
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent-driven chirality for luminescent self-assembled structures: experiments and theory†
Charles
Lochenie
a,
Alberto
Insuasty
a,
Tommaso
Battisti
a,
Luca
Pesce
 b,
Andrea
Gardin
d,
Claudio
Perego
b,
Mike
Dentinger
a,
Di
Wang
c,
Giovanni M.
Pavan
*bd,
Alessandro
Aliprandi
*a and
Luisa
De Cola
*ae
b,
Andrea
Gardin
d,
Claudio
Perego
b,
Mike
Dentinger
a,
Di
Wang
c,
Giovanni M.
Pavan
*bd,
Alessandro
Aliprandi
*a and
Luisa
De Cola
*ae
aLaboratoire de Chimie et des Biomatériaux Supramoléculaires, Institut de Science et d'Ingénierie Supramoléculaires (UMR 7006), Université de Strasbourg & CNRS, 8, allée Gaspard Monge, 67000 Strasbourg, France. E-mail: decola@unistra.fr; aliprandi@unistra.fr
bDepartment of Innovative Technologies, University of Applied Sciences and Arts of Southern Switzerland, CH-6928 Manno, Switzerland
cKarlsruhe Nano Micro Facility (KNMF), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldschaffen, Germany
dDepartment of Applied Science and Technology, Politecnico di Torino, Corso Duca degli Abruzzi 24, 10129 Torino, Italy. E-mail: giovanni.pavan@polito.it
eInstitut für Nanotechnologie (INT), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldschaffen, Germany
First published on 16th October 2020
Abstract
We describe, for a single platinum complex bearing a dipeptide moiety, a solvent-driven interconversion from twisted to straight micrometric assembled structures with different chirality. The photophysical and morphological properties of the aggregates have been investigated as well as the role of the media and concentration. A real-time visualization of the solvent-driven interconversion processes has been achieved by confocal microscopy. Finally, atomistic and coarse-grained simulations, providing results consistent with the experimental observations, allow to obtain a molecular-level insight into the interesting solvent-responsive behavior of this system.
Introduction
The controlled construction of supramolecular aggregates is still an attractive issue in the development of new functional materials. Indeed the supramolecular systems may possess new properties that do not exist in the corresponding molecular entity.1–4 Square-planar Pt(II) d8 complexes with filled dz2 orbitals are useful building blocks in the creation of supramolecular nanostructures due to their high tendency to stack through weak non-covalent metal–metal and/or ligand–ligand interactions.5–7 Moreover, the establishment of PtII⋯PtII metallophilic interactions (distances in the range of 3.0–3.5 Å) leads to a dramatic change in their photophysical properties8–10 making them good candidates for a variety of applications such as liquid crystals,11,12 metallogelators,13,14 and sensors.15,16 Since many of the Pt(II) complexes exhibit high emission quantum yields and different emission energies once aggregated, these dramatic changes in the photophysical properties have been used to probe supramolecular self-assembly pathways in real-time.17In natural systems self-assembly is a constant motif for life. Many biological molecules tend to assembly in functional structures and amongst them peptides have been heavily investigated due to the resulting stable structure that can also cause severe disease such as Alzheimer, Parkinson, or Creutzfeldt–Jakob disease.18–21 Even short peptides sequences can be used to trigger self-assembly, create chiral objects and small molecules allow an easier control of the desired morphologies.22–25 Not only the chemical design of the peptide is relevant but as has been shown different supramolecular structures can be obtained depending on the preparation protocol. For example Stupp, Meijer, and co-workers have described how an amphiphilic peptide can form either long filaments containing β-sheets or smaller aggregates containing peptide segments in random coil conformation.26 In addition, thermodynamic and kinetic studies have revealed the presence of competing pathways in the self-assembly, which can lead to a switch in chirality of the resulting aggregate27 such as an opposite helicity.28 An “artificial infection” that mimic the nucleation–elongation mechanisms, observed in misfolded proteins such as amyloid-β, has been reported by Takeuchi et al., who described two aggregation pathways of a porphyrin as model system.29
Following in real-time the folding or assembly of biomolecules is always challenging experimentally, while the use of fluorescent dyes can furnish some information on the static assembly. Much more interesting would be the employment of a “reporter template” that could participate or not in the assembly process but would be able to change the properties of the assembly.30 In this perspective, an assembler that can follow in real time the peptides assembly or disassembly through a change in the absorption or emission properties can offer an interesting experimental hang to study the assembly. Herein we demonstrate that luminescent Pt(II) complexes can be used to probe supramolecular self-assembly pathways of peptides and even to observe the switching behavior from nanoribbons to chiral twisted fibers as a function of the media, revealing a key effect of the solvent in triggering major structural transitions in the system. A molecular-level understanding of the behavior of such self-assembling systems which is typically difficult to reach has been obtain by molecular models.
Results and discussion
The Pt(II) complex investigated in this work (Fig. 1, Pt-PyAG) consists of an aromatic chromophoric tridentate ligand and an ancillary pyridine functionalized with a dipeptide moiety (Ala-Gly-OEt). The dipeptide is known to have high tendency to form β-sheets31 while the Pt(II) complex head is known to form self-assembled structures with distinct luminescent properties.5,6,11,14,17,32–36 From the molecular design we expected the two components to give an orthogonal contribution to the self-assembly in different media, in fact the square-planar Pt(II) complex head should give π–π stacking and establish metallophilic interactions in polar media while the dipeptide should establish hydrogen bonds between the Ala-Gly dipeptidic groups in non-polar media. The dipeptide ancillary ligand PyAG was obtained in two steps by first coupling Boc-L-Ala-OH and Gly-OEt using HBTU as coupling reagent, then reacting the deprotected Ala-Gly-OEt with isonicotinic acid in the same conditions (see Fig. S1†). The complex Pt-PyAG was obtained by reaction of Pt-DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 37 with PyAG and purified by flash column chromatography and recrystallization from acetonitrile (ACN) (see Fig. S2†). The complex Pt-PyAG was characterized with 1H and 19F-NMR, HR-MS and elemental analysis.
37 with PyAG and purified by flash column chromatography and recrystallization from acetonitrile (ACN) (see Fig. S2†). The complex Pt-PyAG was characterized with 1H and 19F-NMR, HR-MS and elemental analysis.
 | ||
| Fig. 1 Chemical structure of the Pt(II) complex Pt-PyAG. | ||
Upon recrystallization from ACN, a blue emitting yellow material was obtained. Scanning Electron Microscopy (SEM) study has revealed that the complex Pt-PyAG aggregates as ribbon-like fibres, as shown in Fig. 2A, B and Fig. S6.†
 | ||
| Fig. 2 SEM images of the nanoribbons (A & B) and of the twisted fibres (C & D) of Pt-PyAG. | ||
Annular dark field (ADF) scanning transmission electron microscopy (STEM) images of the nanoribbons were recorded and show that the ribbons are composed of several layers, with an interlayer distance of ≈2.4 nm (Fig. S17†). The fibres could also be observed with confocal fluorescence microscopy in solution, as shown in Fig. S15.† The aggregates have a length ranging from 5 to 200 μm, and a width of 300–500 nm. The photophysical properties of the monomeric species in ACN in dilute solution (10−5 M) have been studied and the emission is not detectable at this concentration (see below for the study of the concentration dependent emission). The behaviour at higher concentration, where nanoribbons in (aerated) ACN are formed, is summarized in the absorption, excitation, and emission spectra, depicted in Fig. 3, and the photophysical parameters are reported in Table 1. The aggregates present the typical structured-emission pattern corresponding to a Ligand-Centered (3LC) transition with a triplet character with maxima at 460, 490, and 521 nm.16,17,33 The long excited-state lifetime (see Fig. S8†) of 2.33 μs confirms the triplet nature of the transition.6,14,17,38–40 The photoluminescence quantum yield (ΦPL) of the nanoribbons was measured to be 0.065. We must note that the emission quantum yield is affected by the absorption of the monomeric species, always present in solution which however does not display emission except in TCE where the luminescence is extremely weak (see Fig. S11†). Indeed, the emission quantum yield of the crystalline fibers was determined to be 0.22. The absence of characteristic signal in the CD spectra in the region around 220 nm (Fig. 4),41 indicate the absence of β-sheet domains. On the other hand, an interaction between the metal complexes, and most likely the coordinated ligands, is observed in the excited state (see excitation spectrum) that increases the emission quantum yield and elongate the excited state lifetime (3LC) as reported above.
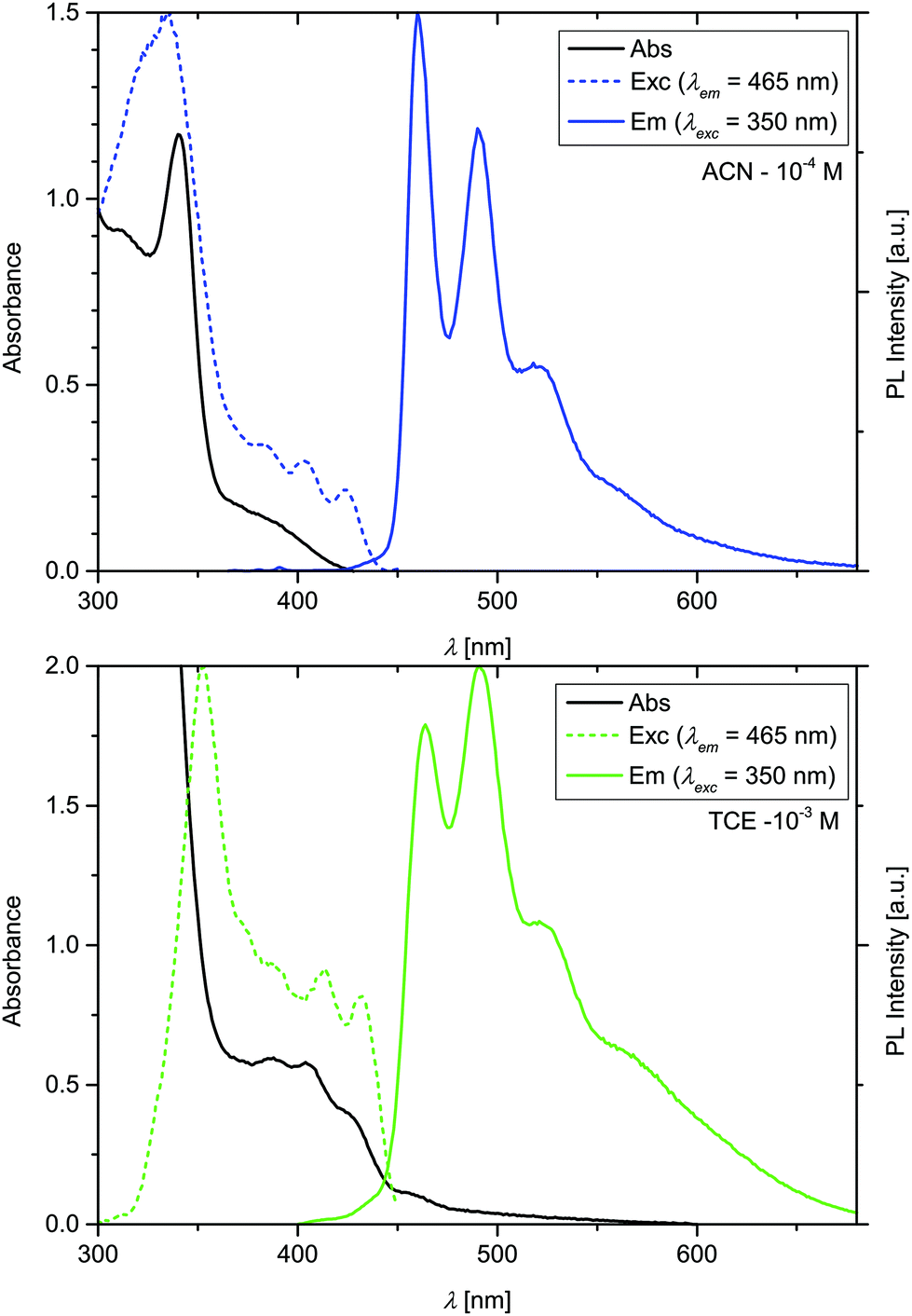 | ||
| Fig. 3 Absorption, excitation, and emission spectra of Pt-PyAG in ACN at 10−4 M (top) and in TCE at 10−3 M (bottom). | ||
 | ||
| Fig. 4 Absorption spectra of a solution or suspensions of PyAG in ACN at 10−3 M (black), Pt-PyAG nanoribbons in ACN (blue) at 10−4 M and of twisted fibers in TCE (green) at 10−3 M and CD (dashed lines) spectra of their respective films. | ||
If the nanoribbons of Pt-PyAG are dissolved in 1,1,2,2-tetrachloroethane (TCE) at 65 °C at a concentration of 10−3 M, and cool down to room temperature, the complex aggregates as twisted fibres. SEM images of the aggregates are presented in Fig. 2C, D and Fig. S7.† Interestingly, the observed fibres consist of only left-handed helixes, suggesting that the chirality present at the molecular level is now transcribed into the mesoscale. ADF-STEM images of the twisted fibres were also recorded and are shown in Fig. S17,† however, no packing structure could be observed due to the high beam sensitivity of the fibres. The twisted fibres have a length ranging from 5 to 50 μm, a width of 75–100 nm, and a height of ≈25 nm, and therefore are much smaller than the ribbons. Indeed 1,1,2,2-tetrachloroethane (TCE) is known to favour H-bonds formations and consequently the formation of β-sheet domains.31,42 It is important to note that the dipeptide pyridine ligand in the same conditions do not form aggregates, demonstrating the need of the platinum complex to initiate the self-assembly process. The absorption, excitation, and emission spectra of the twisted fibres are presented in Fig. 3 and the photophysical parameters are reported in Table 1. They display a slightly red-shifted emission if compared to the nanoribbons peaking at 468, 498, and 522 nm and a more marked difference in terms of relative intensity. In contrast to the ACN nanoribbons that are characterized by a single mono exponential decay, the excited-state lifetime of twisted fibres obtained in TCE is biexponential with a 4.41 μs and a 0.75 μs components. These two excited state lifetimes are due to different excited states: the first one referred to the highest energy structured emission, is ligand centred in nature (3LC), similarly to the ACN nanoribbons, while the shortest component is due to the low lying 3MMLCT emissive state rising from Pt–Pt interactions as already observed for this class of compounds.17 Such assignment has been corroborated by recording the excited state lifetimes at different wavelength of the emission spectrum (Fig. S8†). The photoluminescence quantum yields of a suspension of the fibres in air-equilibrated TCE at room temperature at 10−3 M and of the filtered fibres were measured to be 0.06 and 0.34, respectively. The circular dichroism (CD) in solution of suspended nanoribbons in ACN (10−4 M) showed no CD signal, while the twisted fibres in TCE (10−3 M) showed a CD positive band with an onset at 450 nm (Fig. S9†). The sharpness of the peak and the absence of features below 400 nm is due to the saturation of the detector that occurs because of the strong absorption of the monomeric form whose electronic transitions involve the achiral tridentate ligand and the metal (1LC and 1MLCT) as already reported in a previous paper.32 The CD spectrum of the isolated twisted fibres film shows two features, one broad band from 225 to 370 nm, and one band centred at 425 nm, which corresponds to the transitions of the aggregated complexes. Such result suggests that the CD arise from the supramolecular structure and not from the ancillary ligand, which presents a different CD signal and does not absorb above 400 nm (Fig. 4). Powder X-ray diffraction on the drop-casted has further confirmed that ribbons and twisted fibres are structurally different (see Fig. S14†) even if it was not possible to determine the actual packing of the system.
In order to understand the mechanism of formation of the supramolecular structures we have performed temperature-dependent emission spectroscopy at different concentration (either ACN or TCE).43 Starting from a hot solution of the monomer Pt-PyAG, the cooling curve was recorded at a rate of 1 °C min−1 and emission spectrum measured every 5 °C, with an equilibration time of 20 min. The ribbons were measured at 4 different concentrations (5 × 10−4 M, 2.5 × 10−4 M, 10–4 M, and 7.5 × 10−5 M). In Fig. S12A,† the integrated emission intensity is plotted against the temperature, and it can be seen that upon cooling the emission intensity is constant until reaching a critical temperature at which the intensity grows exponentially suggesting a cooperative process. This was observed for all concentrations except at the lowest (7.5 × 10−5 M) where this phenomenon is either not happening, or could not be measured. For the other concentrations, the integrated emission intensity was normalized, and an exponential fit was applied to the data sets, with a rather good agreement (see Fig. S12B†). For the twisted fibres, it was noticed that upon aggregation, the relative intensities of the emission maxima are changing; in particular the band at 575 nm becomes more and more intense as the temperature is lowered (see normalized temperature-dependent spectra in Fig. S10D†). Such band can be ascribed to 3MMLCT transition of interacting Pt centres. If the emission intensity at 575 nm is plotted against the temperature, Fig. S12C,† a non-linear increase of the emission intensity is observed suggesting the absence of cooperativity.44 Unfortunately we could not confirm such hypothesis by temperature dependent UV-Vis spectroscopy due to the large scattering of the fibres (see Fig. S16†). To prove that it is possible to convert one structure into the other a suspension of the twisted fibres (10−3 M) in TCE was diluted stepwise with ACN. The conversion of the fibres was followed by measuring the photoluminescence quantum yield (ΦPL) of the system using an integrating sphere setup. After each addition of the solvent, the mixture was stirred until ΦPL was stable indicating that the system had equilibrated. In Fig. 5, the ΦPL is plotted against the solvent composition (indicated as percentage of ACN in TCE) as well as the variation in concentration. It is important to note that the starting emission quantum yield is lower than the solid state measurements (0.34) and is function of the aggregation degree that depends on the total concentration. Upon addition of the second solvent, dissolution of the fibres is observed and the value reach zero when 40% of ACN is added. The disassembly of the fibres can be only marginally correlated to the dilution effect (see Fig. S13†). Above 50% ACN the ΦPL starts to rise again, and the molecularly dissolved complexes begin to aggregate as ribbons. The abrupt increase of the emission further points toward a cooperative mechanism in the self-assembly of the ribbon. Those experiments allow us to state that, at equilibrium, the two aggregates cannot coexist because for a range of solvent composition (ACN/TCE) no aggregate formation is observed (molecularly dissolved state).
 | ||
| Fig. 5 Emission quantum yield (λexc = 350 nm) vs. solvent composition (TCE/ACN) upon dilution (upper plot); snapshots at 0, 45, and 90 min of the Movie 1† visualizing the real time conversion from chiral to achiral structures. | ||
A similar experiment has been performed using instead of the pure acetonitrile solvent an equimolar dispersion of the ribbons to the TCE fibres suspension. The results shown in Fig. S13† are consistent with the previous observations.
The visualization of the above reported experiment was realized observing the transition from twisted fibres to nanoribbons in real time, using confocal fluorescence microscopy. A suspension of twisted fibres in TCE was suddenly diluted with ACN, leading to the simultaneous dissolution of the twisted fibres and growth of the nanoribbons. The phenomenon is presented in Fig. 5 and in the ESI Movie 1.† At t = 0, an aggregate of twisted fibres is shown, and after addition of ACN, it is observed that the aggregates dissolve while, at the same time, ribbons grow. In the movie, the two different colours, green for the chiral structures and blue for the crystalline ribbons, are related to the different emission spectra. After a lag time of about 45 min, the growth of the ribbons suddenly started, and after another approximatively 35 min, the system reached equilibrium and no more changes were observed. The observed lag time and the rapid growth of the ribbons are also typical features of a cooperative growth mechanism as described above. To gain a multiscale molecular view of the assembly process, molecular simulations have been performed in different solvents.45 We started from building a fully atomistic (all atom: AA) model for monomer Pt-PyAG (Fig. 6A). We inserted 20 initially disassembled AA-monomers into two simulation boxes filled respectively with TCE or ACN explicit solvent molecules. AA molecular dynamics simulations (AA-MD) provided a first insight into the key type of interactions involved in the self-assembly in the two environments. As seen in Fig. 6B (left), the number of molecular clusters in the system decreases over time in both cases, indicating self-assembly. However, during self-assembly the tail–tail H-bonding increases in time in TCE (Fig. 6B, right: green), while in ACN this is found substantially negligible. This is consistent with the fact that ACN molecules can form H-bonds with the monomer tails, preventing the H-bonding network between the monomer tails, similar to what seen recently in the case of benzene-1,3,5-tricarboxamide self-assembly in water–ACN co-solvents.46 The core–core interactions are present in both cases, although relatively stronger in ACN (see also below). As AA models are limited in size of the system that can be effectively simulated, we also developed consistent coarse-grained (CG) model for Pt-PyAG monomers (Fig. 6C), which allowed us to study self-assembly on a larger scale (see Methods and the ESI†).47 We inserted 400 initially dispersed Pt-PyAG CG-monomers in a simulation box filled with different types of solvents. Together with TCE and ACN, we also found interesting to simulate, for comparison, the self-assembly of the monomer in a case where the solvophobic interactions are increased, such as in octane (OCT). Starting from this latter case, during the CG-MD simulation, the CG-monomers were seen to self-assemble forming helical fibres resulting from the hierarchical aggregation of ∼5–6 stacks (Fig. 6D). The monomers interact both via core–core stacking and via interaction between the peptidic tails. Shown in Fig. S18,† the core–core interactions are found to be a stronger primary interaction between the monomers, present in all cases (stronger in ACN). On the other hands, the tail–tail interaction (stronger in OCT and TCE) dictates as a secondary interaction the structure of the fibres, where the monomer tails are gathered in the helix interior and surrounded by the monomer cores (see e.g., Fig. 6E, in red). The tilting angle between the stacked monomers gives rise to a helical assembly with pitch p ∼ 30 nm. The stability of such an arrangement was also proven by replicating one of the self-assembled helices into a long helix that has been then relaxed in OCT solvent by means of CG-MD simulation. Interestingly, when changing the solvent from OCT to TCE, the CG-MD simulation provides similar helices of ∼5 stacks, but having increased helical pitch of p ∼ 120 nm, which is in good qualitative agreement with the pitch seen in the experimental images (Fig. 2D: ∼9 helical turns per 1 μm of fibre length). Considering the reduced/increased solvophobic interaction between the Pt-PyAG monomers in TCE/OCT solvents, these results suggest that solvophobic effects play a major role in the helical arrangement of these fibres, where the helicity of the assembly may increase as a result of the stronger tendency to reduce the interactions between the hydrophilic tails and the hydrophobic solvent. This is consistent with the folding behaviour seen recently in other types of supramolecular polymers in water.48
 | ||
| Fig. 6 Multiscale molecular simulations of Pt-PyAG in the various solvents. (A) AA model for monomer Pt-PyAG. (B) Self-assembly of 20 initially dispersed Pt-PyAG AA-monomers seen along 500 ns of AA-MD simulations (left: drop of the number of disassembled clusters in solution over time). Right: The AA-MD simulations demonstrate that while the number of tail–tail H-bonds increases as the self-assembly proceeds in TCE (green), H-bonding is negligible in ACN. (C) CG model for monomer Pt-PyAG, which was used to simulate self-assembly in TCE, ACN and octane (OCT) solvents. (D and E) Self-assembly of 400 initially dispersed Pt-PyAG CG-monomers in apolar solvents seen along 1 μs of CG-MD simulation. The CG-MD shows that in OCT (D) the monomers self-assemble into helical fibres composed of ∼5–6 stacks where the helix pitch is found p ∼ 30 nm. While in these fibres the peptide tails are gathered in the fibre interior (E, in red), in TCE the helical pitch is increased to ∼120 nm (in good agreement with the experiments). (F) CG-MD simulations of 400 dispersed Pt-PyAG CG-monomers in CG-ACN that self-assemble into long filaments interacting hierarchically via core–core interactions. (G) CG model of a Pt-PyAG bilayer, showing very high stability during a CG-MD in CG-ACN. | ||
Analogous CG-MD simulations in a CG-ACN (see Methods for details) showed the formation of very long fibres in solution, while the individual fibres tend to interact in a mirror fashion via the cores exposed to the ACN solvent (Fig. 6F). The hierarchical lateral self-assembly of such fibre pairs could give rise to the 2D growth of ribbons similar to those seen in the experiments. To test this hypothesis in a more efficient way, we took one of these fibre pairs and replicated it both along the main axis and laterally, obtaining a preformed 2D bilayer of Pt-PyAG CG-monomers. This system was proved very stable during a CG-MD simulation, supporting the hypothesis that the spontaneous formation of flat ribbons in ACN may be due to similar types of aggregates, thus also controlled by solvophobic effects.
Experimental section
Synthesis
All the reactions were performed under an inert atmosphere of nitrogen. All the chemicals were of analytical grade, and used as received from Sigma Aldrich or Alfa Aesar. Solvents were purchased from Sigma Aldrich and used as received. Pt-DMSO was synthesized as described in literature.371H, 13C, and 19F-NMR spectra were recorded on a Bruker Avance 400 spectrometer. Unless otherwise indicated, chemical shifts (δ) are reported in ppm downfield from tetramethylsilane (TMS) as internal standard at room temperature using deuterated solvents. All coupling constants (J) are given in Hertz (Hz). Electrospray ionization mass spectrometry (ESI-TOF-MS) was recorded on a MicroTOF (Bruker) mass spectrometer equipped with an electrospray source by the mass spectrometry service of the Faculté de Chimie at the University of Strasbourg.Synthesis of the pyridine dipeptide ancillary ligand PyAG
:1 methanol (MeOH)) to give 1 as yellow oil. Yield: 205 mg (61%).
1H-NMR (400 MHz, CDCl3, ppm): δ = 5.09–4.92 (m, 1H, >CH–), 4.23 (q, 3J = 7.1 Hz, 2H, –CH2–), 4.10–3.98 (m, 2H, –CH2–), 1.47 (s, 9H, –C(CH3)3), 1.40 (d, 3J = 7 Hz, 3H, –CH3), 1.30 (t, 3J = 7.1 Hz, 3H, –CH3).
:1 MeOH) to give PyAG as yellow oil. Yield: 105 mg (51%).
1H-NMR (400 MHz, MeOD, ppm): δ = 8.69 (d, J = 6.1 Hz, 2H, Ar–H), 7.83 (d, J = 6.1 Hz, 2H, Ar–H), 4.64 (q, J = 7.3 Hz, 1H, >CH–), 4.19 (q, J = 7.1 Hz, 2H, –CH2–), 4.02–3.90 (m, 2H, –CH2–), 1.51 (d, J = 7.3 Hz, 3H, –CH3), 1.25 (t, J = 7.1 Hz, 3H, –CH3). 13C-NMR (100 MHz, MeOD, ppm): δ = 175.1, 174.8, 171.3, 148.2, 143.6, 124.7, 61.9, 50.8, 42.1, 18.7, 14.3. ESI-MS (m/z): 279.1 ([M]+, C13H17N3O4).
Synthesis of the Pt(II) complex Pt-PyAG
Pt-PyAG: Pt-DMSO (89 mg, 0.14 mmol) was solved at 70 °C in dry acetonitrile (ACN, 5 mL). Once completely solved, a solution of PyAG (40 mg, 0.14 mmol) in ACN (2 mL) was added at once, and the heating was turned off. The reaction mixture was let to stir overnight. Precipitation of yellowish needles happens overnight. The solvent were removed in vacuo, and giving a mixture of yellow (Pt-PyAG) and red (Pt-DMSO) material. The complex was purified with flash chromatography (SiO2/AcOEt), and then recrystallized from acetonitrile to yield Pt-PyAG as a pure yellow solid. Yield 37 mg (33%).1H-NMR (400 MHz, C2D2Cl4, ppm): δ = 9.11 (d, J = 6.7 Hz, 2H), 7.44 (t, J = 8.1 Hz, 1H), 7.26 (d, J = 6.7 Hz, 2H), 7.18 (d, J = 8.1 Hz, 2H), 6.90 (d, J = 7.1 Hz, 1H), 5.90 (t, J = 5.7 Hz, 1H), 4.18–4.09 (m, 1H), 3.60 (q, J = 7.0 Hz, 2H), 3.63–3.42 (m, 2H), 0.96 (d, J = 7.2 Hz, 3H), 0.67 (t, J = 7.0 Hz, 3H). 19F-NMR (400 MHz, C2D2Cl4, ppm): δ = −65.2 (s, –CF3). HR-MS (ESI-TOF, m/z): 822.1290 (calcd 822.1299, [M + H]+. Elemental analysis: calcd (found) C 35.09% (35.17%), H 2.45% (2.65%), N 17.05% (16.95%).
Photophysical measurements
Suprasil (type 111-QS) emission cuvettes with a light path of 10 × 10 mm from Hellma-Analytics were utilized for photophysical experiments. Absorption spectra were measured on a Shimadzu UV-3600 double-beam UV–VIS–NIR spectrophotometer and baseline corrected. Steady-state emission spectra were recorded on a Horiba Jobin–Yvon IBH FL-322 Fluorolog 3 spectrometer equipped with a 450 W xenon arc lamp, double-grating excitation, and emission monochromators (2.1 nm mm−1 of dispersion; 1200 grooves per mm) and a TBX-04 single photon-counting detector. Emission and excitation spectra were corrected for source intensity (lamp and grating) and emission spectral response (detector and grating) by standard correction curves. Time-resolved measurements were performed by using either the time-correlated single-photon counting (TCSPC) electronics PicoHarp300 or the Multi-Channel Scaling (MCS) electronics NanoHarp 250 of the PicoQuant FluoTime 300 (PicoQuant GmbH, Germany), equipped with a PDL 820 laser pulse driver. A pulsed laser diode LDH-P-C-405 (λ = 405 nm, pulse FWHM < 70 ps, repetition rate 200 kHz to 40 MHz) was used to excite the sample and mounted directly on the sample chamber at 90°. The photons were collected by using a PMA-C-192 photomultiplier (PMT) single-photon-counting detector. The data was acquired by using the commercially available software EasyTau (Pico-Quant GmbH, Germany), whereas data analysis was performed by using the commercially available software FluoFit (PicoQuant GmbH, Germany. The absolute photoluminescence quantum yields (PLQY) were measured on a Hamamatsu Quantaurus-QY integrating sphere in air-equilibrated condition using an empty quartz tube as a reference.Confocal fluorescence microscopy
The samples as suspension were placed in a quartz Petri dish, which was then closed with the corresponding quartz lid. Fluorescence confocal images were recorded by using a Zeiss LSM 710 confocal microscope system with a 10× magnification objective. The samples were excited by continuous wave laser at λexc = 355 or 405 nm. The emission of the assemblies was collected in the range from 414 to 721 nm with 9.7 nm resolution by using the lambda-mode option. The raw data recorded by means of the lambda-mode were processed by using linear un-mixing tool option available in the ZEN 2011 software package (Zeiss GmbH, Germany).Scanning electron microscopy
Scanning Electron Microscopy (SEM) images were recorded with a FEI Quanta FEG 250 instrument (FEI corporate, Hillsboro, Oregon, USA) with an acceleration voltage of 20 kV. The samples were prepared by drop-casting onto a glass cover slip, subsequently sputter coated with Au (Emitech K575X Peltier cooled) for 30 s at 60 mA prior to fixation on an Al support.Transmission electron microscopy
Transmission electron microscopy (TEM) was performed on a FEI Titan 80-300 electron microscope (FEI corporate, Hillsboro, Oregon, USA), operating at an accelerating voltage of 300 kV. ADF-STEM images were acquired in nanoprobe mode with Fischione model 3000 STEM detector. The samples were prepared by directly dropping the solvent on Au TEM grids coated with holey carbon film and additional a complete ultrathin carbon film (3 nm thick) and drying before being loaded into TEM.Powder X-ray diffraction
The X-ray diffraction (XRD) patterns were recorded on a Bruker D2 Phaser (Bruker, USA) using CuKα1 radiation.Molecular models
The AA model of Pt-PyAG was parametrized according to the general AMBER force field (GAFF),50 by using the AMBERtools 17.51 The parameters related to the atoms coordinating to the Pt atom were obtained using the Metal Center Parameter Builder (MCPB).52 The CG model for monomer Pt-PyAG was developed based on the MARTINI coarse-grained force field.53 Similar to what recently done for the modelling of similar self-assembling systems, the CG model has been optimized in order to be consistent with the AA model in terms of monomer–monomer interaction.47,54,55 In particular, the CG beads of the dipeptide moiety were chosen according to the standard MARTINI amino-acids models,56 while the CG parametrization of the rest of the monomer core was optimized to match the free-energy ΔG of core–core dimerization obtained at the AA level using metadynamics (MetaD) simulations (see Fig. S19†).57 More details on the parametrization are provided in the ESI.†Molecular simulations
All MD and metadynamics simulations have been conducted using the GROMACS-2018.4 software58 patched with PLUMED-2.4.59 The AA-MD self-assembly simulations were conducted starting from 20 initially disassembled Pt-PyAG monomers in explicit TCE and ACN solvent molecules. The AA systems were simulated for 500 ns of AA-MD at 298 K of temperature and 1 atm of pressure in NPT conditions (constant N: number of particles, P: pressure and T: temperature). All simulations conducted herein used the v-rescale thermostat60 and the Berendsen barostat.61 A timestep of 2 fs was used in the AA-MD simulations. The electrostatic interactions were treated using particle mesh Ewald (PME).62 The cutoff lengths of the real summation and of the VdW were set to 1.0 nm. The dynamics of the hydrogens was constrained using the LINCS algorithm.63The CG-MD self-assembly simulation started from 400 initially disassembled Pt-PyAG molecules immersed into explicit OCT, TCE and ACN solvent molecules. Details on the CG parameters are provided in the ESI.† All CG-systems were run for at least 1 μs of CG-MD using a timestep of 20 fs and isotropic pressure scaling at room temperature. The helical pitch of the twisted fibers in OCT and ACN was estimated by quantifying the average rotation angle of neighbor stacked Pt atoms around the main axis of the longer self-assembled filament obtained during the self-assembling CG-MD runs. The stability of supramolecular helical fibers (TCE and OCT) was tested by replicating such twisted aggregate along its main axis – until reaching a complete helix composed of 432 Pt-PyAG monomers – and equilibrating the helices by CG-MD simulations. The stability of the bilayer configuration of the system in ACN was tested in a similar way. We built a preformed bilayer composed by the lateral packing of mirror-assembled fibers pairs – for a total of 800 monomers – which was then equilibrated in explicit ACN CG-molecules (see Fig. 6G). All these preformed configurations were found stable/persistent over 2.5 μs of CG-MD simulations. Due to the shape of these assemblies, semi-isotropic pressure scaling was used in all these NPT CG-MD simulations.
Conclusion
We have synthetized and investigated the solvent dependent self-assembly properties of a luminescent Pt(II) complex bearing a dipeptide moiety. Two chiral structures with distinct chiral, optical and morphological properties have been identified. In particular chiral nanoribbons are formed in ACN while twisted chiral fibres are formed in TCE. The interconversion of the twisted into the straight structure has been realized and the process has been followed in real time by confocal microscopy. The aggregation process of the ribbons seems to follow a cooperative mechanism (seeding-elongation) while the formation of the twisted fibres seems to take place with a different mechanism experimentally difficult to prove. Finally, atomistic and coarse-grained simulations have provided results consistent with the experimental observations, allowing a molecular-level insight into the interesting solvent-responsive behaviour of this system. Such findings can be used to further elucidate the self-assembly properties of biologically relevant proteins such as amyloid-β.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Charles Lochenie thanks the Deutsche Forschungsgemeinschaft DFG (LO 2486/1-1) for financial support. LDC thanks IUF and Axa funding for financial support. G. M. P. acknowledges the funding received by the Swiss National Science Foundation (SNSF grant 200021_175735) and by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 818776 – DYNAPOL). The authors also acknowledge the computational resources provided by the Swiss National Supercomputing Center (CSCS) and by CINECA.References
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS.
- F. Wurthner, C. R. Saha-Moller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef.
- X. Zhang, S. Rehm, M. M. Safont-Sempere and F. Wurthner, Nat. Chem., 2009, 1, 623–629 CrossRef CAS.
- R. Hu, N. L. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494–4562 RSC.
- C. A. Strassert, C. H. Chien, M. D. Galvez Lopez, D. Kourkoulos, D. Hertel, K. Meerholz and L. De Cola, Angew. Chem., Int. Ed., 2011, 50, 946–950 CrossRef CAS.
- M. Mauro, A. Aliprandi, C. Cebrian, D. Wang, C. Kubel and L. De Cola, Chem. Commun., 2014, 50, 7269–7272 RSC.
- M. E. Robinson, A. Nazemi, D. J. Lunn, D. W. Hayward, C. E. Boott, M. S. Hsiao, R. L. Harniman, S. A. Davis, G. R. Whittell, R. M. Richardson, L. De Cola and I. Manners, ACS Nano, 2017, 11, 9162–9175 CrossRef CAS.
- B. Ma, J. Li, P. I. Djurovich, M. Yousufuddin, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2005, 127, 28–29 CrossRef CAS.
- K. Zhang, M. C. Yeung, S. Y. Leung and V. W. Yam, J. Am. Chem. Soc., 2018, 140, 9594–9605 CrossRef CAS.
- Y. Ai, Y. Li, H. L.-K. Fu, A. K.-W. Chan and V. W.-W. Yam, Chem. – Eur. J., 2019, 25, 5251 CrossRef CAS.
- V. N. Kozhevnikov, B. Donnio and D. W. Bruce, Angew. Chem., 2008, 120, 6382–6385 CrossRef.
- M. Krikorian, S. Liu and T. M. Swager, J. Am. Chem. Soc., 2014, 136, 2952–2955 CrossRef CAS.
- A. Y. Tam and V. W. Yam, Chem. Soc. Rev., 2013, 42, 1540–1567 RSC.
- D. Genovese, A. Aliprandi, E. A. Prasetyanto, M. Mauro, M. Hirtz, H. Fuchs, Y. Fujita, H. Uji-I, S. Lebedkin, M. Kappes and L. De Cola, Adv. Funct. Mater., 2016, 26, 5271–5278 CrossRef CAS.
- M. C.-L. Yeung and V. W.-W. Yam, Chem. – Eur. J., 2011, 17, 11987–11990 CrossRef CAS.
- S. Sinn, F. Biedermann, M. Vishe, A. Aliprandi, C. Besnard, J. Lacour and L. De Cola, ChemPhysChem, 2016, 17, 1829–1834 CrossRef CAS.
- A. Aliprandi, M. Mauro and L. De Cola, Nat. Chem., 2016, 8, 10–15 CrossRef CAS.
- G. Wei, Z. Su, N. P. Reynolds, P. Arosio, I. W. Hamley, E. Gazit and R. Mezzenga, Chem. Soc. Rev., 2017, 46, 4661–4708 RSC.
- C. Soto, G. P. Saborio and L. Anderes, Trends Neurosci., 2002, 25, 390–394 CrossRef CAS.
- P. J. Thomas, B.-H. Qu and P. L. Pedersen, Trends Biochem. Sci., 1995, 20, 456–459 CrossRef CAS.
- C. Soto, FEBS Lett., 2001, 498, 204–207 CrossRef CAS.
- R. M. Capito, H. S. Azevedo, Y. S. Velichko, A. Mata and S. I. Stupp, Science, 2008, 319, 1812–1816 CrossRef CAS.
- C. Chen, J. Tan, M. C. Hsieh, T. Pan, J. T. Goodwin, A. K. Mehta, M. A. Grover and D. G. Lynn, Nat. Chem., 2017, 9, 799–804 CrossRef CAS.
- N. M. Casellas, S. Pujals, D. Bochicchio, G. M. Pavan, T. Torres, L. Albertazzi and M. Garcia-Iglesias, Chem. Commun., 2018, 54, 4112–4115 RSC.
- G. Molev, Y. Lu, K. S. Kim, I. C. Majdalani, G. Guerin, S. Petrov, G. Walker, I. Manners and M. A. Winnik, Macromolecules, 2014, 47, 2604–2615 CrossRef CAS.
- P. A. Korevaar, C. J. Newcomb, E. W. Meijer and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 8540–8543 CrossRef CAS.
- Q. Wan, X.-S. Xiao, W.-P. To, W. Lu, Y. Chen, K.-H. Low and C.-M. Che, Angew. Chem., Int. Ed., 2018, 57, 17189–17193 CrossRef CAS.
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. Smulders, P. A. Hilbers, A. P. Schenning, T. F. De Greef and E. W. Meijer, Nature, 2012, 481, 492–496 CrossRef CAS.
- S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188–195 CrossRef CAS.
- A. S.-Y. Law, L. C.-C. Lee, M. C.-L. Yeung, K. K.-W. Lo and V. W.-W. Yam, J. Am. Chem. Soc., 2019, 141, 18570–18577 CrossRef CAS.
- J. L. Lopez, C. Atienza, A. Insuasty, J. Lopez-Andarias, C. Romero-Nieto, D. M. Guldi and N. Martin, Angew. Chem., Int. Ed., 2012, 51, 3857–3861 CrossRef CAS.
- A. Aliprandi, C. M. Croisetu, M. Mauro and L. Cola, Chemistry, 2017, 23, 5957–5961 CrossRef CAS.
- A. Aliprandi, D. Genovese, M. Mauro and L. De Cola, Chem. Lett., 2015, 44, 1152–1169 CrossRef CAS.
- N. K. Allampally, M. Bredol, C. A. Strassert and L. De Cola, Chem. – Eur. J., 2014, 20, 16863–16868 CrossRef CAS.
- N. K. Allampally, C. A. Strassert and L. De Cola, Dalton Trans., 2012, 41, 13132–13137 RSC.
- Y. Atoini, E. A. Prasetyanto, P. Chen, S. Silvestrini, J. Harrowfield and L. De Cola, Chem. – Eur. J., 2018, 24, 12054–12060 CrossRef CAS.
- S. Sinn, F. Biedermann and L. De Cola, Chem. – Eur. J., 2017, 23, 1965–1971 CrossRef CAS.
- J. A. G. Williams, Photochemistry and Photophysics of Coordination Compounds Ii, 2007, vol. 281, pp. 205–268 Search PubMed.
- M. Mauro, A. Aliprandi, D. Septiadi, N. S. Kehr and L. De Cola, Chem. Soc. Rev., 2014, 43, 4144–4166 RSC.
- D. Septiadi, A. Aliprandi, M. Mauro and L. De Cola, RSC Adv., 2014, 4, 25709–25718 RSC.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2876–2890 CrossRef CAS.
- A. Insuasty, C. Atienza, J. L. Lopez and N. Martin, Chem. Commun., 2015, 51, 10506–10509 RSC.
- M. M. Smulders, M. M. Nieuwenhuizen, T. F. de Greef, P. van der Schoot, A. P. Schenning and E. W. Meijer, Chem. – Eur. J., 2010, 16, 362–367 CrossRef CAS.
- T. F. De Greef, M. M. Smulders, M. Wolffs, A. P. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS.
- D. Bochicchio and G. M. Pavan, Adv. Phys.: X, 2018, 3, 1436408 Search PubMed.
- R. P. M. Lafleur, X. Lou, G. M. Pavan, A. R. A. Palmans and E. W. Meijer, Chem. Sci., 2018, 9, 6199–6209 RSC.
- D. Bochicchio and G. M. Pavan, ACS Nano, 2017, 11, 1000–1011 CrossRef CAS.
- M. B. Baker, L. Albertazzi, I. K. Voets, C. M. A. Leenders, A. R. A. Palmans, G. M. Pavan and E. W. Meijer, Nat. Commun., 2015, 6, 6234 CrossRef.
- C. Spezzacatena, T. Perri, V. Guantieri, L. B. Sandberg, T. F. Mitts and A. M. Tamburro, Eur. J. Org. Chem., 2002, 95–103 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS.
- P. F. Li and K. M. Merz, J. Chem. Inf. Model., 2016, 56, 599–604 CrossRef CAS.
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. de Vries, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS.
- D. Bochicchio, M. Salvalaglio and G. M. Pavan, Nat. Commun., 2017, 8, 147 CrossRef.
- D. Bochicchio, S. Kwangmettatam, T. Kudernac and G. M. Pavan, ACS Nano, 2019, 13, 4322–4334 CrossRef CAS.
- D. H. de Jong, G. Singh, W. F. D. Bennett, C. Arnarez, T. A. Wassenaar, L. V. Schäfer, X. Periole, D. P. Tieleman and S. J. Marrink, J. Chem. Theory Comput., 2013, 9, 687–697 CrossRef CAS.
- A. Laio and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12562–12566 CrossRef CAS.
- H. J. C. Berendsen, D. Vanderspoel and R. Vandrunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- M. Bonomi, D. Branduardi, G. Bussi, C. Camilloni, D. Provasi, P. Raiteri, D. Donadio, F. Marinelli, F. Pietrucci, R. A. Broglia and M. Parrinello, Comput. Phys. Commun., 2009, 180, 1961–1972 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- E. Ulrich, P. Lalith, M. L. Berkowitz, T. Darden, L. Hsing and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr04524a |
| This journal is © The Royal Society of Chemistry 2020 |