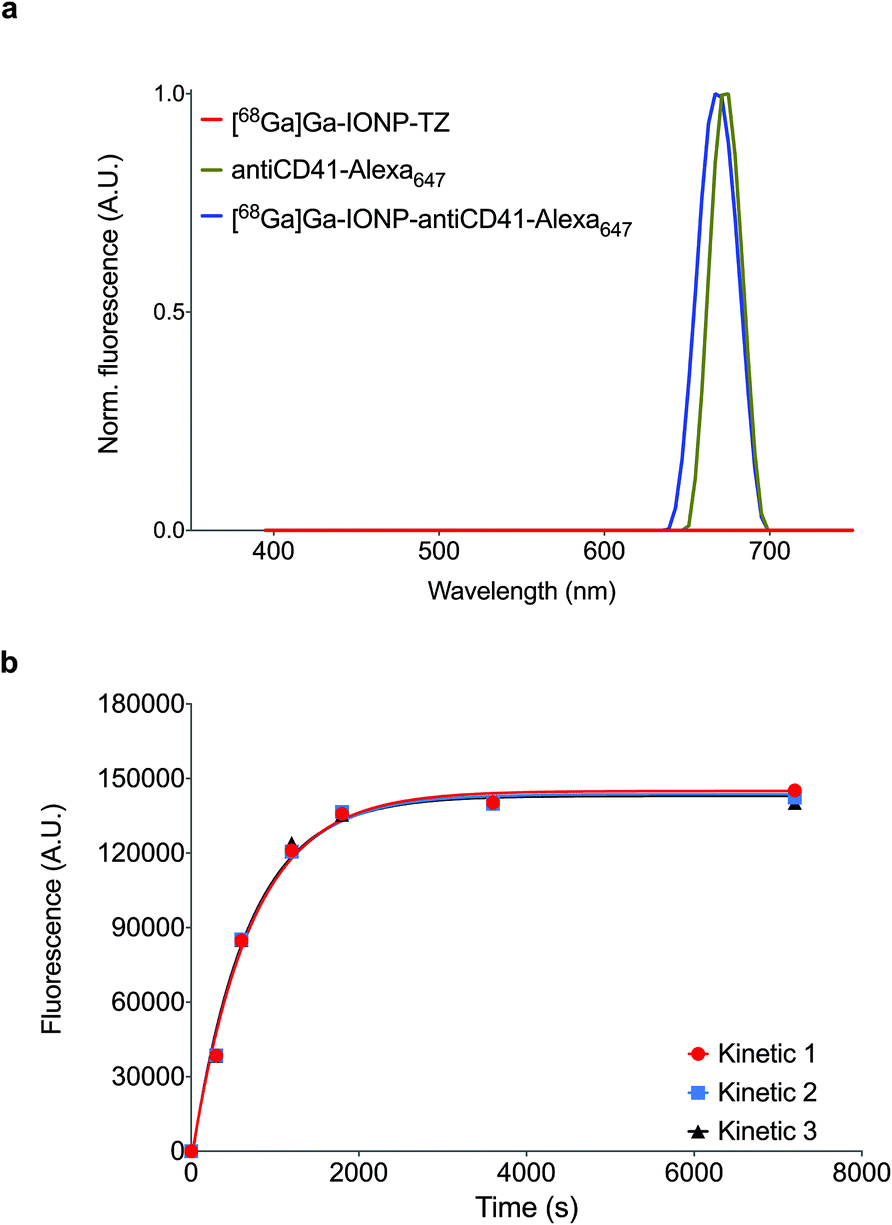 | ||
| Fig. 3 (a) Normalized fluorescence spectra for [68Ga]Ga-IONP-TZ, antiCD41-Alexa647 and [68Ga]Ga-IONP-antiCD41-Alexa647; (b) reaction rates for [68Ga]Ga-IONP-TZ and TCO-antiCD41 tetrazine ligation (N = 3). | ||
To test the specificity of the thrombo-tag nanotracer, we first used a transfusion-related acute lung injury (TRALI) animal model.20 In this model, LPS-primed Balb/c mice are challenged with an antibody against the major histocompatibility complex (MHC-I), leading to neutrophil activation and extensive deposition of activated platelets in the affected lungs, which is a hallmark of the disease (Fig. 4a).21Ex vivo nanotracer biodistribution analyses in organs 1 h after injection of the tracer, expressed as the percentage of injected dose per gram of lung tissue (%ID g−1), showed a marked increase in nanotracer after TRALI induction (Fig. 4b, Fig. S1 and S3†).
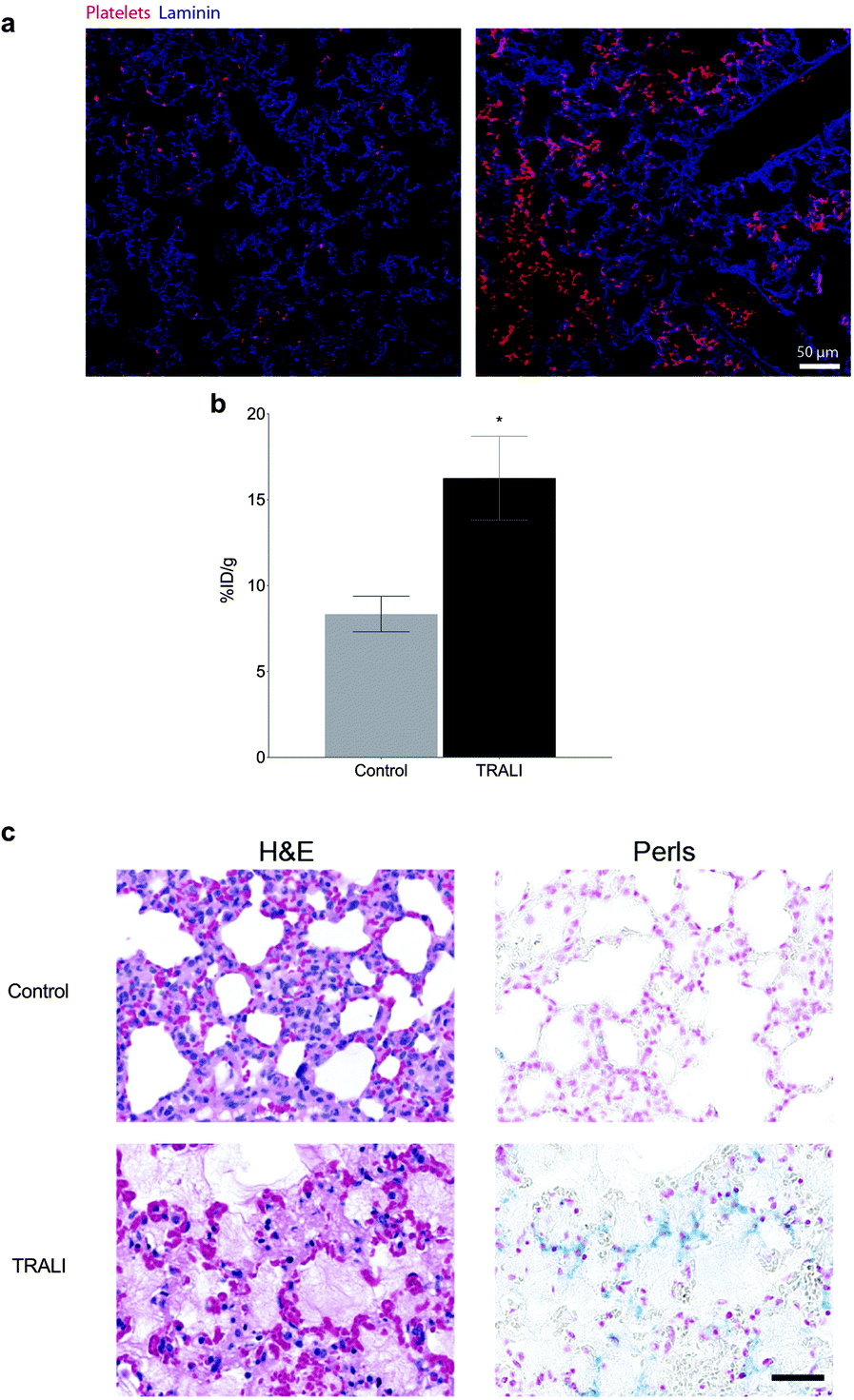 | ||
| Fig. 4 Thrombo-tag in acute lung injury. (a) Immunofluorescence staining of lungs sections from control (left) and TRALI-subjected mice (right) showing platelet deposition in TRALI (in red). (b) Biodistribution of thrombo-tag in the lungs in mice subject to acute lung injury (TRALI) or control, expressed as the percentage of injected dose per gram of lung tissue (%ID g−1). n = 3 mice per condition. (c) Histological sections of mouse lungs stained for hematoxylin and eosin (H&E, left) or Perls Prussian blue (right) to mark the thrombo-tag nanoparticles. Representative image of n = 3 mice per condition. Bars show mean ± s.e.m. * p < 0.05 as determined by unpaired two-tailed t-test analysis. | ||
Consistently, when we stained the lungs with Perls Prussian blue, which reacts with the iron ions of the IONP, we found increase in platelet-bound nanoparticles in the lungs of mice subjected to TRALI, relative to control mice (Fig. 4c). Importantly, the method allowed us to detect traces of platelets deposited in muscle and kidney, with a trend also in the heart and brain of treated mice (Fig. S1†). Thus, thrombo-tag was able to specifically detect platelets immediately after binding to a target organ in the context of acute thrombo-inflammatory disease, and additionally identified platelet deposition in other potentially affected organs.
We next sought to test the nanotracer in a mouse model of acute myocardial infarction (AMI), induced by occlusion of the left anterior descending (LAD) coronary artery, that leads to overt inflammation and death of the cardiac tissue.22 In this model, mice typically recover rapidly and most survive the procedure for at least 24 hours.22 Importantly, however, our previous studies demonstrated that mice whose neutrophils are deficient in CXCR4 (referred to as CXCR4ΔN mice) are extremely susceptible to death upon cardiac ischemia (Fig. 5a), with most mutant mice perishing minutes after the ischemic insult.22
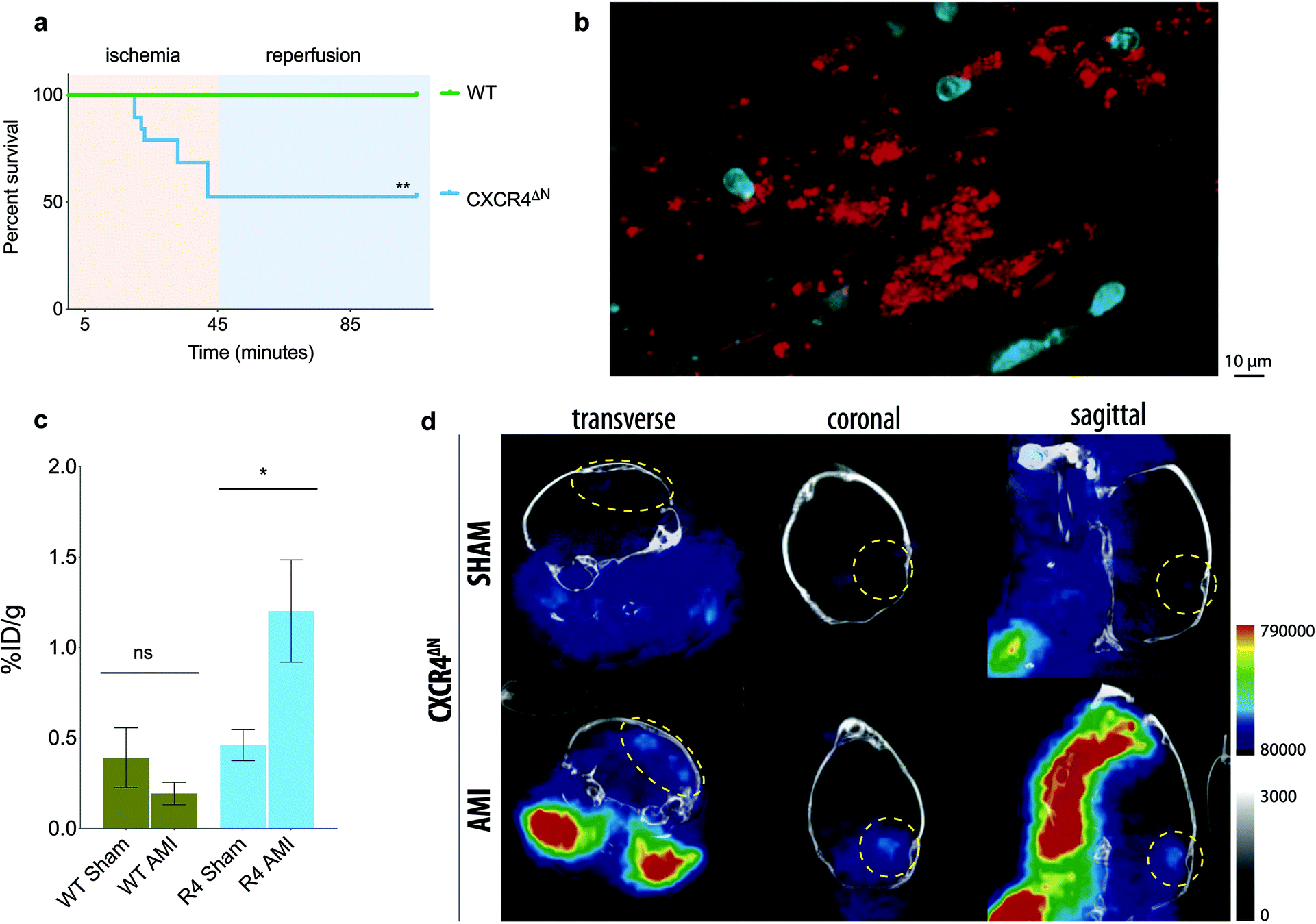 | ||
| Fig. 5 Thrombo-tag detects thrombi secondary to acute myocardial infarction in vivo. (a) Survival of WT and CXCR4ΔN mice upon AMI induction. (b) Intravital images of the cremaster muscle microvasculature of CXCR4ΔN mice under ischemia/reperfusion injury shows disseminated intravascular platelet deposition. Representative of n = 3 imaged mice. (c) Biodistribution of thrombo-tag in the brains of WT and CXCR4ΔN mice subjected to acute myocardial infarction (AMI) or sham-operated, expressed as the percentage of injected dose per gram of tissue (%ID g−1). n = 3–5 mice per condition. (d) Representative PET/CT image of the CXCR4ΔN mice quantified in (c). The bars show mean ± s.e.m. * p < 0.05; ** p < 0.01 as determined by one-way ANOVA with the Sidak's multiple comparisons test (c) or the Mantel-Coxt Log-rank test (a). | ||
Given the rapid lethality, we hypothesized that this poor survival might be caused by rapid thrombi formation and secondary cerebral ischemia. This contention was supported by the rapid appearance of disseminated intravascular microthrombi in these mice upon ischemia/reperfusion injury, as assessed by intravital microscopy of the cremaster microvasculature (Fig. 5b).
We analyzed the biodistribution of the thrombo-tag nanotracer 1 h post-injection of the tracer in WT or CXCR4ΔN mice, comparing sham-operated mice with mice subject to ischemia. While no significant differences in CD41 signal distribution was found between the sham and AMI groups in WT mice (which are normally protected from death; Fig. S2a†), the brains of CXCR4ΔN mice subjected to AMI showed consistent increases of thrombo-tag levels, and we found an increasing trend for the heart and lungs (Fig. 5c and Fig. S2b†). Confirming this observation, we detected signal in the PET/CT images of the brains of CXCR4ΔN mice subjected to AMI, but not in SHAM-operated mice (Fig. 5d). This was consistent with our prediction that CXCR4ΔN mice succumbed to acute brain stroke secondary to the AMI injury. The thrombo-tag signal was also visible in the carotids after AMI injury (Fig. 5d), a phenomenon most likely due to the deposition of platelets in the vasculature, this was also found in the cremasteric venules by intravital microscopy (Fig. 5b).
To verify the nature of the signal, we serially cut the brains of CXCR4ΔN mice subjected to AMI and adjacent sections were stained with hematoxylin/eosin and Perl Prussian blue staining to detect iron accumulation due to the thrombo-tag uptake. We confirmed our PET/CT imaging data since thrombi were present in the brains of CXCR4ΔN mice. In addition, Prussian blue staining, which labels nanoparticles, was found specifically in thrombi (Fig. 6). These data show that the thrombo-tag nanotracer specifically binds to platelets in vivo, and that it enables accurate visualization and identification of thrombi in vivo despite the rapid kinetics of this model.
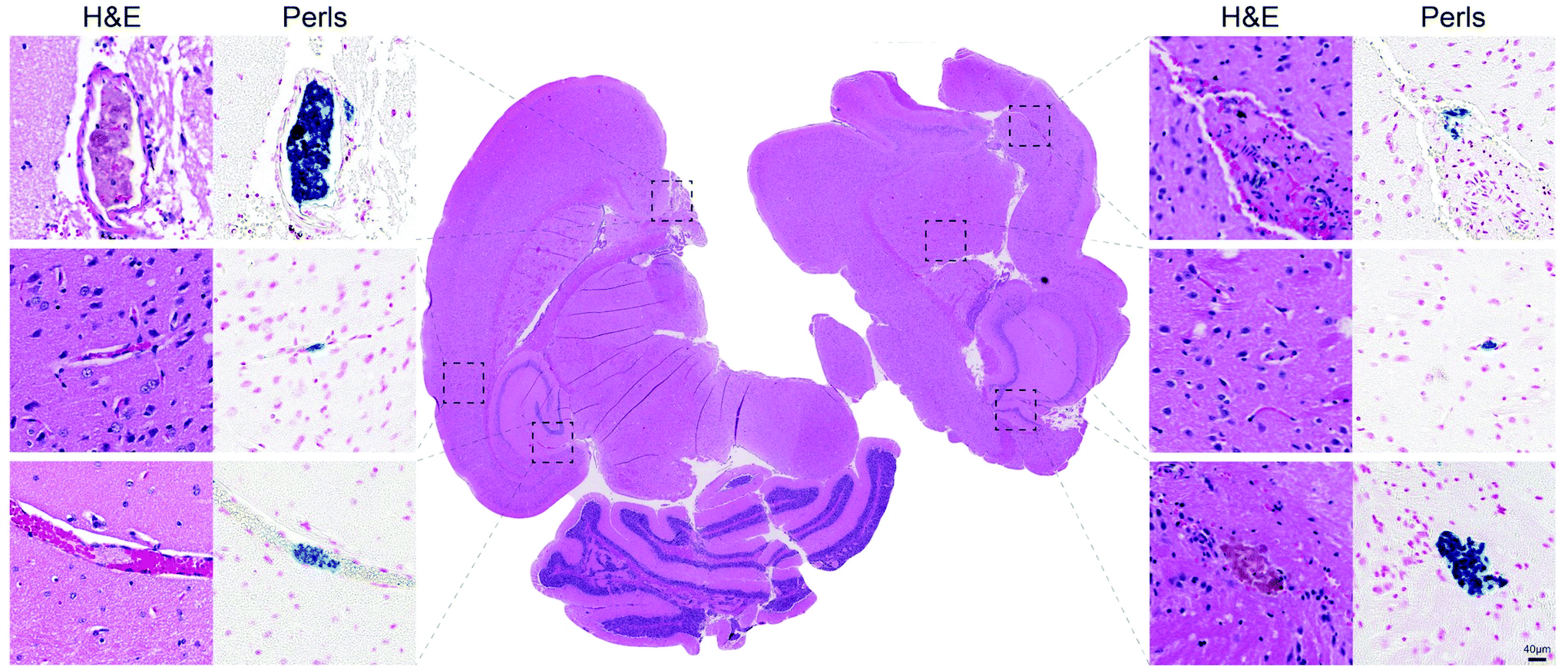 | ||
| Fig. 6 Thrombo-tag co-localization with brain thrombi. Serial sections of brains from AMI-induced CXCR4ΔN mice (quantified in Fig. 4c) stained for H&E (left columns and center) and Perls Prussian blue (right columns) confirm the colocalization of thrombi with thrombo-tag nanoparticles. Representative of n = 3 mice. | ||
Conclusions
[68Ga]Ga-IONP is a versatile nanotracer combining the tunable physicochemical properties of iron oxide nanoparticles with the unparalleled sensitivity of PET imaging. The incorporation of 68Ga isotope in the iron oxide core allows multiple applications without the drawbacks associated with the use of chelators, as in the case of transmetallation.23 Using a microwave-driven synthesis, we obtained extremely small 68Ga doped iron oxide nanoparticles with a hydrodynamic size of 7.6 nm, excellent colloidal stability and −36 mV of zeta potential. [68Ga]Ga-IONP consist on an extremely small iron oxide core and a very thick organic layer composed of citrate molecules. This makes it difficult to visualize them using traditional transmission electron microscopy techniques. However, the use of scanning transmission electron microscopy facilitates to obtain information on the size and shape of the core. In this way, we could confirm the formation of extremely small iron oxide cores of about 2.9 nm, hence a thick organic layer of 4.7 nm. In terms of radiolabeling, we obtained the [68Ga]Ga-IONP-TZ with an activity of 188 MBq that permits the injection of small amounts of IONP for normal image acquisitions (1.5 mg kg−1).We tested the covalent addition of the tetrazine moiety using a number of techniques: zeta potential measurement confirmed the modification of the citrate surface showing an increase from −36 mV to −14 mV, the hydrodynamic size also increased due to the in vitro tetrazine ligation with the antibody and, finally, the in vitro tetrazine ligation between the [68Ga]Ga-IONP-TZ and the dye-labelled TCO-antiCD41 confirmed the reaction and therefore the successful covalent modification. The hydrodynamic size for the in vitro conjugation of the antibody and the nanoparticles indicated a large extent of cross-linking. This was expected since there are many TZ molecules on the IONP able to bind a large amount of the anti-CD41 antibody, which can subsequently react with more [68Ga]Ga-IONP-TZ (cross-linking). Contrary to this, in vivo dilution of the compounds and the instantaneous binding of the antibody to platelets, exposing the constant region and blocking the variable region, minimizes the cross-linking observed in vitro. Finally, we quantified the amount of TZ in the [68Ga]Ga-IONP and of TCO in the antibody by fluorescence. Nanoparticles show 675 nmol of TZ per mmol of Fe as previously demonstrated.17
The efficacy of thrombo-tag platelet targeting was evaluated in a well-established model of transfusion-related acute lung injury (TRALI20). In this model platelets are known to accumulate in the lung, where they play a key role in the pathophysiology.21,24Ex vivo biodistribution in a gammacounter depicted significant differences in lung uptake between control and TRALI-induced mice. These results were further confirmed by histology with the IONP clearly stained with Prussian Blue. We also studied the applicability of thrombo-tag biodistribution in the context of cardiac ischemia/reperfusion, in a genetic background (CXCR4ΔN) prone to very rapid and highly penetrant death after ischemia, for reasons that were previously unknown.22 Remarkably, thrombo-tag allowed us to identify differential signal in the brain when comparing sham or wild-type animals with ischemic CXCR4ΔN mice. The high accumulation in the brain appeared as clearly defined spots instead of as disseminated signal through the organ, a behavior that is consistent with the possibility that premature death in CXCR4ΔN mice is caused by large thrombi affecting critical organs. These defined spots are clearly observed in the histology of the brain, with a remarkable correlation between the presence of platelets and thrombo-tag uptake. Other organs potentially affected by thrombosis (lungs and heart; Fig. S2 and S4†) might help explain the dramatic mortality rates in these mice. These findings in the context of disseminated thrombosis in the lungs, or remote thrombus formation in the brain, validate what, to our knowledge, is one of the most efficient non-invasive imaging methods reported to identify thrombus to date, at least in small rodents. Because in both models platelet aggregation on vessels or formation of thrombi occurs within minutes of the initiating insult (anti-MHCI antibody or ischemia), thrombo-tag demonstrates robust applicability for events with very fast kinetics, which are a hallmark of thromboinflammatory events. Direct radiolabeling of the antibody could be considered an alternative approach to thrombo-tag; however, this has several limitations. First, the antibody must be modified twice in vitro, first for the covalent addition of a chelator and then radiolabeled with the appropriate radioisotope. This would increase the risk of variable region modification, thereby reducing the in vivo activity of the antibody. Secondly, due to the possibility of multifunctionalization of nanoparticles, our results open up the possibility of therapies targeting thrombi, an approach impossible to develop with traditional probes.
Experimental section
Antibody functionalization with trans-cyclooctene (Ab-TCO)
AntiCD41 antibody (200 μg) is added to an Eppendorf tube and volume adjusted with PBS 1× to a final volume of 0.5 ml. The pH of the solution was adjusted to 8.9–9.1 with 90 μl of Na2CO3 0.1 M. Then, 10 μl of TCO-NHS (1 mg ml−1) was added and the reaction was kept at room temperature for 60 min in the vortex. Afterwards, the reaction was purified by ultra-filtration through 100 kDa cut off filters and dissolved in PBS 1× to reach 0.5 ml.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 350g through Amicon 30 kDa centrifugal filters for 4 min to remove excess reagents. The nanoparticles were resuspended in 1.5 mL of HEPES buffer 0.5 M, pH = 8, and 1 mg of Benzylamino tetrazine hydrochloride dissolved in 50 μL DMSO was added to the solution. The mixture was maintained at r.t. for 60 min with stirring. Finally, another ultrafiltration step was performed to eliminate unreacted tetrazine. The nanoparticles were resuspended in saline solution, yielding 188 MBq (measured 120 min after starting reaction) of [68Ga]Ga-IONP-TZ, a radiolabeling yield of 98.9%.
350g through Amicon 30 kDa centrifugal filters for 4 min to remove excess reagents. The nanoparticles were resuspended in 1.5 mL of HEPES buffer 0.5 M, pH = 8, and 1 mg of Benzylamino tetrazine hydrochloride dissolved in 50 μL DMSO was added to the solution. The mixture was maintained at r.t. for 60 min with stirring. Finally, another ultrafiltration step was performed to eliminate unreacted tetrazine. The nanoparticles were resuspended in saline solution, yielding 188 MBq (measured 120 min after starting reaction) of [68Ga]Ga-IONP-TZ, a radiolabeling yield of 98.9%.
Quantification of TZ in [68Ga]Ga-IONP-TZ
To calculate the amount of TZ per IONP after conjugation, fluorescent probe Cy5-TCO (Click Chemistry Tools) was used. A calibration curve was computed by measuring fluorescence signal of different Cy5-TCO concentrations (8, 6, 4, 2, 0.5 and 0.1 μM) at λ = 725 nm. Then, 250 μl (0.917 mg ml−1 Fe) of [68Ga]Ga-IONP-TZ dissolved in saline solution was mixed with Cy5 TCO to reach a final concentration of 300 μM. The resulting solution was vortexed for 6 h at r.t. Once this step was completed, sample was purified by ultrafiltration with Amicon® 100 K. The final product was resuspended in saline solution and fluorescence signal measured at λ = 725 nm.In vitro bioorthogonal reaction
To perform the in vitro bioorthogonal reaction, 50 μg of Alexa 647-NHS ester fluorophore was added in the conjugation step of the antiCD41 antibody to the TCO molecule. Once conjugation was complete, 200 μl of IONP-TZ (2 mg ml−1) and 200 μl of antiCD41-A647-TCO (0.4 mg ml−1 antiCD41) were mixed and allowed to react under vigorous stirring. Aliquots were taken at different times (0 min, 5 min, 10 min, 20 min, 30 min, 60 min and 120 min). Samples were purified through ultrafiltration 300 kDa filters to eliminate unreacted antiCD41-A647-TCO and posteriorly, fluorescence signal at 665 nm was measured.Animals and ethics
All experiments were performed in 7 to 14-week-old male C57BL/6 (acute myocardial infraction model) or Balb/c (acute lund injury model) mice kept in a specific pathogen-free facility at Centro Nacional de Investigaciones Cardiovasculares (CNIC) under a 12 h light/12 h dark schedule (lights on at 7 am, off at 7 pm), with water and chow available ad libitum. For acute myocardial infraction, we also analysed CXCR4ΔN mutants in the C57BL/6 background (lacking CXCR4 specifically in neutrophils, described in ref. 22). All experimental procedures were approved by the Animal Care and Ethics Committee of CNIC and Madrid regional authorities (PROEX277/16 and PROEX101/18).Acute lung injury (TRALI) model
A two-event model of transfusion-related ALI was adopted for our studies as described.25 Male Balb/c mice were injected intraperitoneally with 0.1 mg kg−1 LPS. 24 hours later, mice received an intravenous injection of 0.5 mg kg−1 anti-H2d (clone 34-1-2s; BioXcell) antibody.Acute myocardial infarction (AMI) model
Male 8 to 12-week-old C57BL/6 (WT or CXCR4ΔN) mice were subjected to 45 min occlusion of the left anterior descending (LAD) coronary artery followed by 1 h reperfusion (for survival experiments). The I/R procedure was performed as previously described.26 Briefly, fully anesthetized animals were intubated and temperature controlled throughout the experiment at 36.5°. Thoracotomy was then performed and the LAD was ligated with a nylon 8/0 monofilament suture. At this point, mice were ready for PET/CT imaging. For survival analysis, mice were kept ligated for 45 min. At the end of the ischemia, the chest was closed, and animals were kept with 100% O2 and analgesized with buprenorphine (subcutaneous injection, 0.1 mg kg−1).Intravital microscopy of the cremaster muscle upon ischemia/reperfusion injury
Mouse cremaster were prepared as previously described.22 Upon exteriorization of the muscle we intravenously injected fluorescent antibodies to label cellular populations. Ly6G-APC (clone 1A8; BioXcell) for neutrophils and CD41-PE (eBioscience) for platelets. Ischemia was achieved by occlusion of the incoming and outgoing vessels by clamping the tissue connecting the muscle and the animal's body with a 15 mm Micro Serrefine clamp (Fine Science Tools, Heidelberg, Germany) for 45 minutes. Reperfusion was achieved by removal of the clamp.PET/CT imaging
In vivo PET/CT imaging in mice was performed with a nanoPET/CT small-animal imaging system (Mediso Medical Imaging Systems, Budapest, Hungary). List-mode PET data acquisition commenced immediately after injection of a bolus of 5.5 ± 2 MBq of thrombo-tag through the tail vein and continued for 60 minutes. At the end of PET, microCT was performed for attenuation correction and anatomic reference. The dynamic PET images in a 105 × 105 matrix (frame rates: 2 × 15 min, 2 × 30 min, 2 × 60 min) were reconstructed using a Tera-Tomo 3D iterative algorithm. Images were acquired and reconstructed with proprietary Nucline software (Mediso, Budapest, Hungary). Images were analyzed using Amide.Biodistribution quantification
Biodistribution was conducted in a Wizard 1470 gammacounter (PerkinElmer). Mice were euthanized 1 h after injection of the probe. Deaths of animals were assured by cervical dislocation and the blood extracted by cardiac puncture. Then, organs of interest were extracted and counted in the gammacounter for 1 min each. Readings were decay corrected presented as the percentage injected dose per gram (%ID g−1).Statistical methods
Unless otherwise indicated, data are represented as mean values ± s.e.m. Paired or unpaired two-tailed t-test was used when two groups were compared, and comparison of more than two datasets was done using one-way analysis of variance (ANOVA) with Turkey's posttest. The specific test applied in a figure is stated in the figure legend. Where applicable, normality was estimated using the D'Agostino–Pearson or Shapiro–Wilk normality test. Log-rank analysis was used for Kaplan–Meier survival curves. All statistical analyses were performed using Prism v.7 (GraphPad Software). A P value below 0.05 was considered statistically significant. * p < 0.05; ** p < 0.01.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Spanish Ministry of Science, grants number (SAF2016-79593-P, SAF2017-84494-C2-R and RED2018-102469-T), and from the Gobierno Vasco, Dpto. Industria, Innovación, Comercio y Turismo under the ELKARTEK Program (Grant No. KK-2019/bmG19). JR-C received funding from the BBVA Foundation (Ayudas a Equipos de investigación científica Biomedicina 2018).The study was also supported by Intramural grants from the Severo Ochoa program (IGP-SO), a grant from Fundació La Marató de TV3 (120/C/2015-20153032), grant SAF2015-65607-R from Ministerio de Ciencia e Innovacion (MICINN) with cofunding from Fondo Europeo de Desarrollo Regional, grant RTI2018-095497-B-I00 from MICINN, and HR17_00527 from Fundación La Caixa (to A. H.), and fellowship BES-2013-065550 from MICINN (to J. M. A.). The CNIC is supported by the MICINN and the Pro-CNIC Foundation, and is a Severo Ochoa Center of Excellence (MICINN award SEV-2015-0505). CIC biomaGUNE is supported by the Maria de Maeztu Units of Excellence Program from the Spanish State Research Agency – Grant No. MDM-2017-0720.
The authors wish to thank the staff at the Advance Imaging Unit in the Spanish Cardiovascular Research Centre for their help in the imaging studies. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI). We thank Dr. M. Eugenio Vázquez for cover design.
Notes and references
- E. J. Benjamin, P. Muntner, A. Alonso, M. S. Bittencourt, C. W. Callaway, A. P. Carson, A. M. Chamberlain, A. R. Chang, S. Cheng, S. R. Das, F. N. Delling, L. Djousse, M. S. V. Elkind, J. F. Ferguson, M. Fornage, L. C. Jordan, S. S. Khan, B. M. Kissela, K. L. Knutson, T. W. Kwan, D. T. Lackland, T. T. Lewis, J. H. Lichtman, C. T. Longenecker, M. S. Loop, P. L. Lutsey, S. S. Martin, K. Matsushita, A. E. Moran, M. E. Mussolino, M. O'Flaherty, A. Pandey, A. M. Perak, W. D. Rosamond, G. A. Roth, U. K. A. Sampson, G. M. Satou, E. B. Schroeder, S. H. Shah, N. L. Spartano, A. Stokes, D. L. Tirschwell, C. W. Tsao, M. P. Turakhia, L. B. VanWagner, J. T. Wilkins, S. S. Wong and S. S. Virani, Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association, 2019, vol. 139 Search PubMed.
- B. Bikdeli, M. V. Madhavan, D. Jimenez, T. Chuich, I. Dreyfus, E. Driggin, C. Der Nigoghossian, W. Ageno, M. Madjid, Y. Guo, L. V. Tang, Y. Hu, J. Giri, M. Cushman, I. Quéré, E. P. Dimakakos, C. M. Gibson, G. Lippi, E. J. Favaloro, J. Fareed, J. A. Caprini, A. J. Tafur, J. R. Burton, D. P. Francese, E. Y. Wang, A. Falanga, C. McLintock, B. J. Hunt, A. C. Spyropoulos, G. D. Barnes, J. W. Eikelboom, I. Weinberg, S. Schulman, M. Carrier, G. Piazza, J. A. Beckman, P. G. Steg, G. W. Stone, S. Rosenkranz, S. Z. Goldhaber, S. A. Parikh, M. Monreal, H. M. Krumholz, S. V. Konstantinides, J. I. Weitz and G. Y. H. Lip, J. Am. Coll. Cardiol., 2020, 75 DOI:10.1016/j.jacc.2020.04.031.
- J. Kim, J. E. Park, M. Nahrendorf and D. E. Kim, J. Stroke, 2016, 18, 286–296 CrossRef.
- G. G. Gasparian, N. Sanossian, M. S. Shiroishi and D. S. Liebeskind, Int. J. Stroke, 2015, 10, 298–305 CrossRef.
- T. Peisker, B. Koznar, I. Stetkarova and P. Widimsky, Trends Cardiovasc. Med., 2017, 27, 59–66 CrossRef.
- K. L. Ciesienski and P. Caravan, Curr. Cardiovasc. Imaging Rep., 2011, 4, 77–84 CrossRef.
- M. R. Dweck, E. Aikawa, D. E. Newby, J. M. Tarkin, J. H. F. Rudd, J. Narula and Z. A. Fayad, Circ. Res., 2016, 119, 330–340 CrossRef CAS.
- J. Pellico, J. Ruiz-Cabello, M. Saiz-Alía, G. del Rosario, S. Caja, M. Montoya, L. Fernández de Manuel, M. P. Morales, L. Gutiérrez, B. Galiana, J. A. Enríquez and F. Herranz, Contrast Media Mol. Imaging, 2016, 11, 203–210 CrossRef CAS.
- J. Pellico, A. V. Lechuga-Vieco, E. Almarza, A. Hidalgo, C. Mesa-Nuñez, I. Fernández-Barahona, J. A. Quintana, J. Bueren, J. A. Enríquez, J. Ruiz-Cabello and F. Herranz, Sci. Rep., 2017, 7, 13242 CrossRef.
- I. Fernández-Barahona, L. Gutiérrez, S. Veintemillas-Verdaguer, J. Pellico, M. del P. Morales, M. Catala, M. A. del Pozo, J. Ruiz-Cabello and F. Herranz, ACS Omega, 2019, 4, 2719–2727 CrossRef.
- J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13–21 CrossRef CAS.
- D. H. Dube, J. A. Prescher, C. N. Quang and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4819–4824 CrossRef CAS.
- M. Tera and N. W. Luedtke, Bioconjugate Chem., 2019, 30, 2991–2997 CrossRef CAS.
- H. Merten, F. Brandl, A. Plückthun and U. Zangemeister-Wittke, Bioconjugate Chem., 2015, 26, 2176–2185 CrossRef CAS.
- N. K. Devaraj, ACS Cent. Sci., 2018, 4, 952–959 CrossRef CAS.
- R. Rossin, P. Renart Verkerk, S. M. van den Bosch, R. C. M. Vulders, I. Verel, J. Lub and M. S. Robillard, Angew. Chem., Int. Ed., 2010, 49, 3375–3378 CrossRef CAS.
- J. Pellico, I. Fernández-Barahona, M. Benito, Á. Gaitán-Simón, L. Gutiérrez, J. Ruiz-Cabello and F. Herranz, Nanomedicine, 2019, 17, 26–35 CrossRef CAS.
- M. Altai, R. Membreno, B. Cook, V. Tolmachev and B. M. Zeglis, J. Nucl. Med., 2017, 58, 1553–1559 CrossRef CAS.
- B. M. Zeglis, K. K. Sevak, T. Reiner, P. Mohindra, S. D. Carlin, P. Zanzonico, R. Weissleder and J. S. Lewis, J. Nucl. Med., 2013, 54, 1389–1396 CrossRef CAS.
- M. R. Looney, B. M. Gilliss and M. A. Matthay, Curr. Opin. Hematol., 2010, 17, 418–423 CrossRef.
- V. Sreeramkumar, J. M. Adrover, I. Ballesteros, M. I. Cuartero, J. Rossaint, I. Bilbao, M. Nacher, C. Pitaval, I. Radovanovic, Y. Fukui, R. P. McEver, M.-D. Filippi, I. Lizasoain, J. Ruiz-Cabello, A. Zarbock, M. A. Moro and A. Hidalgo, Science, 2014, 346, 1234–1238 CrossRef CAS.
- J. M. Adrover, C. del Fresno, G. Crainiciuc, M. I. Cuartero, M. Casanova-Acebes, L. A. Weiss, H. Huerga-Encabo, C. Silvestre-Roig, J. Rossaint, I. Cossío, A. V. Lechuga-Vieco, J. García-Prieto, M. Gómez-Parrizas, J. A. Quintana, I. Ballesteros, S. Martin-Salamanca, A. Aroca-Crevillen, S. Z. Chong, M. Evrard, K. Balabanian, J. López, K. Bidzhekov, F. Bachelerie, F. Abad-Santos, C. Muñoz-Calleja, A. Zarbock, O. Soehnlein, C. Weber, L. G. Ng, C. Lopez-Rodriguez, D. Sancho, M. A. Moro, B. Ibáñez and A. Hidalgo, Immunity, 2019, 50, 390–402 CrossRef CAS.
- W. Gibby, W. Parish, R. M. Merrill, D. Fernandez, C. R. Anderson, E. Merchel and R. Parr, Magn. Reson. Imaging, 2019, 58, 76–81 CrossRef CAS.
- A. Caudrillier, K. Kessenbrock, B. M. Gilliss, J. X. Nguyen, M. B. Marques, M. Monestier, P. Toy, Z. Werb and M. R. Looney, J. Clin. Invest., 2012, 122, 2661–2671 CrossRef CAS.
- V. Sreeramkumar, J. M. Adrover, I. Ballesteros, M. I. Cuartero, J. Rossaint, I. Bilbao, M. Nácher, C. Pitaval, I. Radovanovic, Y. Fukui, R. P. McEver, M.-D. Filippi, I. Lizasoain, J. Ruiz-Cabello, A. Zarbock, M. A. Moro and A. Hidalgo, Science, 2014, 346(6214), 1234–1238 CrossRef CAS.
- J. García-Prieto, R. Villena-Gutiérrez, M. Gómez, E. Bernardo, A. Pun-García, I. García-Lunar, G. Crainiciuc, R. Fernández-Jiménez, V. Sreeramkumar, R. Bourio-Martínez, J. M. García-Ruiz, A. S. del Valle, D. Sanz-Rosa, G. Pizarro, A. Fernández-Ortiz, A. Hidalgo, V. Fuster and B. Ibanez, Nat. Commun., 2017, 8, 14780 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Further biodistribution images. See DOI: 10.1039/d0nr04538a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |