
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlatinum nanoparticle decorated vertically aligned graphene screen-printed electrodes: electrochemical characterisation and exploration towards the hydrogen evolution reaction†
Jessica
Scremin
ab,
Isabella V.
Joviano dos Santos
ac,
Jack P.
Hughes
ac,
Alejandro
García-Miranda Ferrari
 ac,
Enrique
Valderrama
d,
Wei
Zheng
de,
Xizhou
Zhong
e,
Xin
Zhao
de,
Elen J. R.
Sartori
b,
Robert D.
Crapnell
ac,
Samuel J.
Rowley-Neale
*ac and
Craig E.
Banks
*ac
ac,
Enrique
Valderrama
d,
Wei
Zheng
de,
Xizhou
Zhong
e,
Xin
Zhao
de,
Elen J. R.
Sartori
b,
Robert D.
Crapnell
ac,
Samuel J.
Rowley-Neale
*ac and
Craig E.
Banks
*ac
aFaculty of Science and Engineering, Manchester Metropolitan University, Chester Street, Manchester M1 5GD, UK. E-mail: S.rowley-neale@mmu.ac.uk; c.banks@mmu.ac.uk; Fax: +(0)1612476831; Tel: +(0)1612474622 Tel: +(0)1612471196
bDepartamento de Química, Centro de Ciências Exatas, Universidade Estadual de Londrina, CEP 86057-970, Londrina, PR, Brazil
cManchester Fuel Cell Innovation Centre, Manchester Metropolitan University, Chester Street, Manchester M1 5GD, UK
dWilliam and Mary Research Institute, The College of William and Mary, Jamestown Street, Williamsburg, Virginia, 23817, USA
eShenzhen Yick Xin Technology Development Ltd Co., Fl. 5, Block B, Boton Technology Park, Chaguang Road, Nanshan District, Shenzhen, 518055, China
First published on 21st August 2020
Abstract
We present the fabrication of platinum (Pt0) nanoparticle (ca. 3 nm average diameter) decorated vertically aligned graphene (VG) screen-printed electrodes (Pt/VG-SPE) and explore their physicochemical characteristics and electrocatalytic activity towards the hydrogen evolution reaction (HER) in acidic media (0.5 M H2SO4). The Pt/VG-SPEs exhibit remarkable HER activity with an overpotential (recorded at −10 mA cm−2) and Tafel value of 47 mV (vs. RHE) and 27 mV dec−1. These values demonstrate the Pt/VG-SPEs as significantly more electrocatalytic than a bare/unmodified VG-SPE (789 mV (vs. RHE) and 97 mV dec−1). The uniform coverage of Pt0 nanoparticles (ca. 3 nm) upon the VG-SPE support results in a low loading of Pt0 nanoparticles (ca. 4 μg cm−2), yet yields comparable HER activity to optimal Pt based catalysts reported in the literature, with the advantages of being comparatively cheap, highly reproducible and tailorable platforms for HER catalysis. In order to test any potential dissolution of Pt0 from the Pt/VG-SPE surface, which is a key consideration for any HER catalyst, we additively manufactured (AM) a bespoke electrochemical flow cell that allowed for the electrolyte to be collected at regular intervals and analysed via inductively coupled plasma optical emission spectroscopy (ICP-OES). The AM electrochemical cell can be rapidly tailored to a plethora of geometries making it compatible with any size/shape of electrochemical platform. This work presents a novel and highly competitive HER platform and a novel AM technique for exploring the extent of Pt0 nanoparticle dissolution upon the electrode surface, making it an essential study for those seeking to test the stability/catalyst discharge of their given electrochemical platforms.
1. Introduction
In order to support a paradigm shift within the world's energy economy away from fossil fuels (FF) to less polluting renewable sources, sustainable energy vectors must be developed to store and transport the energy generated from renewable sources.1 Green hydrogen is potentially one such vector due to its high energy density (140 MJ kg−1), which exceeds that of coal (24 MJ kg−1) and petrol (44 MJ kg−1).2 Currently a limiting factor to the ubiquitous use of green hydrogen is the cost of its production, which as of this report, is £11.40 per kg (ref. 3) compared to 1 gallon of diesel which is £4.47.4 (in automotive applications these are equivalent quantities).5,6 Therefore, if green hydrogen is to become a viable energy vector the cost of its production has to be significantly decreased. Water splitting within a electrolyser, when its required energy is being drawn from renewable sources, wind, wave, etc., is a promising method of generating green hydrogen gas, as it has no associated carbon emissions (CO, CO2 or CH4).7,8 Hydrogen is produced on the cathode of an electrolyser device via the Hydrogen Evolution Reaction (HER): 2H+ + 2e− → H2.9,10 Currently, Platinum (Pt) based materials are utilised to catalyse the HER as Pt has a near zero H+ binding energy, which results in the HER having a negligible overpotential.11–13 The most common commercially available HER catalyst is Pt, with a 20–40% mass loading, on graphitised carbon (Pt/C).14 Pt is a precious metal in high demand with relatively low earth abundance, which results in it having a very high cost.15 Consequently, current research is focused on improving/maintaining the catalytic efficiency exhibited by Pt/C whilst lowering the content of Pt. See ESI Table 1† that contains numerous studies that utilise Pt based catalysts towards the HER.16–19 For example, Devadas et al.20 loaded Pt nanoparticles (PtNPs) upon carbon nanotubes (CNTs) by chemical linkage to achieve a loading of 1.03 wt%. The CNT/PtNPs exhibited a low HER onset potential of −16 mV (vs. RHE), which is comparable to the HER onset potential exhibited by Pt/C, with a 40 wt% Pt content, of 0 mV (vs. RHE). Anchoring PtNPs to chemically resistive supports, such as carbon nanotubes or graphene has been reported to prevent agglomeration and electrical disconnection of the catalyst material.21 Additionally, corrosion resistance of Pt based catalysts is enhanced by strong surface interactions between PtNP's and a chemically resistive support, which also prevents migration of energetically unstable Pt nanoparticles.14,22Vertically aligned graphene (VG) has been reported within the literature as a high performing support material due to its high conductivity, large surface area and good chemical stability.23–25 A single graphene sheet consists of 2D-hexagonally arranged carbon atoms. The vertical orientation of these 2D graphene sheets beneficially alters the morphology by reducing agglomeration and increasing the number of inter-sheet open channels and exposed active edge sites.10 One study that utilises VG as a surface to disperse Pt nanoparticles is by Zhang et al.26 who supported 3D-VG nanosheet arrays on carbon cloth (CC). The Pt-VGNAs/CC, synthesised by a microwave plasma-enhanced chemical vapour deposition (CVD) method, were explored towards the HER. The Pt-VGNAs/CC were shown to exhibit an overpotential of 60 mV (vs. RHE) at −10 mA cm−2, which is superior to that of commercial Pt/C, where the observed over potential at −10 mA cm−2 was 85 mV (vs. RHE). It is important to note that the respective Pt loadings of the Pt-VGNAs/CC and commercial 20% Pt/C were 41.92 and 44.00 μg cm−2. The superior HER catalysis of the Pt-VGNAs/CC is a result of short ion diffusion pathways and fast heterogeneous electron transfer (HET) upon thin Pt nanoplates, coupled with highly conductive VG. Literature has shown that the immobilisation of PtNP's upon a VG platform results in a highly electrocatalytic electrode. However, if the application of this catalyst is to become scalable within commercial electrolysers, the loading of the PtNPs must be reduced and an efficient, reliable and tailorable technique has to be employed to incorporate the material into the cathodic catalyst layer. Given the above, this work will describe a unique methodology for depositing a minimal amount of Pt onto a VG substrate, and the subsequent incorporation of the Pt/VG into the bulk of screen-printed electrodes (Pt/VG-SPEs). SPEs have numerous advantages over traditional carbon electrodes such as being highly reproducible, scalable and easily tailorable, allowing research to be translated from the laboratory to commercial applications.27–30 ESI Table 1† highlights that the majority of studies use a drop casting technique to modify a GC electrode with electrocatalytic material, which needs to be mechanically polished between each measurement, where the reproducibility is dependent upon the skill of the researcher undertaking the task. In the case of SPEs this step is not required, due to screen printing being a highly reproducible and automated deposition technique. Drop-casting is impractical for the production of commercial catalyst coated membranes, therefore we report a screen printing procedure which is directly transferable to commercial applications, especially where roll-to-roll technologies are employed. The Pt/VG-SPEs produced within this study exhibit excellent HER catalysis, which is derived from the significant distribution of Pt0 nanoparticles mimicking the response of a macro-polycrystalline Pt electrode, whilst having a comparatively low loading of costly Pt0. The Pt/VG-SPEs therefore have the potential to be cheap, highly/mass reproducible and tailorable alternatives to Pt/C as the cathodic material within commercial electrolysers in order to lower the production costs of green hydrogen.
2. Experimental section
2.1. Chemicals and materials
All chemicals (analytical grade or higher) were used as received from Sigma-Aldrich without any further purification. A commercially available catalyst (20% Pt/C) was utilised to benchmark the fabricated VG-SPE variants. Note that the Pt nanoparticles reported within the commercial Pt/C catalyst are >5 nm in diameter.31 A high purity Pt0 Magnetron target (>99.99%) was utilised in the production of the Pt0 nanoparticles found on the Pt/VG-SPEs. Solutions were prepared with deionised water of resistivity not less than 18.2 MΩ cm and were vigorously degassed prior to electrochemical measurements with high purity, oxygen free nitrogen.32,33 Measurements were performed in 0.5 M H2SO4 and the sulfuric acid solution utilised was of the highest possible grade available from Sigma-Aldrich (99.999%, double distilled for trace metal analysis).2.2. Design and fabrication of the vertically aligned graphene screen-printed electrodes (VG-SPEs) and Pt nanoparticle doped VG-SPEs (Pt/VG-SPEs)
The fabrication of the Pt/VG-SPEs is summarised in Fig. 1(A). First the SPE strip, Ag/AgCl reference and dielectric layer were produced using a bespoke conductive graphitic ink and an automated screen-printing technique previously described34 (summarized in stage 1, 2 and 3). Note that the addition of the Ag/AgCl has been included to highlight the versatility of the SPEs as self-contained mobile electrochemical platforms however, an external saturated calomel electrode (SCE) reference was utilised within this study. Stage 4 of the Pt/VG-SPE fabrication involved a radio frequency (RF) plasma enhanced chemical vapour deposition (PECVD) technique, which was utilised to grow the vertically aligned graphene onto flexible graphite paper of 100 μm thickness. This involved inducing an RF electromagnetic wave (1000–2000 W) via a quartz portal into a vacuum chamber, the heated (600 to 900 °C, controlled via a thermocouple) sample stage was located ca. 2 cm from the quartz portal. Methane (with a flow rate of 5–10 SCCM) was utilised as a carbon source, varying from 5 to 100% against a H2 gas atmosphere. Note the pressure of the chamber was kept at between 6 to 12 Pa. A growth time of 20 min was utilised.35 The VG can be observed in Fig. 2. The final stage of the Pt/VG-SPE production (stage 5) was the decoration of the VG-SPE with Pt0 nanoparticles that were grown on the VG nanosheets via a conventional DC magnetron sputtering physical vapour deposition process, a.k.a. MS-PVD. The PVD process used a standard industry plasma source for sputtering, which has a 6-inch-diameter sputtering target made of a high purity Pt0 foil material. Using a deposition rate calculation of the Pt0 magnetron target it was possible to calculate the average weight loading of Pt0 on the Pt/VG-SPEs surface, which was determined to be ca. 4 μg cm−2 with the Pt0 nanoparticles having a dimeter of ca. 3 nm. The defined (geometric) working area of the Pt/VG-SPE was designed to be 0.0314 cm2 for the Pt/VG-SPEs, this can be easily tailored from μm to m in order to suite a particular application. The use of SPEs in order to optimise the deposition of the Pt/VG is essential as they are mass-producible and can be easily scaled to commercial applications, which is not possible using glassy carbon electrodes, typically utilised within the literature. | ||
| Fig. 1 (A) Illustration showing an overview of the fabrication steps required to produce the Pt/VG-SPEs. (B) Diagram of the additively manufactured electrochemical flow cell; the inset shows the actual flow cell utilised. | ||
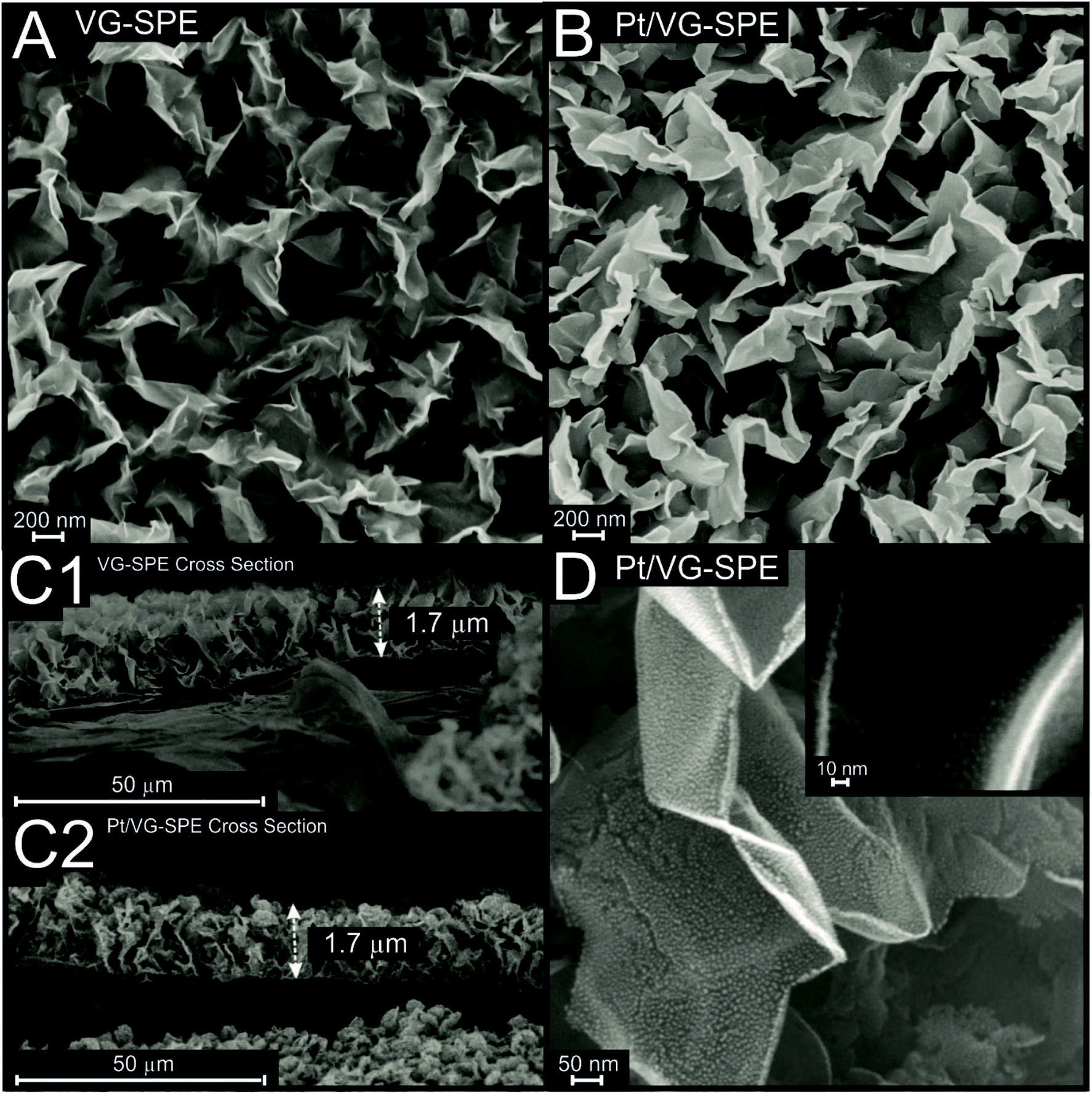 | ||
| Fig. 2 Scanning Electron Microscope (SEM) images from the top and cross-section images of the VG-SPEs (A and C1) and Pt/VG-SPEs (B, C2 and D), respectively. | ||
2.3. Electrochemical measurements
Cyclic voltammetry (CV) and linear sweep voltammetry (LSV) measurements were carried out using an AUTOLAB PGSTAT101 driven by NOVA 2.0 software. The VG-SPE variants acted as working electrodes whilst an external saturated calomel electrode (SCE) reference was utilized rather than the onboard pseudo Ag/AgCl. This allows for a direct comparison between the data collected within this study and other studies within the literature. The inclusion of an onboard pseudo Ag/AgCl is beneficial for non-lab based applications where the use of a larger/more expensive/fragile reference is inappropriate; however during this study its use was not necessary. A large area graphitic SPE acted as the counter electrode. Note that the obtained potential values have been converted to a reversible hydrogen electrode (ERHE = ESCE + 0.059 pH + 0.242), using a measured pH of 0.3 for the 0.5 M H2SO4. The electroactive areas of the Pt/VG-SPEs and VG-SPEs were calculated using the quasi-reversible Randles–Ševćik equation and cyclic voltammetry (CV).36 The scan rates utilized for the CV were (5, 10, 15, 25, 50, 75, 100, 150, 250, and 350 mV s−1) and the final area values were determined from an average of (N = 5). The CVs pertaining to the commercially procured 20% Pt/C used the same reference and counter as described above, whilst the working electrode was a classical glassy carbon electrode (GCE) drop-cast with 1 μg cm−2 of the Pt/C. The HER onset is determined as the potential at which the observed current density deviates from the background current density by the value of 25 μA cm−2.37Electrochemical impedance spectroscopy (EIS) of the VG-SPEs and Pt/VG-SPEs was performed in 0.5 M H2SO4 under their respective HER voltages. ESI Fig. S1† shows the Nyquist's plot for both VG-SPEs and Pt/VG-SPEs. As can be seen, the series resistance (Rs) for VG-SPE and Pt/VG-SPE is 12.7 and 9.13 Ω, respectively. After the introduction of Pt on the VG surface, a reduction in the series resistance can be observed which indicates that Pt facilitates the electron transport process. Also, in the case of the Pt/VG-SPE the formation of two semi-circles is observed, which confirms the presence of two phases in the system (Pt and VG-SPE).
2.4. Surface characterisation
The morphologies of VG-SPE variants were analyzed by scanning electron microscope (SEM) using a Zeiss SUPRA 40 (Carl Zeiss Ltd, Germany). Composition of the electrodes were investigated by energy dispersive X-rays spectroscopy (EDS) using an Apollo 40 SDD (EDAX Inc., USA), coupled to the SEM. Raman spectra were recorded using a Renishaw inVia Raman microscope spectrometer (Renishaw PLC, UK) equipped with an argon laser (λ = 514.3 nm). Atomic force microscopy (AFM) measurement was obtained using a HQ:NSC19/Al BS silicon tip back coated with aluminium (Mikromasch, France) connected to a Smart SPM1000 coupled to an XploRa PLUS V1.2 (using Omegascope AIST-NT v3.5 and LabSepc 6, respectively; Horiba, France) and a vibration isolation table, all carried out using AC mode. Samples were attached to magnetic disks using double-sided tape. High-resolution transmission electron microscopy (HRTEM) images were collected using a JEM-2100 coupled with a OI Aztec 80 mm X-max EDS detector. The X-ray photoelectron spectroscopy (XPS) data was acquired using an AXIS Supra (Kratos, UK), equipped with an Al X-ray source (1486.6 eV) operating at 300 W in order to perform survey scans and 450 W for narrow scans. All X-rays were mono-chromated using a 500 mm Rowland circle quartz crystal X-ray mirror. The angle between X-ray source and analyser was 54.7° with an electron energy analyser: 165 mm mean radius hemispherical sector analyser operating in fixed analyser transmission mode, pass energy 160 eV for survey scans and 40 eV narrow scans. A detector with a delay line detector with multichannel plate was utilised.2.5. Additive manufactured (AM) scanning flow cell
The electrochemical scanning flow cell (see Fig. 1(B)) was designed to have a main working electrode chamber that has a diameter of 9.5 mm. This was sealed with an o-ring (RS Components, UK) and AM base plate using 5 mm M3 Pan Head screws (RS Components, UK). The channels to and from the working electrode were positioned at 60° from each other.38 An external reference electrode was located downstream from the working electrode in a 7.6 mm inlet, sealed with Parafilm (Sigma, UK) as common within the literature. The inlets and outlets to the cell were fitted with 0.79 mm inner diameter Tygon® tubing (Cole Palmer, US), that could connect to the syringe and collector, respectively.The flow cell STL files were designed using Fusion 360 (Autodesk, USA) and converted into build files using PreForm (FormLabs, USA). The designs were produced on a FORM 2 stereolithography 3D-Printer (FormLabs, USA) using FORM 2 Clear Resin (GPCLO4). The flow cells were post-processed by washing with isopropanol and the rough edges removed with Wetordry paper (P2000). Post-curing was performed at 60 °C for 10 min using a FormLabs FORM Cure (405 nm, FormLabs, USA).
2.6. ICP-OES measurements using the bespoke additive manufactured (AM) scanning flow cell
The electrolyte was fully degassed with high purity, oxygen free nitrogen prior to use. This was then injected into the electrochemical scanning flow cell at a constant rate of 4 mL h−1 using an IPS-12 syringe pump (Inovenso Technology Ltd, Turkey) during the course of the electrochemical stability measurements, in order to collect solution quantities large enough for inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis. ICP-OES data was acquired using a Thermo Scientific iCAP6300 Duo in axial mode, where the wavelength and order for Pt are 214.42 and 457, respectively. A 1000 mg L−1 Pt (in HCl) ICP standard was used for benchmarking.39 It should be noted that this technique can be easily performed using the SPE variants developed within this study given their size and geometry, where traditional carbon electrodes would be limited.3. Results and discussion
3.1. Physicochemical characterisation
A thorough physicochemical characterisation of the VG-SPE and Pt/VG-SPEs was performed in order to determine their quality, purity and uniformity of the electrodes surface. This involved using Energy-dispersive X-ray spectroscopy (EDS), Raman spectroscopy, Scanning Electron Microscopy (SEM), Atomic Force Microscopy (AFM) and high-resolution Transmission Electron Microscopy (HR-TEM).Raman characterisation of the VG-SPE and Pt/VG-SPEs was performed, with the obtained spectra being given in ESI Fig. S2,† where the ratios, intensities and full width half-maximum (FWHM) values are provided in Table 1. The Raman spectra of the graphene electrodes confirms the characteristic D (ca. 1380 cm−1), G (ca. 1582 cm−1), 2D (ca. 2706 cm−1) and D + D′ (ca. 2954 cm−1) bands to be consistent with that of vertical aligned graphene.40,41 Graphene's D band is usually referred to disorder/defects in the lattice, the G band is linked to graphitic structures and the 2D band is related to stacking order of the graphene layers.42 In vertically aligned graphene, it is expected to have a prominent D band due to defects and/or the vertical orientation of the graphene presented herein.43 The respective Raman fingerprints of the graphene samples reveals that the intensity ratio D/G (ID/IG) are 0.86 and 1.23 for VG- and Pt-VG-SPE, respectively. The results indicate a higher presence of defects/edges/in-plane stretching in the graphene framework in the Pt-doped sample,44–46 likely due to the high presence of edges and the destruction of the graphene sheets by the Pt0.47 The ratio of I2D/IG is also changed from 0.92 to 0.69 when the VG-SPE is doped with Pt0. The I2D/IG for the VG-SPE sample indicates that the graphene is few-layer graphene,48 as expected due to having vertical graphene in this sample. The increase in the I2D/IG ratio, could indicate an increase in the number of layers, or could be due to the intercalation/doping with Pt0.48 Ratio of D and D′ peaks has been reported to help distinguishing the nature of the defects in graphene (sp3, vacancy-like or graphite boundaries),49 however in VG-SPE samples the ID′ could not be calculated due to the proximity of D′ and G bands (as shown in ESI Fig. S2†). FWHM data of 2D peaks (Table 1) is 60.69 and 120.3 cm−1 for the VG and Pt/VG-SPE, respectively. 60.69 cm−1 for VG-SPE's FWHM confirms the few-layer-like structure/vertical orientation of the graphene, and an increase, since the only difference is the addition of Pt to the VG-SPE surface, to 120.3 cm−1 is likely due to the Pt0 doping of the sample.48 However, it is important to note that the use of FWHM for the estimation the number of layers in graphene has been said to be valid only for AB–stacked few layer graphene.48,50 Next, Raman mapping characterisation of the batches was performed in order to explore the homogeneity of the sample's surface, with the results shown in ESI Fig. S2C and S2D† for VG-SPE and Pt/VG-SPE, respectively. The Raman maps represent the ID/IG ratios for the samples across their surface, showing similar values at every point for both samples.
| Sample | FWHM cm−1 | Peak ratio | |||||||
|---|---|---|---|---|---|---|---|---|---|
| D | G | 2D | I D | I G | I 2D | I D/IG | I 2D/IG | I D/I2D | |
| VG-SPE | 37.82 | 27.62 | 60.69 | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 682 682 |
14756 |
13673 |
0.86 | 0.92 | 0.93 |
| PT/VG-SPE | 63.30 | 45.40 | 120.30 | 9800 | 7961 | 5530 | 1.23 | 0.69 | 1.77 |
Scanning Electron Microscope (SEM) images of the vertical graphene (VG-SPE) and Pt-doped vertical graphene SPEs (Pt/VG-SPE) are presented in Fig. 2, the produced VG has a morphology typical of vertically aligned few-layer graphene.51,52 It is evident from the cross-section images (Fig. 2(C1) and (C2) for VG-SPE and Pt/VG-SPE, respectively) that the both vertical graphene layers exhibit a height of ca. 1.7 μm, which is similar to other reports of VG within the literature.53,54Fig. 2(D) shows that the platinum nanoparticles are uniformly distributed on the surface of the VG edge and basal planes, it can be visually assessed that the particles have an average particle diameter of ca. 3 nm. ESI Fig. S3† shows typical Atomic Force Microscope (AFM) topographic 2D and 3D maps of the Pt/VG-SPE sample. AFM analysis shows that the irregular vertical domains merge at the top of them,55 not allowing the AFM tip to measure the depth of them; however, they map a rough surface that corresponds with the ones seen via SEM (Fig. 2), that covers the entire surface with a network of vertical structures.
High Resolution Transmission Electron Microscope (HRTEM) images of the Pt/VG-SPEs are presented in ESI Fig. S4,† where it can be observed a highly packed uniform distribution of Pt0 particles of ca. 3 nm average diameter, which is similar to the ones reported in the literature.56 The Fourier transforms shown within ESI S4 (D and E)† display cubic and pseudo hexagonal diffraction patterns, which correspond to the 100 and 111 lattice faces.57 This shows the Pt0 nanoparticles to have a highly quality crystalline structure. Additionally, EDS analysis shown in ESI Fig. S5† confirms the presence of Pt0 on the surface of the Pt/VG-SPEs. X-ray photoelectron spectroscopy (XPS) analysis confirmed the Pt0 decorated vertically aligned graphene to be comprised of high quality graphene sheets and polycrystalline Pt0 nanoparticles with the obtained spectra being depicted in ESI Fig. S6.† There is a shift in the C 1s peaks for the Pt against the non-decorated sample this is likely due to a fermi-level change caused by newly decorated/doped (via MS-PVD) metal(Pt)-graphene.58 The characteristic peaks associated with metallic Pt0 (4f) are observed in ESI Fig. S6(A)† from the surface of the Pt/VG-SPE at ca. 70.9 and 67.6 eV.59 The C 1s carbon regions of the Pt/VG-SPEs and VG-SPEs are given in ESI S6(B and C),† the main peak at ca. 284.64 eV corresponds to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C sp2 bonded graphitic structures, and the one ∼285.74 eV corresponds to edge plane like-sites/defects related to C–H.60–62 In addition, peaks related to C–O, CO and O–CO bonding are likely to be due to adsorbed impurities.43
C sp2 bonded graphitic structures, and the one ∼285.74 eV corresponds to edge plane like-sites/defects related to C–H.60–62 In addition, peaks related to C–O, CO and O–CO bonding are likely to be due to adsorbed impurities.43
3.2. Electrochemical characterisation
The cyclic voltammetric responses (CVs) of the VG-SPEs and Pt/VG-SPEs were explored using the near ideal outer-sphere redox probe 1 mM [Ru(NH)6]3+/0.1 M KCl and are shown in ESI Fig. S7.† A plot of peak current verses the square-root of scan rate exhibited a linear response for both the VG-SPEs and Pt/VG-SPEs indicating the electrochemical processes is diffusional in nature. Analysis of the CVs (see ESI Fig. S7†) using the Nicholson's method63 allows the heterogeneous rate constants, k0 to be deduced. Both the VG-SPEs and Pt/VG-SPEs display similar k0 values at 4.09 × 10−3 and 4.06 × 10−3 cm s−1, respectively. Note that as the Pt0 nanoparticles are significantly dispersed across the surface of the VG-SPE, their closeness to each other demonstrates that each Pt0 nanoparticle is not diffusional independent. Such that significantly diffusional overlap occurs over the timescales of the electrochemical measurements meaning that the CV response is characteristic of linear diffusion with peak shaped responses expected of a Pt macroelectrode even though the overall amount of Pt upon the surface is less than that of a solid Pt macroelectrode. Note in ESI Fig. S7,† the magnitude of the voltammetric peak heights of the Pt/VG-SPEs over that of the VG-SPEs, which is as expected given the increase in the surface area, e.g. the coverage of the Pt0 nanoparticles. The estimated electroactive areas (Areal) for VG-SPEs and Pt/VG-SPEs using the quasi-reversible Randles–Ševćik equation,64 are found to correspond to 0.077 and 0.109 cm2, respectively, which are significantly larger than the physical geometric (Ageo) working area (0.0314 cm2) of the screen-printed electrodes, which is as expected. For comparison, the Areal and Ageo for the Pt/C was calculated to be 0.055 and 0.071 cm2 respectively. The electroactive area (Areal) of the Pt/VG-SPEs was determined more accurately using the hydrogen adsorption methodology described by Rodríguez et al.65 The obtained electroactive area for the Pt/VG-SPE using this method was determined to be 0.096 cm2, which closely matches the electroactive areas obtained from the analysis of the CVs using the quasi-reversible Randles–Ševćik equation (see above). Note that the current densities given within this manuscript have been normalised against the geometric area of the electrodes used to derive them.3.3. Application of the VG-SPEs and Pt/VG-SPEs towards the hydrogen evolution reaction (HER)
Next, the electrochemical characterisation of the VG-SPE and Pt/VG-SPEs was explored towards the HER using linear sweep voltammetry (LSV) in 0.5 M H2SO4. Firstly, it was important to benchmark the HER activity of a polycrystalline solid Pt electrode and 20% Pt/C in order for an accurate comparison between the VG-SPEs and Pt/VG-SPEs (see Fig. 3). As expected the polycrystalline Pt electrode and the Pt/C displayed highly efficient HER catalysis with HER over potentials (recorded at −10 mA cm−2) of 73 mV (vs. RHE) and 84 mV (vs. RHE), respectively. Next, the VG-SPE and Pt/VG-SPEs were explored towards the HER with the obtained LSVs displayed in Fig. 3. The VG-SPE exhibited a large overpotential at 789 mV (vs. RHE) that is significantly more electronegative than the polycrystalline Pt electrode or Pt/C (see Fig. 3A), which is characteristic of unmodified carbon based electrode materials.27 The Pt/VG-SPE outperforms the polycrystalline Pt electrode and the Pt/C, in regards to their HER overpotential with a value of 47 mV (vs. RHE). Note that the Pt/VG-SPEs on a Pt per cm2 basis have ×10 less Pt, (ca. 4 μg cm−2) than the typical commercially available 20% Pt/C catalyst (ca. 44.00 μg cm−2), value reported by Zhang et al.26 This obtained HER overpotential was utilised to gauge the intra-repeatedly of the Pt/VG-SPEs; the relative standard deviation (%RSD) for the Pt/VG-SPEs was found to be 0.9% (N = 5). The small %RSD attests to the repeatability of the Pt/VG-SPE production method and reliability of their HER signal output. | ||
| Fig. 3 (A) Typical linear sweep voltammograms (LSVs) showing the HER activity of a polycrystalline platinum macroelectrode, 20% Pt/Vulcan carbon, VG-SPE and Pt/VG-SPE. Solution composition: 0.5 M H2SO4. Scan rate: 25 mV s−1. (B) Tafel slopes corresponding to the Faradaic regions of the LSVs shown in (A). | ||
In order to determine the rate limiting step of the HER reaction mechanism occurring at each of the explored electrodes, Tafel analysis was performed on the faradaic sections of the LSVs shown in Fig. 3(A) with the obtained Tafel curves being shown in Fig. 3(B).66 The VG-SPE, Pt/VG-SPE, polycrystalline Pt0 and Pt/C exhibited Tafel slopes of 97, 27, 31 and 30 mV dec−1, respectively. These Tafel values indicate that for the VG-SPE the initial Volmer H+ adsorption step is the rate-limiting step and for the Pt/VG-SPE, polycrystalline Pt and Pt/C it is the desirable Tafel discharge step that limits the reaction rate. With the Pt/VG-SPEs having ca. ×10 lower loading of Pt0 than the Pt/C and yet exhibiting more beneficial HER catalysis it is clear that the Pt/VG-SPEs have potential to be utilised as the cathodic material within electrolyser devices. The excellent intrinsic HER electrocatalysis exhibited by the Pt/VG-SPE may be further confirmed via calculations of hydrogen turnover frequency (ToF), mass activity (MA) and specific activity (SA), which are displayed in the ESI.† The Pt/VG-SPE exhibited an exceptional ToF value of 8.77 s−1, higher than optimal HER catalysts reported in the wider literature.67–70 The Pt/VG-SPE also exhibited high mass and specific activity values of 52.31 A mg−1 and 6.24 A m−2, respectively, in comparison to other state of the art HER catalysts (in acidic media).71 The efficient HER activity displayed by the Pt/VG-SPE which is competitive with other Pt based catalysts reported within the literature is likely derived from the Pt0 nanoparticle deposition technique, which ensures the Pt0 is located upon the high surface area VG support. This results in a very large surface area of exposed Pt0 active sites to catalyse the HER, when compared to Pt/C or a macro polycrystalline Pt electrode, thus explaining the marginal increase in HER activity. It is worth noting that this study was performed using a typical three electrode system as is common within the literature, however, future work will seek to deposit the Pt/VG-SPE onto a membrane where the catalyst can be tested within a functional PEM electrolyser.
An important consideration for the applicability of a catalyst as the cathode material within an electrolyser is its stability and durability regarding the HER electrochemical signal output. In order to investigate the stability of the Pt/VG-SPEs HER signal output in an acidic solution, chronoamperometry at −0.1 V (vs. RHE) for 36000 seconds and 1000 repeat CV scans were carried out (see Fig. 4). The Pt/VG-SPE exhibited a significant decrease in HER activity over the course of 1000 repeat scans, with the observed current decreasing from −28.8 to −3.85 mA cm−2 from at 0.1 V (vs. RHE) from the 1st to the 1000th scan. There was also a significant decrease in the Pt/VG-SPEs HER signal output during the chronoamperometry test (see Fig. 4(B)). Fig. 4(B) shows that there was a rapid decrease in observed current density from −23.5 to −8.24 mA cm−2 in the initial 355 seconds (magnified in ESI Fig. 8†), after which there was a more gradual decline to −5 mA cm−2 at 4275 seconds. The HER signal then remained somewhat stable with only a small decline in HER activity to −3.73 mA cm−2 by 36000 seconds. It is clear from the data presented above that the Pt/VG-SPEs exhibit poor stability in acidic conditions. In order to investigate whether the Pt/VG-SPEs exhibited poor stability due to the discharging of Pt0 nanoparticles from the surface of the Pt/VG-SPEs, ICP-OES was utilised.72,73 ICP-OES coupled with electrochemical stability measurements (chronoamperometry for 36000 seconds at −0.1 V (vs. RHE) whilst the electrolyte (0.5 M H2SO4) was flowed over the electrodes surface at 4 mL h−1), in order to assess whether the Pt0 nanoparticles were detaching from the Pt/VG-SPEs surface, and thus causing the observed decrease in HER activity. Fig. 4(A inset) exhibits the rate of Pt0 detachment (ng mL−1 h−1) from the surface of the Pt/VG-SPE within 0.5 M H2SO4 over a 10 h period. It is evident that a minimal quantity of Pt0 has detached from the Pt/VGE-SPE after 5 h, where the concentration of Pt0 is 0.01 ng mL−1. After 6 h the concentration of Pt0 increases to 253.32 ng mL−1, then levels out at ca. 285 ng mL−1 until 10 h. It is likely that the dramatic increase in Pt0 concentration within the electrolyte is a result of damage to the structural integrity of the VG support, leading to a degradation of the VG sheets and detachment of the Pt0 from the VG to the solution. The decoration of the VG with Pt nanoparticles via MS-PVD likely leads to weak chemical bonds between the graphene sheets and Pt0, which contributes to the observed detachment under these conditions. It has been previously described by Borup et al.74 that Pt0 dissolution is a factor of particle size with Pt0 particles that have a diameter less than 5 nm having a dissolution rate significantly greater than Pt0 particles with a diameter greater than 5 nm (i.e. 1–2 nm particles were observed to have a ×20 greater dissolution rate than 5 nm Pt0 nanoparticles). Further work is currently underway that is attempting to prevent the Pt dissolution from the VG surface. The degradation of graphene sheets as a result of the HER occuring, has been previously reported in pristine horizontal CVD grown graphene.75 From the above data we deduce that it is likely that the observed decrease in HER activity is due to a combination of Pt0 dissolution and VG degradation. This is supported by Fig. 4(C) that displays a stability test (1000 repeat CV scans as above) for a VG-SPE. The VG-SPEs’ HER signal output remained relatively stable in terms of its achievable current from scan 1 to 100, however from 100 to 1000 there is a drastic decrease from 179.01 mA to 48.22 mA by −1.2 V (vs. RHE). This degradation in HER signal output is likely a result of the VG sheets structurally breaking due to the mechanical force of hydrogen bubbling and electrochemical perturbation, which has been seen in the case of horizontally alligned graphene.75 This reduces the number of electrical pathways within the VG-SPE with contributions from the weak van der Waals forces of the Pt0 upon the graphene surface; this likely explains the discharge of the Pt0 observed using ICP-OES after 5 h of chronoamperometry (see Fig. 4(A)).
 | ||
| Fig. 4 (A) Cyclic stability examination of a Pt/VG-SPE via LSV (scan rate: 100 mV s−1 (vs. RHE)) was performed between the potential range of +0.4 to −0.1 V, repeated for 1000 cycles, inset showing Pt0 dissolution from a Pt/VG-SPE recorded using ICP-OES. Solution composition: 0.5 M H2SO4. (B) shows chronoamperometry using a Pt/VG-SPE for 36000 seconds, solution composition: 0.5 M H2SO4. (C) Cyclic stability examination of a VG-SPE via LSV (scan rate: 100 mV s−1 (vs. RHE)) was performed between the potential range of +0.4 to −0.1 V, repeated for 1000 cycles. Solution composition: 0.5 M H2SO4. | ||
4. Conclusions
We have explored the electrocatalytic performance of VG-SPEs and Pt/VG-SPEs towards the HER in an acidic electrolyte (0.5 M H2SO4) and revealed the Pt/VG-SPEs to exhibit remarkable HER activity with a comparatively low loading of Pt0 per cm2 (×10 lower than that of 20% commercial Pt/C). The competitive performance of Pt/VG-SPEs can be attributed to the ca. 3 nm Pt0 nanoparticles that are significantly dispersed upon a high surface area 3D VG support, resulting in maximal exposure of the Pt0 active sites for HER catalysis. The screen-printing technique utilised within this study more closely mimics the manufacturing techniques implemented in the production of commercial PEM electrolyser membranes when compared to deposition techniques used in wider literature, where drop-casting is typically employed to modify given electrochemical platforms. The Pt/VG-SPEs utilised within this study are easily tailored and cheaply mass manufactured, demonstrating them to be promising cathodic catalyst systems for application within commercial electrolysers. That is on the proviso that their electrochemical stability in acidic media can be improved in subsequent work. The ICP-OES technique utilised within this study allowed us to accurately measure the extent of Pt0 nanoparticle dissolution from the electrodes surface. The described technique can be further adapted to measure a plethora of different electrode designs and materials, offering opportunities to benchmark the stability of novel catalyst materials for electrolysers and other electrochemical devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding is also from the Engineering and Physical Sciences Research Council (Reference: EP/P007767/1 and EP/N0011877/1). The Manchester Fuel Cell Innovation Centre is funded by the European Regional Development Fund.References
- A. Mohammadi and M. Mehrpooya, Energy, 2018, 158, 632–655 CrossRef CAS.
- G. Thomas , presented in part at the Hydrogen Program Review, Livermore, CA, May, 2000.
- P. E. L. o. b. o. Cadent, HyMotion - Network-supplied hydrogen unlocks low carbon transport opportunities, 2019 Search PubMed.
- Global Petrol Prices, https://www.globalpetrolprices.com/United-Kingdom/diesel_prices/ (accessed July 5, 2020).
- O. J. Guerra, J. Eichman, J. Kurtz and B.-M. Hodge, Joule, 2019, 3, 2425–2443 CrossRef.
- A. A. Janovic and R. Haas, Energy Policy, 2018, 123, 280–288 CrossRef.
- D. Xu, L. Dong and J. Ren, in Hydrogen Economy, ed. A. Scipioni, A. Manzardo and J. Ren, Academic Press, 2017, pp. 35–54, DOI:10.1016/B978-0-12-811132-1.00002-X.
- F. M. Sapountzi, J. M. Gracia, C. J. Weststrate, H. O. A. Fredriksson and J. W. Niemantsverdriet, Prog. Energy Combust. Sci., 2017, 58, 1–35 CrossRef.
- J. Benson, M. Li, S. Wang, P. Wang and P. Papakonstantinou, ACS Appl. Mater. Interfaces, 2015, 7, 14113–14122 CrossRef CAS PubMed.
- F. Wang, X. Yang, B. Dong, X. Yu, H. Xue and L. Feng, Electrochem. Commun., 2018, 92, 33–38 CrossRef CAS.
- M. Liu, Y. Niu, L. Yang, L. Yao, D. Lin, C. Shao, D. Sun, L. Li and C. Wang, Diamond Relat. Mater., 2018, 90, 79–83 CrossRef CAS.
- M. Bernt, A. Siebel and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, F305–F314 CrossRef CAS.
- L. Yin, T. Yang, X. Ding, M. He, W. Wei, T. Yu and H. Zhao, Electrochem. Commun., 2018, 94, 59–63 CrossRef CAS.
- M. Karuppannan, Y. Kim, S. Gok, E. Lee, J. Y. Hwang, J.-H. Jang, Y.-H. Cho, T. Lim, Y.-E. Sung and O. J. Kwon, Energy Environ. Sci., 2019, 12, 2820–2829 RSC.
- M. Wang, L. Jiang, Q. Li and X. Zhou, Int. J. Hydrogen Energy, 2019, 44, 31062–31071 CrossRef CAS.
- X. Shang, Z.-Z. Liu, S.-S. Lu, B. Dong, J.-Q. Chi, J.-F. Qin, X. Liu, Y.-M. Chai and C.-G. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 43561–43569 CrossRef CAS PubMed.
- A. H. Ghanim, J. G. Koonce, B. Hasa, A. M. Rassoolkhani, W. Cheng, D. W. Peate, J. Lee and S. Mubeen, Front. Chem., 2018, 6, 523 CrossRef CAS PubMed.
- Z. Jiang, J. Ren, Y. Li, X. Zhang, P. Zhang, J. Huang, C. Du and J. Chen, Dalton Trans., 2019, 48, 8920–8930 RSC.
- S. H. Ahn, S. J. Yoo, H.-J. Kim, D. Henkensmeier, S. W. Nam, S.-K. Kim and J. H. Jang, Appl. Catal., B, 2016, 180, 674–679 CrossRef CAS.
- B. Devadas and T. Imae, Electrochem. Commun., 2016, 72, 135–139 CrossRef CAS.
- Y. Shao, S. Zhang, C. Wang, Z. Nie, J. Liu, Y. Wang and Y. Lin, J. Power Sources, 2010, 195, 4600–4605 CrossRef CAS.
- S. Sun, G. Zhang, D. Geng, Y. Chen, R. Li, M. Cai and X. Sun, Angew. Chem., Int. Ed., 2011, 50, 422–426 CrossRef CAS PubMed.
- K. Mouralova, R. Zahradnicek and J. Bednar, Diamond Relat. Mater., 2019, 97, 107439 CrossRef CAS.
- J. Zhan, S. Deng, Y. Zhong, Y. Wang, X. Wang, Y. Yu, X. Xia and J. Tu, Nano Energy, 2018, 44, 265–271 CrossRef CAS.
- Z. Zhang, W. Li, M. F. Yuen, T.-W. Ng, Y. Tang, C.-S. Lee, X. Chen and W. Zhang, Nano Energy, 2015, 18, 196–204 CrossRef CAS.
- H. Zhang, W. Ren, C. Guan and C. Cheng, J. Mater. Chem. A, 2017, 5, 22004–22011 RSC.
- S. J. Rowley-Neale, D. A. C. Brownson, G. C. Smith, D. A. G. Sawtell, P. J. Kelly and C. E. Banks, Nanoscale, 2015, 7, 18152–18168 RSC.
- S. J. Rowley-Neale, M. Ratova, L. T. N. Fugita, G. C. Smith, A. Gaffar, J. Kulczyk-Malecka, P. J. Kelly and C. E. Banks, ACS Omega, 2018, 3, 7235–7242 CrossRef CAS PubMed.
- S. J. Rowley-Neale, C. W. Foster, G. C. Smith, D. A. C. Brownson and C. E. Banks, Sustainable Energy Fuels, 2017, 1, 74–83 RSC.
- S. J. Rowley-Neale, D. A. C. Brownson and C. E. Banks, Nanoscale, 2016, 8, 15241–15251 RSC.
- Sigma-Aldrich, https://www.sigmaaldrich.com/catalog/product/aldrich/738549?lang=en®ion=GB, (accessed 07/01/2019).
- A. B. Laursen, A. S. Varela, F. Dionigi, H. Fanchiu, C. Miller, O. L. Trinhammer, J. Rossmeisl and S. Dahl, J. Chem. Educ., 2012, 89, 1595–1599 CrossRef CAS.
- D. Marin, F. Medicuti and C. Teijeiro, J. Chem. Educ., 1994, 71, A277 CrossRef CAS.
- S. J. Rowley-Neale, G. C. Smith and C. E. Banks, ACS Appl. Mater. Interfaces, 2017, 9, 22539–22548 CrossRef CAS PubMed.
- J. Wang, M. Zhu, R. A. Outlaw, X. Zhao, D. M. Manos and B. C. Holloway, Carbon, 2004, 42, 2867–2872 CrossRef CAS.
- A. G.-M. Ferrari, C. W. Foster, P. Kelly, D. A. C. Brownson and C. E. Banks, Biosensors, 2018, 8, 53 CrossRef CAS PubMed.
- L. Cheng, W. Huang, Q. Gong, C. Liu, Z. Liu, Y. Li and H. Dai, Angew. Chem., Int. Ed., 2014, 53, 7860–7863 CrossRef CAS PubMed.
- S. O. Klemm, A. Karschin, A. K. Schuppert, A. A. Topalov, A. M. Mingers, I. Katsounaros and K. J. J. Mayrhofer, J. Electroanal. Chem., 2012, 677, 50–55 CrossRef.
- Sigma-Aldrich, https://www.sigmaaldrich.com/catalog/product/sial/19078?lang=en®ion=GB&cm_sp=Insite-_-caContent_prodMerch_cooccuranceModel-_-prodMerch10-1, (accessed 20/01/2020).
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- D. Graf, F. Molitor, K. Ensslin, C. Stampfer, A. Jungen, C. Hierold and L. Wirtz, Nano Lett., 2007, 7, 238–242 CrossRef CAS PubMed.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- S. Ghosh, K. Ganesan, S. R. Polaki, T. R. Ravindran, N. G. Krishna, M. Kamruddin and A. K. Tyagi, J. Raman Spectrosc., 2014, 45, 642–649 CrossRef CAS.
- O. Akhavan, Carbon, 2010, 48, 509–519 CrossRef CAS.
- S. Li, Y. Hu, Q. Xu, J. Sun, B. Hou and Y. Zhang, J. Power Sources, 2012, 213, 265–269 CrossRef CAS.
- E. Sandoz-Rosado, W. Page, D. O'Brien, J. Przepioski, D. Mo, B. Wang, T.-T. Ngo-Duc, J. Gacusan, M. W. Winter, M. Meyyappan, R. D. Cormia, S. Takahashi and M. M. Oye, J. Mater. Res., 2014, 29, 417–425 CrossRef CAS.
- G. Jiahao, Z. Jianguo, Z. Hanqing, F. Yongshuang, M. Kun, H. Hao, C. Junming and W. Xuchun, R. Soc. Open Sci., 2018, 5, 180925 CrossRef PubMed.
- Y. Hao, Y. Wang, L. Wang, Z. Ni, Z. Wang, R. Wang, C. K. Koo, Z. Shen and J. T. L. Thong, Small, 2010, 6, 195–200 CrossRef CAS PubMed.
- A. Eckmann, A. Felten, A. Mishchenko, L. Britnell, R. Krupke, K. S. Novoselov and C. Casiraghi, Nano Lett., 2012, 12, 3925–3930 CrossRef CAS PubMed.
- S. Latil, V. Meunier and L. Henrard, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 201402 CrossRef.
- A. Malesevic, R. Vitchev, K. Schouteden, A. Volodin, L. Zhang, G. V. Tendeloo, A. Vanhulsel and C. V. Haesendonck, Nanotechnology, 2008, 19, 305604 CrossRef PubMed.
- A. Malesevic, R. Kemps, A. Vanhulsel, M. P. Chowdhury, A. Volodin and C. V. Haesendonck, Jpn. J. Appl. Phys., 2008, 104, 084301 CrossRef.
- N. Soin, S. S. Roy, T. H. Lim and J. A. D. McLaughlin, Mater. Chem. Phys., 2011, 129, 1051–1057 CrossRef CAS.
- M. Zhu, R. Outlaw, M. Bagge-Hansen, H. Chen and D. Manos, Carbon, 2011, 49, 2526–2531 CrossRef CAS.
- L. Cui, K. Cui, H. Ci, K. Zheng, H. Xie, X. Gao, Y. Zhang and Z. Liu, Nanomaterials, 2019, 9, 558 CrossRef CAS PubMed.
- Y. Xin, J.-G. Liu, Y. Zhou, W. Liu, J. Gao, Y. Xie, Y. Yin and Z. Zou, J. Power Sources, 2011, 196, 1012–1018 CrossRef CAS.
- S. Yang, N.-Y. Park, J. W. Han, C. Kim, S.-C. Lee and H. Lee, Chem. Commun., 2012, 48, 257–259 RSC.
- D. Arjun, A. Rafik, C.-D. Horacio, L. James and B. Matthias, APL Mater., 2013, 1, 042107 CrossRef.
- J. Song, C. Zhu, B. Xu, S. Fu, M. Engelhard, R. Ye, D. Du, S. Beckman and Y. Lin, Adv. Energy Mater., 2017, 7, 1601555 CrossRef.
- N. G. Shang, P. Papakonstantinou, M. McMullan, M. Chu, A. Stamboulis, A. Potenza, S. S. Dhesi and H. Marchetto, Adv. Funct. Mater., 2008, 18, 3506–3514 CrossRef CAS.
- H. Medina, Y.-C. Lin, C. Jin, C.-C. Lu, C.-H. Yeh, K.-P. Huang, K. Suenaga, J. Robertson and P.-W. Chiu, Adv. Funct. Mater., 2012, 22, 2123–2128 CrossRef CAS.
- A. Nikitin, L.-A. Näslund, Z. Zhang and A. Nilsson, Surf. Sci., 2008, 602, 2575–2580 CrossRef CAS.
- R. S. Nicholson, Anal. Chem., 1965, 37, 1351–1355 CrossRef CAS.
- A. G.-M. Ferrari, C. W. Foster, P. J. Kelly, D. A. C. Brownson and C. E. Banks, Biosensors, 2018, 8, 53 CrossRef CAS PubMed.
- J. M. D. Rodríguez, J. A. H. Melián and J. P. Peña, J. Chem. Educ., 2000, 77, 1195 CrossRef.
- M. C. Tavares, S. A. S. Machado and L. H. Mazo, Electrochim. Acta, 2001, 46, 4359–4369 CrossRef CAS.
- J. P. Hughes, P. L. dos Santos, M. P. Down, C. W. Foster, J. A. Bonacin, E. M. Keefe, S. J. Rowley-Neale and C. E. Banks, Sustainable Energy Fuels, 2020, 4, 302–311 RSC.
- K. Jiang, B. Liu, M. Luo, S. Ning, M. Peng, Y. Zhao, Y.-R. Lu, T.-S. Chan, F. M. F. de Groot and Y. Tan, Nat. Commun., 2019, 10, 1743 CrossRef PubMed.
- A. Kalasapurayil Kunhiraman, M. Ramasamy and S. Ramanathan, Int. J. Hydrogen Energy, 2017, 42, 9881–9891 CrossRef CAS.
- K. Li, Y. Li, Y. Wang, J. Ge, C. Liu and W. Xing, Energy Environ. Sci., 2018, 11, 1232–1239 RSC.
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. L. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Adv. Mater., 2019, 31, 1806296 CrossRef PubMed.
- Y. Ding, H. Zheng, J. Li, S. Zhang, B. Liu and C. Ekberg, Materials, 2019, 12, 1205 CrossRef CAS PubMed.
- W. R. Reynolds, P. Plucinski and C. G. Frost, Catal. Sci. Technol., 2014, 4, 948–954 RSC.
- R. L. Borup, A. Kusoglu, K. C. Neyerlin, R. Mukundan, R. K. Ahluwalia, D. A. Cullen, K. L. More, A. Z. Weber and D. J. Myers, Curr. Opin. Electrochem., 2020, 21, 192–200 CrossRef CAS.
- A. G.-M. Ferrari, D. A. C. Brownson and C. E. Banks, Sci. Rep., 2019, 9, 15961 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr04336b |
| This journal is © The Royal Society of Chemistry 2020 |