
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicroneedles loaded with anti-PD-1–cisplatin nanoparticles for synergistic cancer immuno-chemotherapy†
Xinmiao
Lan
a,
Wangyong
Zhu
a,
Xinshuo
Huang
bc,
Yingjie
Yu
d,
Haihua
Xiao
ef,
Lijian
Jin
 g,
Jingya Jane
Pu
a,
Xi
Xie
b,
Juncong
She
b,
Vivian Wai Yan
Lui
h,
Hui-Jiuan
Chen
*bc and
Yu-xiong
Su
*a
g,
Jingya Jane
Pu
a,
Xi
Xie
b,
Juncong
She
b,
Vivian Wai Yan
Lui
h,
Hui-Jiuan
Chen
*bc and
Yu-xiong
Su
*a
aDivision of Oral and Maxillofacial Surgery, Faculty of Dentistry, The University of Hong Kong, Hong Kong SAR, China. E-mail: richsu@hku.hk
bState Key Laboratory of Optoelectronic Materials and Technologies, School of Electronics and Information Technology, Sun Yat-Sen University, Guangzhou, China. E-mail: chenhuix5@mail.sysu.edu.cn
cThe First Affiliated Hospital of Sun Yat-Sen University, Guangzhou, China
dInstitute of Translational Medicine, The First Affiliated Hospital of Shenzhen University, Shenzhen Second People's Hospital, Shenzhen, China
eBeijing National Laboratory for Molecular Sciences, State Key Laboratory of Polymer Physics and Chemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing, China
fUniversity of Chinese Academy of Sciences, Beijing, China
gDivision of Periodontology and Implant Dentistry, Faculty of Dentistry, The University of Hong Kong, Hong Kong SAR, China
hSchool of Biomedical Sciences, Faculty of Medicine, The Chinese University of Hong Kong, Hong Kong S.A.R., China
First published on 9th September 2020
Abstract
Programmed cell death protein-1 (PD-1) on T-cells combined with programmed cell death ligand-1 (PD-L1) critically accounts for tumor immune evasion. Anti-PD-1 (aPD-1) blocks the binding of PD-1 to PD-L1, thus allowing T-cell activation for tumor cell eradication. Currently, the major challenges for cancer immunotherapy are how to improve the response rate and overcome drug resistance. Dermal administration turns out to be a promising route for immunotherapy since skin is a highly active immune organ containing a large population of resident antigen-presenting cells. Microneedle arrays can pierce the immune-cell-rich epidermis, leading to a robust T-cell response in the microenvironment of tumor cells. Herein, we successfully developed a microneedle patch loaded with pH-responsive tumor-targeted lipid nanoparticles (NPs), which allows local delivery of aPD-1 and cisplatin (CDDP) precisely to cancer tissues for cancer therapy. For in vivo studies, aPD-1/CDDP@NPs delivered through microneedles effectively boosted the immune response, thereby a remarkable effect on tumor regression was realized. Synergistic anticancer mechanisms were therefore activated through robust microneedle-induced T-cell response, blockage of PD-1 in T-cells by aPD-1, and an increase in direct cytotoxicity of CDDP in tumor cells. Strikingly, transdermal delivery using MNs increased the response rate in the animal model unresponsive to aPD-1 systemic therapy. This exhibited promise in the treatment of immunotherapy-unresponsive cancers. Taken together, microneedle-mediated local delivery of nano-encapsulated chemotherapeutic and immunotherapeutic agents at tumor skin sites provides a novel treatment strategy and insights into cancer therapy.
1. Introduction
Cancer is the second leading cause of death after cardiovascular diseases. Although chemotherapy remains the main strategy for cancer therapy, immune checkpoint inhibitors have led to a paradigm shift in cancer treatment recently. Programmed cell death protein-1 (PD-1) expressed on T-cells binds to its receptor programmed cell death ligand-1 (PD-L1) on tumor cells, leading to inactivation of T-cell immune responses and contributing to tumor immune evasion.1 The immune checkpoint inhibitor anti-PD-1 (aPD-1) can block the binding of PD-1 to PD-L1, thereby activating the T-cell immune responses to tumor cells.2 The application of immune checkpoint inhibitors has rapidly increased for a wide spectrum of cancers and has demonstrated encouraging treatment outcomes.3 However, the limitations of immune checkpoint inhibitors hinder their further clinical application. The overall objective response rate to aPD-1 was reported to be approximately 33% in advanced melanoma,4 and only 20% in head and neck cancer.5 A high percentage of patients exhibited primary resistance or developed adaptive resistance during the treatment course.6–8 Furthermore, the immune response activated by aPD-1 is usually not persistent.2,9 Improving the response rate and overcoming the resistance remain the major challenges in immunotherapy.Immune profiling in the tumor microenvironment plays a critical role in chemotherapy and immunotherapy. Clinical studies demonstrated that stromal fibroblasts in the tumor microenvironment confer cis-diammineplatinum(II) (CDDP) chemo-resistance, and CD8+ T cells abolish the cancer-associated fibroblast-mediated chemo-resistance through CD8+ T-cell-derived interferon (IFN)-γ (a major effector cytokine of CD8+ T-cell), which reveals the interplay between chemotherapy and immunotherapy.10–12 The clonal expansion of T-cells in the tumor microenvironment correlates with a better response to chemotherapy;13 therefore, the effectiveness of chemotherapy relies on the induction of a durable anticancer immune response.14,15 Pre-existing CD8+ T-cells in the tumor microenvironment also predict the efficacy of aPD-1 therapy. This represents a novel treatment strategy in which the outcome of chemotherapy and immunotherapy can be improved by altering the tumor microenvironment. It has been reported that combination therapies are potentially synergistic and more effective than monotherapies to combat resistance, because tumors evade the immune response through multiple pathways.1,16 Nanoparticles have been utilized in our study to exploit the synergistic effect.
A microneedle (MN) is a minimally invasive device that can penetrate the stratum corneum of the skin or mucosa for transdermal delivery. Skin is a highly active immune organ containing a large population of resident antigen-presenting cells. MNs piercing the immune-cell-rich epidermis can provoke robust immune responses by activating T-cells. Recently, several studies have used MNs to assist the immunotherapy for melanoma. Ye et al. developed an MN-based transcutaneous delivery approach for immunotherapy targeting PD-1 and immunosuppressive enzyme indoleamine 2,3-dioxygenase for the treatment of melanoma in mice.17 The device has been demonstrated to facilitate sustained release of drugs and enhance T cell immunity. The same research group further reported that MNs loaded with aPD-1 and glucose oxidase NPs could induce more tumor-infiltrating CD8+ T cells and achieved enhanced anti-melanoma effects.18 Another study reported the use of MN-assisted co-delivery of pTRP-2 and paclitaxel targeting dendritic cells for transcutaneous immunotherapy in a melanoma murine model.19 These studies have demonstrated that an MN is a promising tool for boosting the immune response for cancer therapy.
Our previous study reported MNs loaded with pH-responsive tumor-targeted lipid-coated cisplatin nanoparticles as a promising transdermal delivery system for superficial cancer treatment.20 In this study, aPD-1 delivered by MNs exhibited promise in the treatment of immunotherapy-unresponsive cancers and we further developed a novel MN loaded with aPD-1/CDDP@NPs (Scheme 1) to facilitate synergistic immuno-chemotherapy. Using an immunocompetent murine tumor homograft model, we demonstrated that the robust T-cell response activated by MNs can enhance the efficacy of both aPD-1 and CDDP. Synergistic immuno-chemotherapeutic effects were sustained through robust T-cell response activated by MNs, blockage of PD-1 in T cells by aPD-1, and direct cytotoxicity of CDDP in tumor cells. The MN-mediated host immune response could augment the aPD-1-activated T-cell immunity, which in turn would enhance the direct killing of cancer cells by CDDP. This could increase the response rate in the animal model unresponsive to aPD-1 systemic therapy, which is promising in cancer immunotherapy.
 | ||
| Scheme 1 Schematic illustration of the synergistic effects of immuno-chemotherapy delivered through a microneedle. aPD-1 and CDDP were encapsulated into NPs for the combination of chemotherapy and immunotherapy. Then, the NPs were embedded into the MN for transdermal delivery. aPD-1 could competitively block the binding of PD-L1 to PD-1, leading to the activation of T-cells. Meanwhile, intracellular release of CDDP could induce direct cytotoxicity to the tumor cells. | ||
2. Results and discussion
2.1 Characterization of aPD-1/CDDP@NPs
The aPD-1/CDDP@NPs were synthesized through a reverse-phase microemulsion method. A well-prepared CDDP precursor, cis-[Pt(NH3)2(H2O)2]2(NO3)2, was utilized to increase the solubility and further facilitate the encapsulation of CDDP. 1,2-Dioleoyl-sn- glycero-3-phosphate (DOPA) formed a bilayer to enable linking with other lipids, such as 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (ammonium salt) (DSPE-PEG-AA) and cholesterol. These lipids present the advantages of tumor targeting and selectivity, since DSPE-PEG-AA can bind with a tumor-overexpressed sigma receptor.21–23 Additionally, aPD-1 was loaded onto the outer layer of the NPs because of hydrophobic and electrostatic non-specific interactions at the hydrophobic region on the surface of aPD-1 protein.24 The encapsulation efficiency of aPD-1 was 50%, as determined using an enzyme linked immunosorbent assay (ELISA) kit. No significant antibody release was detected within one month of storage at 4 °C. As depicted in the transmission electron microscopy (TEM) image, NPs exhibited a spherical morphology with a diameter of approximately 57.6 nm (Fig. 1A), which was consistent with the result of dynamic light scattering (DLS) (Fig. 1B). The particle size was double checked by measuring the TEM image and directly reading from the DLS using a Nanotrac Wave II dynamic light scattering particle analyzer. The average diameter detected by DLS was 55.5 nm. To evaluate the release of aPD-1, NPs were incubated at 37 °C in PBS with various pH values. A higher proportion of aPD-1 dissociated from the NPs in an acidic environment within 24 h, whereas Pt in the inner layer of the NPs could release sustainably for 72 h according to our previous study20 (Fig. 1C). The aPD-1/CDDP@NPs facilitated the synergistic delivery of chemotherapeutic agents and immunotherapeutic agents.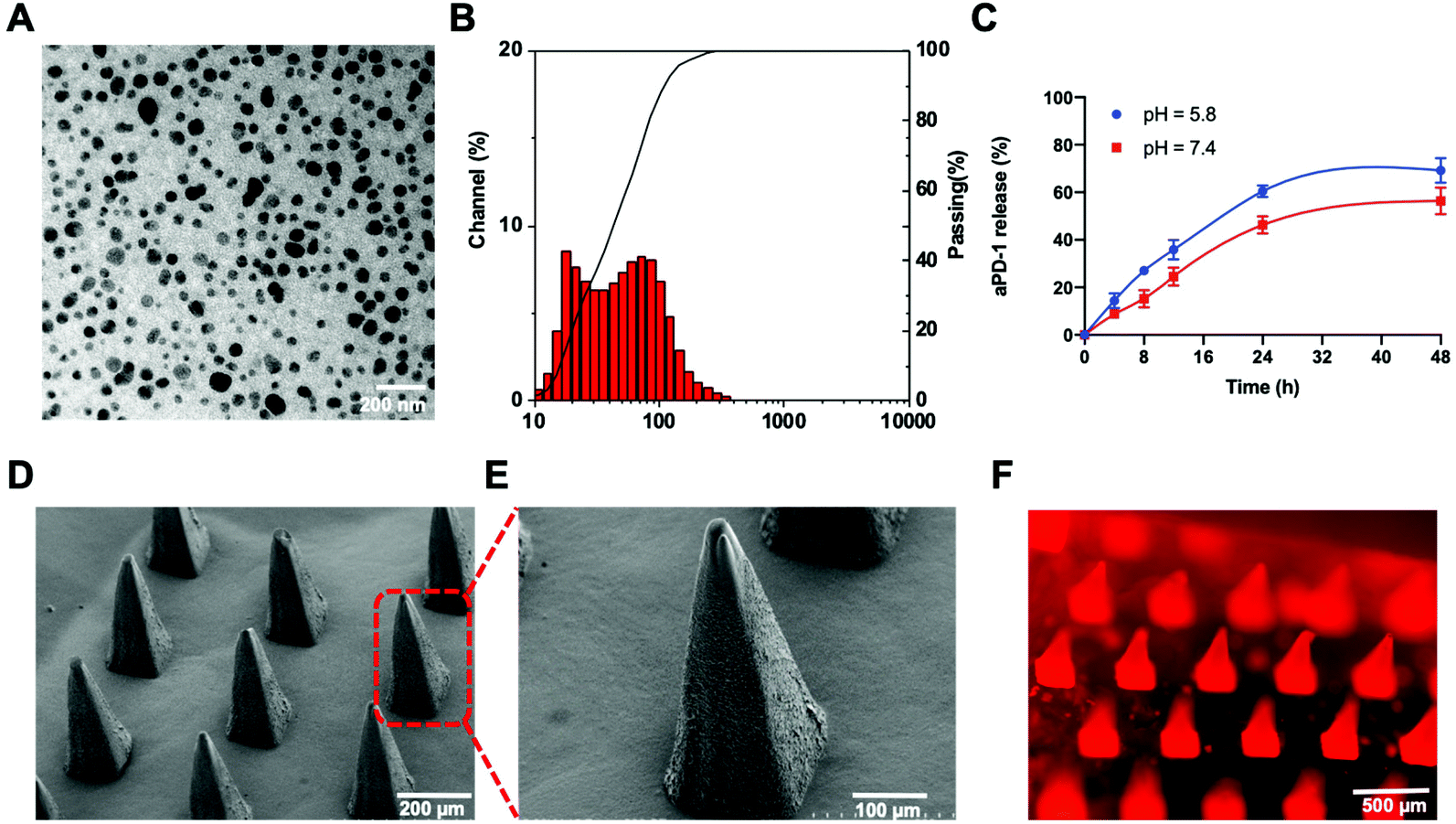 | ||
| Fig. 1 Characterization studies of aPD-1/CDDP@NPs and MNs. (A) aPD-1/CDDP@NPs exhibited a spherical morphology with homogeneous distribution, as illustrated by TEM images. (B) Particle size detected through DLS. (C) aPD-1 release profile from the NPs in PBS with diverse pH values. (D) SEM images of dissolving MNs. (E) Higher magnification of the MN tips. (F) Fluorescence microscopy image of the MNs. The NPs were labeled using Liss Rhod PE lipids. The error bar was the standard deviation (SD) of the samples (n = 3). | ||
2.2 Antitumor activity of aPD1/CDDP@NPs in vitro
To assess the antitumor efficiency of the aPD1/CDDP@NPs, cytotoxicity, cellular uptake and cell apoptosis were detected in FaDu, CAL 27 and SCC VII cell lines. aPD1 alone could not induce any cell cytotoxicity, even when added into CDDP did not enhance its anticancer efficiency in vitro.25 aPD-1/CDDP@NPs were synthesized based on the procedures for synthesizing CDDP@NPs and were conferred the same advantages. The half-maximal inhibitory concentration (IC50) values of NPs for the three cell lines were all significantly lower than that of the free CDDP (Fig. 2A), and more Pt was detected after the cell lines were treated with drugs at 100 μM for 4 h (Fig. 2B). In sum, more drugs were taken up by cells and they exerted higher drug efficiency after lipid coating and nano-encapsulation. NPs increased the solubility of CDDP, and their escape from endosome and avoidance of lysosome degradation due to the encapsulation of DOTAP, further increasing the drug uptake amount by the cell lines.26 aPD-1 was mainly used to inhibit the immune checkpoint blockade (ICB) and evoke the T-cell activity. aPD-1/CDDP@NPs showed no significant difference on both IC50 and cellular uptake compared with the CDDP@NPs, given that aPD-1 was not effective in vitro. In conclusion, aPD-1/CDDP@NPs had the same cytotoxicity for cancer cell lines as CDDP@NPs in vitro. | ||
| Fig. 2 In vitro antitumor profiles. Higher drug efficiency and more Pt uptake were detected after lipid coating and nano-encapsulation. (A) IC50 of the three cell lines (FaDu, CAL 27 and SCC-VII) treated with three drugs (CDDP, CDDP@NPs, and aPD1/CDDP@NPs). (B) Cellular uptake of the cell lines after being treated with a Pt concentration of 100 μM in a 24-well plate for 4 h. (C) The cells were treated with free CDDP or aPD-1-CDDP-NPs with a Pt concentration of 100 μM for 4 h. Cell apoptosis and cell cycle were determined using an APO-BrdU™ TUNEL Assay Kit, and they both were evaluated through flow cytometry. (D) The cell cycle was evaluated based on the florescence intensity of PI. The error bar represents the SD of three independent experiments, and statistical significance was calculated through one-way ANOVA. P value: ***p < 0.01; **** p < 0.0001. | ||
In addition, cell lines were treated with free CDDP or aPD-1/CDDP@NPs respectively, followed with analysis through flow cytometry using APO-BrdU kit. A large number of 3′-hydroxyl ends appeared when DNA strands became fragmented, and they were labeled with BrdUTP and terminal deoxynucleotidyl transferase (TdT) using the TUNEL technique. After DNA incorporation, BrdU could be detected through an anti-BrdU antibody. As shown in Fig. 2C, aPD-1/CDDP@NPs induced 24.5% cell apoptosis, whereas free CDDP induced only 2.79% cell apoptosis. This result indicated that NPs triggered discernable apoptosis in the cell lines. Furthermore, according to cell cycle analysis, the percentage of the G2 phase was remarkably decreased, indicating lower cell growth in the aPD-1/CDDP@NP group (Fig. 2D).
In conclusion, lower IC50 values and higher cellular uptake have been achieved for NPs, and they could induce more apoptosis by reducing the number of cells in the G2 phase. All the data suggested that NPs could significantly enhance the antitumor efficiency, although adding aPD-1 did not internalize the medical effects.
2.3 Characterization of dissolving MNs
To target the immune region on the skin through transdermal delivery, drugs were further encapsulated into dissolving MNs. Dissolving MNs were fabricated following a molding method. Polyvinylpyrrolidone (PVP) was selected as a structural material, because it offers the advantages of biocompatibility, strong mechanical properties and high water solubility.27 As illustrated in scanning electron microscopy (SEM) images (Fig. 1D and E), an MN consisted of 9 × 9 needles with a height of 588 μm and a base diameter of approximately 240 μm. Liss Rhod PE lipids were applied to label the NPs. The fluorescent image of the MNs further confirmed the adequate distribution of the therapeutic agents on MN tips (Fig. 1F). The obtained MNs could be completely dissolved in water within 5 minutes and they could be dissolved into mouse skin in 20 minutes. After the MN-array was pressed on the mouse skin with appropriate strength for 20 minutes, obvious pinholes could be observed, and tips could be dissolved in the process (Fig. S1†). Moreover, the skin region with pinholes could recover in less than one day without causing any skin ulceration. In addition, the bioactivity of the loaded aPD-1 and NPs did not degrade after one month of storage at 4 °C. Taken together, the results indicate that this type of PVP-structured MN may mediate drug delivery with uniform distribution, high solubility in the skin, and strong mechanical properties.2.4 PD-L1 expression in vitro and in vivo
PD-L1 is a logical biomarker of aPD-1 and aPD-L1 therapy.28,29 The SCC VII cell line is a murine squamous cell carcinoma cell line that spontaneously arises in the abdominal wall of a C3H mouse. We determined whether PD-L1 protein and mRNA are expressed in the SCC VII cell lines through western blot and qPCR, respectively. RAW 264.7 cells exhibited higher PD-L1 expression after LPS stimulation; however, no expression was observed in SCC VII cells (Fig. S1A & S2B†). Whether PD-L1 is expressed in an immunocompetent murine tumor homograft model was further detected using immunohistochemistry (IHC). The representative micrographs with PD-L1 positive and negative expression are presented in Fig. S1C.† Partial or complete membrane staining less than 1% is regarded as negative expression of PD-L1. In addition, a staining ratio ranging from 1% to 49% is defined as low PD-L1 expression, whereas high PD-L1 expression represents staining ratios of more than 50%.30 According to the aforementioned criteria, 85% of the tumor positively expressed PD-L1, among which 50% demonstrated high PD-L1 expression.2.5 Antitumor efficiency in vivo
We further investigated tumor inhibition on an immunocompetent murine tumor homograft model. SCC VII cell lines were subcutaneously inoculated into the right flank of C3H/HeJ mice to obtain a superficial tumor. Mice were divided into 8 groups, including (i) control group treated with PBS (indicated as PBS); (ii) intraperitoneal (i.p.) injection of CDDP (indicated as CDDP); (iii) i.p. injection of aPD-1 (indicated as aPD-1); (iv) i.p. injection of both aPD1 and CDDP (indicated as aPD-1 + CDDP); (v) i.p. injection of aPD-1/CDDP@NPs (indicated as aPD1/CDDP@NPs); (vi) CDDP NPs loaded with MN patches (indicated as CDDP@NP MN); (vii) aPD-1 loaded MN patches (indicated as aPD-1 MN); (viii) aPD-1/CDDP@NP loaded MN patches (indicated as aPD-1/CDDP@NP MN). Tumor volume was monitored using a Vernier caliper before the treatment and 3 days after each treatment. Furthermore, tumor growth was double confirmed using bioluminescent images (Fig. 3A & S2†). Mice treated with PBS showed natural tumor growth. Mice treated with CDDP exhibited a limited treatment efficacy, with significantly reduced tumor weight but no difference in tumor volume compared to the control group. Although 85% of tumors positively expressed PD-L1, aPD-1 systemic treatment caused a negligible inhibitory effect. aPD-1 is an immune drug that can block the PD-1/PD-L1 axis to renovate cytotoxic T-cells. The ineffective therapeutic role of aPD-1, however, may be due to the following reasons. First, the renovated T-cells may be too exhausted that they perform impaired effector function with limited proliferative potential.31 Second, cancer antigens may be “masked”; therefore, they cannot be identified by T-cells.32 Third, the tumor microenvironment may contain multiple inhibitory immune cells, such as tumor-associated macrophages, which overwhelm the T-cells and have adverse effects.33 Although the exact reason was unclear, the animal model in this study was unresponsive to systemic aPD-1 immunotherapy. | ||
| Fig. 3 Antitumor efficacy in vivo. MN group exhibited the most notable tumor regression effect. (A) Representative bioluminescent images of mice with various treatments [(i) PBS; (ii) aPD-1; (iii) aPD1 + CDDP; (iv) aPD-1/CDDP@NPs; (v) aPD-1 MNs; and (vi) aPD-1/CDDP@NP MNs] at different time points (T1: 3 days after 1st treatment, T2: 3 days after 2nd treatment, and T3: 3 days after 3rd treatment). Antitumor efficiency was evaluated based on the (B) tumor volume and (C) tumor weight after sacrifice. Each group started treatment when the tumor volume reached 10 mm3. Three treatments were given for each group and lasted for three cycles. Tumor volume was measured before the treatment and 3 times after the treatment. The error bar represents the SD for each groups (n = 7). Statistical analysis was performed using the Mann–Whitney U test. P value: *p < 0.05, ***p < 0.001. | ||
However, aPD-1 delivered by MN patches revealed significant anticancer effects. The tumor volume of the aPD-1 MN groups (418 ± 66 mm3) was significantly reduced compared to that of the aPD-1 systemic injection group (90.252 ± 39.343 mm3), with a p value less than 0.05. Tumor weight of the aPD-1 MN group (0.05 ± 0.017 g) decreased by 8-fold compared to that of the aPD-1 systemic injection group (0.443 ± 0.083 g) (p < 0.001). Skin is a highly active immune organ containing a large population of resident antigen-presenting cells. It has been reported that an MN was applied for increasing vaccine immunogenicity by targeting antigen delivery to skin,34 and an MN could induce immune responses by activating T-cells.35 Our results demonstrated that aPD-1 delivered by an MN could achieve potent antitumor effects in the animal model unresponsive to aPD-1 systemic therapy. This represented a promising treatment strategy for immunotherapy-unresponsive cancers.
Clinical studies have reported an interplay between chemotherapy and immunotherapy.10 In this study, the aPD-1 + CDDP group exhibited a greater tumor regression effect than either the aPD-1 or CDDP groups (p < 0.05, Fig. 3 & Fig. S2†). Notably, aPD-1/CDDP@NPs could synergistically deliver both chemotherapeutic and immunotherapeutic drugs to mice, and tumor growth was inhibited more significantly than in the aPD-1 + CDDP groups (p < 0.01, Fig. 3). Tumor weight of the aPD-1/CDDP@NP group was 0.099 ± 0.03 g, whereas it was 0.219 ± 0.061 g in the aPD-1 + CDDP group (p < 0.01). Tumor volume in the aPD-1 + CDDP group was 192.799 ± 79.824 mm,3 while it was only 93.246 ± 36.017 mm3 in the aPD-1/CDDP@NP group (p < 0.05). Moreover, the aPD-1/CDDP@NP MN group showed the most notable tumor regression effect compared with any other groups. Compared to those in the aPD-1/CDDP@NP group, both tumor volume (18.312 ± 8.286 mm3) and tumor weight (0.012 ± 0.005 g) were significantly decreased in the aPD-1/CDDP@NP MN group (p < 0.05 and p < 0.001, respectively). These results proved that an MN is a highly promising tool for the transdermal co-delivery of chemotherapeutic and immunotherapeutic drugs.
Cell proliferation was further confirmed through IHC in formalin-fixed paraffin-embedded tumor tissue sections. Mice treated with CDDP achieved delayed tumor growth, with 25% of the cells continuing to proliferate, however, mice treated with aPD-1 still exhibited sufficient cell proliferation of approximately 70%. Tumor infiltrating lymphocytes (TILs) such as CD4+ cells and CD8+ cells infiltrated the tumor tissue and started to proliferate after aPD-1 treatment. From Fig. 4A & C, we concluded that CDDP could inhibit cell proliferation, and aPD-1 could enhance T-cell infiltration. aPD-1 delivered through MN patches could amplify the results. TILs attacked the tumor tissues and killed the tumor cells afterwards. Whether these proliferated cells were T-cells or tumor cells were further confirmed through flow cytometry. Additionally, tumor cell apoptosis was assessed through in situ TUNEL assay (Fig. 4B & D). The cell apoptotic indexes in the aPD-1 MN and aPD-1/CDDP@NP MN groups were 61.4% ± 12.1% and 73.2% ± 11.6%, respectively, which were significantly higher than those in the aPD-1 group (5.4% ± 2.7%, p < 0.01). These data further substantiated our hypothesis that an MN could provoke T-cell activity and then induce cell apoptosis through T-cells. These results demonstrated that the tumor inhibition was mediated through tumor cell apoptosis. In sum, an aPD-1/CDDP@NP MN can not only increase the amount of activated T-cells but also kill tumor cells.
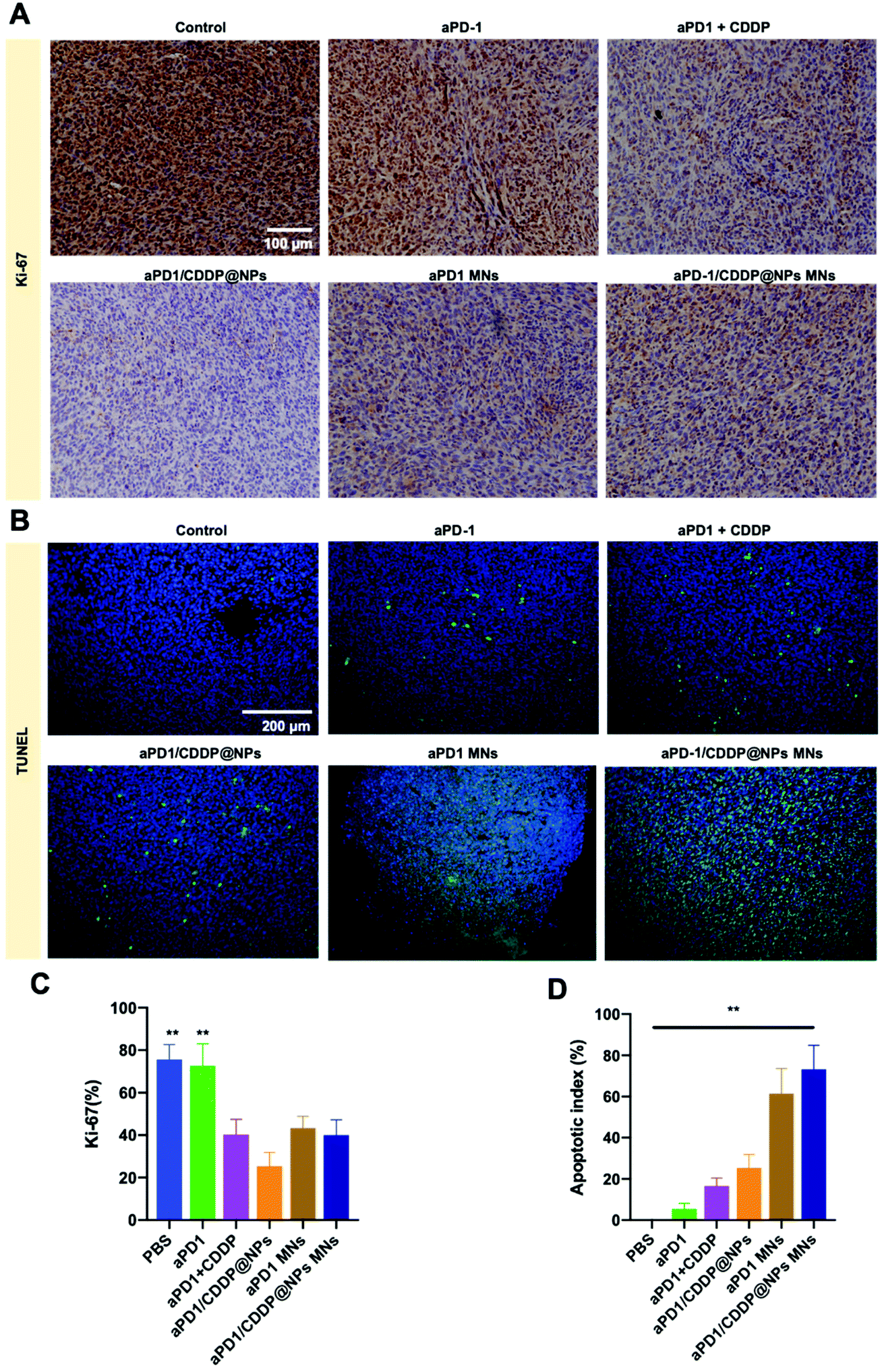 | ||
| Fig. 4 Tumour-specific cell proliferation and induced cell apoptosis. (A) Representative Ki-67 IHC in tumor tissues in various groups [(i) PBS; (ii) aPD-1; (iii) aPD1 + CDDP; (iv) aPD-1/CDDP@NPs; (v) aPD-1 MNs; and (vi) aPD-1/CDDP@NP MNs]. (B) TUNEL images in tumor tissues. (C) Results of the statistical analysis. Ki-67 density was presented as positive area/total area. (D) The apoptotic index was calculated as green cell number/total cell number. Each bar represents the mean ± SD, n = 7. Statistical analysis was performed using the Mann–Whitney U test. P value: **p < 0.01. | ||
2.6 T-cell responses and the immune profile
To evaluate T-cell responses and the immune profile, tumor tissues and blood were harvested after sacrifice. aPD-1, being an ICB, could be a positive regulator of TILs. The infiltration of CD4+ T-cells and CD8+ T-cells into tumor cells was assessed through flow cytometry. As illustrated in Fig. 5D, only 4.92% was detected in the control group. By contrast, mice treated with aPD-1 or aPD-1 + CDDP groups demonstrated infiltration of 4-fold T-cells compared to the control group. In addition, significantly increased activated CD8+ T-cells and CD4+ T-cells were detected in both the aPD-1/CDDP@NP (45.95% for CD8+ and 24.31% for CD4+) and aPD-1 MN groups (47.98% for CD8+ and 37.50% for CD4+), whereas only 12.13% of CD8+ T-cells and 15.0% of CD4+ T-cells were detected in the aPD-1 group, indicating that tumor cells were remarkably infiltrated by T-cells after nano-encapsulation or delivered transdermally. More importantly, MN mediated aPD-1/CDDP@NPs exhibited the highest T-cell infiltration (75.95% of CD8+ T-cells among tumor cells) compared with the other groups. These results were consistent with the IHC-detected Ki-67. The increase in effector T-cells was correlated with the tumor regression efficiency, indicating that activated T-cells were attacking the tumor cells. To reveal the cellular mechanism in vivo, the production of IFN-γ and TNF-α in the mouse serum was determined using an ELISA kit. No significant difference was detected in TNF-α levels; however, IFN-γ demonstrated a positive correlation with CD8+ T-cells (Fig. 5B). Mice treated with PBS showed lower IFN-γ levels than other five groups. aPD-1 resulted in more IFN-γ production after nano-encapsulation. Notably, the MN-mediated delivery system could contribute more to IFN-γ expression; among the groups, the aPD-1/CDDP@NP MN group showed the highest IFN-γ expression. Moreover, the infiltration of CD4+Foxp3+ T-cells was analyzed, and the three MN groups showed a remarkable decrease in the regulatory T-cells (Fig. 5E). | ||
| Fig. 5 T-cell responses and immune profile. MNs effectively boosted the immune response. (A) Representative images of CD4+ T-cell and CD8+ T-cell infiltration in tumor tissues detected using flow cytometry. (B) IFN-γ detected using ELISA in the serum. (C) Quantitative analysis of CD8+ T-cells among tumor cells. (D) Quantitative analysis of CD4+ T-cells among tumor cells. (E) The percentages of CD4+FOXP3+ Tregs within total CD4+ TILs. Each bar represents the mean ± SD, n = 7. Statistical analysis was performed using the Mann–Whitney U test. P value: *p < 0.05, **p < 0.01. | ||
Taken together, increased cytotoxic T-cell responses activated by MNs might explain why MNs loaded with aPD-1 or aPD-1/CDDP@NPs demonstrated potent anticancer effects in the animal model unresponsive to aPD-1 systemic therapy. The aPD-1/CDDP@NPs delivered by MN patches could lead to more robust T-cells and then infiltrate into the tumor sites as well as release chemotherapeutic drugs into the tumor sites, leading to synergistic anticancer efficiency.
2.7 Systemic toxicity and side effects
Side effects of chemotherapeutic agents severely limit their clinical application and further lead to non-ideal outcomes. To assess the systemic toxicity and side effects of these treatments, we recorded the body weight, and determined the blood urea nitrogen (BUN) value and total immunoglobulin G (IgG) value in the serum using a urea nitrogen detection kit and a total IgG ELISA kit, respectively. Mice treated with CDDP showed severe body weight loss no matter whether aPD1 was added or not. Mice injected with PBS or CDDP exhibited body weight loss after the last injection due to cancer cachexia, while mice treated with MNs had no body weight loss during the whole experiment (Fig. 6A). The normal range for the BUN value is 12 mg mL−1 to 33 mg mL−1. As illustrated in Fig. 6B, BUN values in mice treated with CDDP and aPD-1 were out of the normal range. After nano-encapsulation, the BUN values of some mice in the aPD-1/CDDP@NP group were in the normal range, while there was no statistical significance compared to the aPD-1 plus CDDP group. Remarkably, the BUN values in all the MN patch groups were within the normal range, as well as in the control group, indicating that an MN may be a safe delivery system without causing nephrotoxicity. In addition, CDDP caused severely decreased IgG, while the addition of aPD-1 to CDDP could recover the downtrend to some extent. Notably, MN groups showed significantly increased IgG values compared with other groups (p < 0.001) (Fig. 6C). | ||
| Fig. 6 Systemic effects in each group. (A) Mice body weight during the whole experiment. (B) The BUN value and (C) total IgG value in the serum. The error bars were based on the SD of 7 mice. (D) H&E staining of main organs (liver, lungs, kidneys and spleen) in six groups (control, aPD-1, aPD-1 + CDDP, aPD-1/CDDP@NPs, aPD-1 MNs, and aPD-1/CDDP@NP MNs). The CDDP groups exhibited severe side effects, which could be reduced through nano-encapsulation and MN-mediated delivery. Each bar represents the mean ± SD of seven mice. Statistical analysis was performed using the Mann–Whitney U test. P value: *p < 0.05, ***p < 0.001. | ||
Liver, lungs, kidneys and spleen of each group were collected after sacrifice for hematoxylin and eosin (H&E) staining to analyze whether drugs induced toxicity to these organs or not (Fig. 6C & S3†). Light microscopy analysis revealed that in mice liver sections, some alterations were found in the liver parenchyma in CDDP and aPD1 plus CDDP groups. Microscopic views showed some areas of necrotic hepatocytes, inflammatory cell infiltration, bile duct proliferation, and hepatocyte swelling. In addition, congestion was slightly more visible in these two groups than in the control group, and the structure of the hepatic lobule was confused. By contrast, the aPD-1/CDDP@NP group exhibited generally normal structures with slight disintegration of the hepatic cords, and the structure of the hepatic lobule was not entirely clear. In the aPD-1, aPD-1 MNs, and aPD-1/CDDP@NP MN groups, the liver parenchyma was quite comparable with that of the control group. For lung sections, compared with the control group, diffuse damage of the pulmonary alveoli and severe inflammatory infiltration were observed in the CDDP, aPD-1 plus CDDP, and aPD-1/CDDP@NP groups. The other groups exhibited normal microscopic lung structures. Regarding nephrotoxicity, the microscopic examination of kidneys from mice in the CDDP group and the aPD-1 plus CDDP group revealed severe toxic tubular necrosis. This finding was characterized by the glomeruli collapsing and mesangial cells fusing with surrounding tubules, coupled with collapsed Bowman's capsule, disappearance of Bowman's space and infiltration of scattered lymphocytes. By contrast, there was no evidence of renal damage in mice treated with aPD-1, aPD-1 MNs, and aPD-1/CDDP@NP MNs. The morphological features of the kidneys were similar to those of the control group. As for spleen in the CDDP group, the white pulp was evidently reduced, and the periarterial lymphatic sheaths disappeared and were accompanied by more neutrophilic granulocyte infiltration, and the boundary of the red and white pulp was unclear. In the aPD-1 + CDDP and aPD-1/CDDP@NP groups, the white pulp area was reduced, and periarterial lymphatic sheath lymphocytes decreased. In addition, neutrophilic granulocyte infiltration was visible. The microscopic examination of spleens from mice in the aPD-1, aPD-1 MN and NP MN groups did not reveal differences with the control group. In sum, CDDP caused severe side effects to all organs, whereas aPD-1 did not induce toxicity to these organs. After nano-encapsulation, the toxicity of CDDP was reduced to some extent, and no apparent toxicity was observed when aPD-1/CDDP@NPs were delivered using MNs. This demonstrated that the transdermal delivery of aPD-1/CDDP@NPs through an MN is a safe solution for cancer therapy.
3. Conclusions
This pioneering study described a novel strategy for the local co-delivery of chemotherapeutic and immunotherapeutic agents by MNs for synergistic immuno-chemotherapy. Lipid-coated nanoparticles were utilized to facilitate drug release and tumor-targeting. The platform that was integrated with both immune checkpoint blockade and chemotherapy could boost immune response in the tumor microenvironment and further enhance the inhibition efficiency for tumor cells as well as decrease systemic toxicity. Notably, transdermal delivery using MNs increased the response rate in the animal model unresponsive to aPD-1 systemic therapy. Taken together, the synergistic effects of MN-mediated aPD-1/CDDP@NPs render them a powerful tool for cancer therapy. Future studies shall focus on the use of this novel strategy in the treatment of immunotherapy-unresponsive cancers.4. Materials and methods
4.1 Materials
CDDP (cis-diammineplatinum(II) dichloride), cyclohexane, Igepal®-520, Triton™ X-100, hexanol, and silver nitrate were purchased from Sigma-Aldrich (St Louis, MO, USA). The lipids, DOPA (1,2-dioleoyl-sn-glycero-3-phosphate), DOTAP (1,2-dioleoyl-3-trimethylammonium-propane), Liss Rhod PE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt)), DSPE-PEG-AA (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (ammonium salt)), and cholesterol were obtained from Avanti Polar Lipids (Alabaster, AL, USA). The aPD-1 antibody (GoInVivo™ Purified anti-mouse CD279, BioLegend, San Diego, CA, USA) was purchased from BioLegend.4.2 Cell line
FaDu and CAL 27 cell lines, which are human HNSCC cell lines, were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). RAW264.7, a type of mouse macrophage cell line, was also obtained from the ATCC. The SCC VII cell line is derived from murine oral squamous cell carcinoma and can be used to build an immunocompetent murine tumor homograft model tumor model on syngeneic hosts.36 The SCC VII cell line was kindly gifted by Dr Susan J. Knox from Stanford University, USA. FaDu, CAL 27 and SCC VII cell lines were both cultured in Dulbecco's Modified Eagle's Medium (DMEM, Gibco, Grand Island, NY, USA) supplemented with 10% (v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v) fetal bovine serum (FBS, Gibco, USA) and 100 U mL−1 penicillin–streptomycin (Gibco, USA) in a humidified atmosphere with 5% CO2 at 37 °C. The RAW 264.7 cell line was maintained in DMEM with 10% FBS without antibiotics and the SCC VII cell line was tagged with the luciferase gen reporter luc2 using the pGL4.51 [luc2/CMV/Neo] vector (Promega, Madison, WI, USA). The gene reporter was transfected using the Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific, Waltham, MA, USA) following the product protocol. The Luciferase Assay System (Promega, USA) was applied to the detection of firefly luciferase activity rapidly in cell lysates. After 14 days of selection with geneticin (Thermo Fisher Scientific, USA) at a concentration of 500 μg mL−1, the luciferase-tagged SCC VII (SCC VII-luc) cell line could be injected into mice and further detected based on bioluminescent signals.
:v) fetal bovine serum (FBS, Gibco, USA) and 100 U mL−1 penicillin–streptomycin (Gibco, USA) in a humidified atmosphere with 5% CO2 at 37 °C. The RAW 264.7 cell line was maintained in DMEM with 10% FBS without antibiotics and the SCC VII cell line was tagged with the luciferase gen reporter luc2 using the pGL4.51 [luc2/CMV/Neo] vector (Promega, Madison, WI, USA). The gene reporter was transfected using the Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific, Waltham, MA, USA) following the product protocol. The Luciferase Assay System (Promega, USA) was applied to the detection of firefly luciferase activity rapidly in cell lysates. After 14 days of selection with geneticin (Thermo Fisher Scientific, USA) at a concentration of 500 μg mL−1, the luciferase-tagged SCC VII (SCC VII-luc) cell line could be injected into mice and further detected based on bioluminescent signals.
4.3 Synthesis of aPD1/CDDP@NPs
Synthesis of the lipid-coated cisplatin nanoparticles (CDDP@NPs) was done according to previous publications.20,37 Briefly, 0.2 mmol CDDP was dissolved in 1.0 mL deionized (DI) water with 0.39 mmol silver nitrate. After heating at 60 °C for 3 h, the mixture was stirred overnight protected from light subsequently. After centrifugation at 16000 rpm for 15 min, the supernatant was filtered through a 0.22 μm syringe filter. The concentration of platinum (Pt), monitored through inductively coupled plasma optical emission spectrometry (ICP-OES, Spectro Arcos, Kleve, Germany), was then adjusted to 200 mM; thus the cis-[Pt(NH3)2(H2O)2]2(NO3)2 precursor was obtained; 800 mM KCl in water and 200 mM cis-[Pt(NH3)2(H2O)2]2(NO3)2 were added into a microemulsion composed of cyclohexane/Igepal® CO-520 (71%:29%, v:v) and cyclohexane/Triton™ X-100/hexanol (75%:15%:10%, v:v:v) (3:1) respectively. Subsequently, 20 mM DOPA was added into the precursor solution and stirred for 20 minutes before the two solutions were mixed. Thirty minutes later, ethanol was added into the solution and centrifuged at 12000g for more than 15 min. After washing with ethanol at least three times, the particles re-dispersed into chloroform and lipids, such as DOTAP, DSPE-PEG-AA, and cholesterol, were added into the solution. Subsequently, chloroform was evaporated, and the particles were dispersed into DI water. Ultimately, 100 μg aPD-1 was added into 1.0 mL NPs stirred at 4 °C overnight to obtain the aPD-1/CDDP@NPs. The NPs were collected through centrifugation and followed by re-suspending in DI water.
4.4 Characterization of aPD-1/CDDP@NPs
The morphology of aPD-1/CDDP@NPs was observed using a transmission electron microscope (TEM, CM100, Philips Electron Optics, Eindhoven, the Netherlands). The particle size was detected through a dynamic light scattering particle size analyzer (Nanotrac Wave II, Microtrac, Montgomeryville, PA, USA). The CDDP loading capacity was determined based on the Pt content and the Pt content was monitored through ICP-OES. In addition, the loading capacity of aPD-1 was detected using a rat total IgG ELISA kit (Thermo Fisher Scientific, USA). To measure the in vitro aPD-1 release profile, NPs were added to phosphate-buffered saline (PBS) with various pH values of 5.8 and 7.4 at 37 °C. Moreover, 10 μL medium was collected for testing, and 10 μL fresh medium was added at a predetermined time point. The amount of aPD-1 was determined using a rat total IgG ELISA kit (Thermo Fisher Scientific, USA).4.5 Antitumor activity of aPD-1/CDDP@NPs in vitro
4.6 Cell apoptosis and cell cycle
The cells were placed in 6 cm dishes and treated with CDDP and aPD-1/CDDP@NPs, as the Pt concentration was 100 μM. Cell apoptosis was detected using flow cytometry with the APO-BrdU™ tranferase dUTP nick end laveling (TUNEL) Assay Kit (Thermo Fisher Scientific, USA), and the phase of the cell cycle was determined using propidium iodide (PI, Thermo Fisher Scientific, USA) following the manufacturer's protocol. The cells were fixed with 1% paraformaldehyde (PFA) or ethanol before staining, and they were analyzed through flow cytometry (BD FACSVerse™, BD, Franklin Lake, NJ, USA) immediately after staining. All data were analyzed using FlowJo (7.6.1, BD, USA).4.7 Fabrication of dissolving MNs
Poly-di-methyl siloxane (PDMS, Sylgard 184, Dow Corning, MI, USA) was employed to produce the MN mold using the injection molding method. PDMS and its collocational curing agent were blended at a weight ratio of 10:1 and then stirred uniformly. The bubbles in the PDMS mixture solution had to be removed through centrifugation at 7000 rpm for 10 minutes. SU-8 masters were placed in the middle bottom of the PDMS solution without bubbles, and the tip of the needle was upward. The SU-8 masters placed in the PDMS solution were dried at approximately 70 °C for 24 h to form the PDMS mold. The PDMS mold was then detached from the SU-8 master molds and was ready to be used as molds for producing MNs. Thereafter, polyvinyl pyrrolidone (PVP, MW ∼ 360 K, Sigma-Aldrich, USA) used as the microneedle material was pre-sterilized through autoclaving sterilization. 1.5 g sterilized PVP was dissolved in 10 mL the sterilized water. The loading of NPs in MNs were performed in a sterilized environment. aPD-1 or aPD-1/CDDP@NPs powder of a specified weight was added in the 15% (w/w) PVP solution, and the 0.1 mL mixed PVP solution was placed into the MN mold and then centrifuged at 4000 rpm for 5 min to ensure that the mixed solution filled the cavities. The MN patch was dried at 40 °C for 24 h in an oven with a constant temperature air circulation system. The dried MN patch was then ready to be peeled off from the mold for further use.
4.8 Expression of PD-L1 in vitro and in vivo
The PD-L1 expression of the SCC VII cell line in vitro was detected through western blot and quantitative polymerase chain reaction (qPCR). Lipopolysaccharide (LPS, Escherichia coli LPS, Sigma, USA) stimulated RAW 264.7 cells were selected as a positive control. SCC VII and RAW 264.7 cells were treated with 10 μg mL−1 LPS for 4 h. Subsequently, the cells were lysed using RIPA Lysis Buffer with Halt Protease and phosphate inhibitors (Thermo Fisher Scientific, USA). The protein amount was determined using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, USA). A total amount of 30 μg protein was loaded onto SDS-PAGE and transferred to the PVDF membrane. After blocking with 5% non-fat milk in Tris-Buffered Saline (TBS) with 0.1% Tween 20, the membrane was incubated with primary antibodies (anti-PD-L1 and anti-β-actin, Abcam, USA) overnight at 4 °C. The membrane was then washed three times with TBS/Tween 20 followed by incubation in the HRP-conjugated secondary antibody (Cell Signaling Technology, Danvers, MA, USA) at room temperature for 2 h. Finally, the membrane was observed using the ChemiDoc XRS System (BioRad, Hercules, CA, USA) following incubation with the Pierce™ ECL Western Blotting Substrate (Thermo Fisher Scientific, USA). Total RNA in cell lysate was isolated using the RNeasy Mini kit (Qiagen, Hilden, Germany). cDNA was reverse transcribed from RNA following the manufacturer's protocol using SuperScript™ III Reverse Transcriptase (Thermo Fisher Scientific, USA). Then, cDNA was applied to quantitatively synthesize PCR products using a TaqMan® Fast Advanced Master Mix (Thermo Fisher Scientific, USA). PCR amplification was performed using a StepOnePlus™ Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The gene expression of PD-L1 was detected and β-actin was applied to normalize the copy numbers for the target gene.For the in vivo detection of PD-L1 expression, a part of the tumor tissue was harvested after sacrifice and was fixed in 4% PFA. The tissues were then embedded in paraffin and sliced to a thickness of 5 μm. IHC was performed after deparaffinization and rehydration. After antigen retrieval, the sections were incubated with the primary antibody, rabbit anti-PD-L1 (Abcam, USA) at 4 °C overnight. Then, a Rabbit Specific HRP/DAB Detection IHC Kit (Abcam, USA) was applied to detect the expression of PD-L1. Hematoxylin was used for counterstaining. Ultimately, sliced sections were visualized using optical microscopy and photography (ECLIPSE LV100POL, Nikon, Tokyo, Japan), and the images were analyzed using the Image J software (NIH, Bethesda, MD, USA).
4.9 Antitumor activity of MNs in vivo
4.10 Systemic immune effect
:400 ratio, and incubated at room temperature for 15 min protected from light after being added into samples.
4.11 Toxicity and side effect study
The livers, kidneys, spleens and lungs were collected after sacrifice and fixed in 4% PFA. After embedding into paraffin, the tissues were sliced into 5 μm sections for H&E staining. Images of the tissues were collected using a polarized light microscope (ECLIPSE LV100POL, Japan). The BUN and total IgG level in the serum were detected using a urea nitrogen detection kit (Thermo Fisher Scientific, USA) and a mouse IgG total ELISA kit (ThermoFisher Scientific, USA) respectively.4.12 Statistical analysis
All statistical analyses were performed using IBM SPSS Statistics 25.0, including one-way ANOVA, multiple t tests, or the Mann–Whitney U test. The quantitative data are expressed as mean ± standard derivation (SD). A p value of <0.05 was considered statistically significant.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project was supported by the Hong Kong Research Grant Council General Research Fund (No. 17120718), The University of Hong Kong Seed Fund for Basic Research (No. 201611159117 and 201711159117), and was awarded in 2019 The University of Hong Kong Research Output Prize. The authors would like to acknowledge financial support from the National Natural Science Foundation of China (Grant No. 61771498, 61901535, and 31900954), the Science and Technology Planning Project of Guangdong Province for Industrial Applications (Grant No. 2017B090917001), the Guangdong Province Key Area R&D Program (Grant No. 2018B030332001), the Science and Technology Program of Guangzhou, China (Grant No. 201907010038) and the Guangdong Basic and Applied Basic Research Foundation (Grant No. 2019A1515012087 and 2020A151501665). The authors wish to thank the Open Fund of State Key Laboratory of Biocontrol and the 100 Talents Program of Sun Yat-Sen University (2019SKLBC-KF06, 76120-18841213).References
- A. Naiing, Immunotherapy, Springer International Publishing AG, 2017 Search PubMed.
- S. L. Topalian, C. G. Drake and D. M. Pardoll, Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy, Cancer Cell, 2015, 27(4), 450–461 CrossRef CAS.
- L. Ni and C. Dong, New Checkpoints in Cancer Immunotherapy, Immunol. Rev., 2017, 276(1), 52–65 CrossRef CAS.
- A. Ribas, O. Hamid, A. Daud, F. S. Hodi, J. D. Wolchok, R. Kefford, A. M. Joshua, A. Patnaik, W. J. Hwu, J. S. Weber, T. C. Gangadhar, P. Hersey, R. Dronca, R. W. Joseph, H. Zarour, B. Chmielowski, D. P. Lawrence, A. Algazi, N. A. Rizvi, B. Hoffner, C. Mateus, K. Gergich, J. A. Lindia, M. Giannotti, X. N. Li, S. Ebbinghaus, S. P. Kang and C. Robert, Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma, J. Am. Med. Assoc., 2016, 315(15), 1600–1609 CrossRef CAS.
- V. Dogan, T. Rieckmann, A. Munscher and C. J. Busch, Current Studies of Immunotherapy in Head and Neck Cancer, Clin. Otolaryngol., 2018, 43(1), 13–21 CrossRef CAS.
- S. Aspeslagh, S. Postel-Vinay, S. Rusakiewicz, J. C. Soria, L. Zitvogel and A. Marabelle, Rationale for Anti-OX40 Cancer Immunotherapy, Eur. J. Cancer, 2016, 52, 50–66 CrossRef CAS.
- P. Sharma, S. Hu-Lieskovan, J. A. Wargo and A. Ribas, Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy, Cell, 2017, 168(4), 707–723 CrossRef CAS.
- Y. Li, T. Yang, Y. Yu, N. Shi, L. Yang, Z. Glass, J. Bolinger, I. J. Finkel, W. Li and Q. Xu, Combinatorial library of chalcogen-containing lipidoids for intracellular delivery of genome-editing proteins, Biomaterials, 2018, 178, 652–662 CrossRef CAS.
- A. C. Huang, M. A. Postow, R. J. Orlowski, R. Mick, B. Bengsch, S. Manne, W. Xu, S. Harmon, J. R. Giles, B. Wenz, M. Adamow, D. Kuk, K. S. Panageas, C. Carrera, P. Wong, F. Quagliarello, B. Wubbenhorst, K. D'Andrea, K. E. Pauken, R. S. Herati, R. P. Staupe, J. M. Schenkel, S. McGettigan, S. Kothari, S. M. George, R. H. Vonderheide, R. K. Amaravadi, G. C. Karakousis, L. M. Schuchter, X. Xu, K. L. Nathanson, J. D. Wolchok, T. C. Gangadhar and E. J. Wherry, T-cell Invigoration to Tumour Burden Ratio Associated with Anti-PD-1 Response, Nature, 2017, 545(7652), 60–65 CrossRef CAS.
- W. Wang, I. Kryczek, L. Dostal, H. Lin, L. Tan, L. Zhao, F. Lu, S. Wei, T. Maj, D. Peng, G. He, L. Vatan, W. Szeliga, R. Kuick, J. Kotarski, R. Tarkowski, Y. Dou, R. Rattan, A. Munkarah, J. R. Liu and W. Zou, Effector T Cells Abrogate Stroma-Mediated Chemoresistance in Ovarian Cancer, Cell, 2016, 165(5), 1092–1105 CrossRef CAS.
- H. Xiao, R. Qi, T. Li, S. G. Awuah, Y. Zheng, W. Wei, X. Kang, H. Song, Y. Wang, Y. Yu, M. A. Bird, X. Jing, M. B. Yaffe, M. J. Birrer and P. P. Ghoroghchian, Maximizing Synergistic Activity When Combining RNAi and Platinum-Based Anticancer Agents, J. Am. Chem. Soc., 2017, 139(8), 3033–3044 CrossRef CAS.
- Y. Yu, Q. Xu, S. He, H. Xiong, Q. Zhang, W. Xu, V. Ricotta, L. Bai, Q. Zhang, Z. Yu, J. Ding, H. Xiao and D. Zhou, Recent advances in delivery of photosensitive metal-based drugs, Coord. Chem. Rev., 2019, 387, 154–179 CrossRef CAS.
- J. H. Park, M. Jang, Y. E. Tarhan, T. Katagiri, M. Sasa, Y. Miyoshi, K. R. Kalari, V. J. Suman, R. Weinshilboum, L. Wang, J. C. Boughey, M. P. Goetz and Y. Nakamura, Clonal Expansion of Antitumor T cells in Breast Cancer Correlates with Response to Neoadjuvant Chemotherapy, Int. J. Oncol., 2016, 49(2), 471–478 CrossRef CAS.
- W. H. Fridman, L. Zitvogel, C. Sautes-Fridman and G. Kroemer, The Immune Contexture in Cancer Prognosis and Treatment, Nat. Rev. Clin. Oncol., 2017, 14(12), 717–734 CrossRef CAS.
- Z. Tao, M. D. Muzumdar, A. Detappe, X. Huang, E. S. Xu, Y. Yu, T. H. Mouhieddine, H. Song, T. Jacks and P. P. Ghoroghchian, Differences in Nanoparticle Uptake in Transplanted and Autochthonous Models of Pancreatic Cancer, Nano Lett., 2018, 18(4), 2195–2208 CrossRef CAS.
- S. L. Topalian, G. J. Weiner and D. M. Pardoll, Cancer Immunotherapy Comes of Age, J. Clin. Oncol., 2011, 29(36), 4828–4836 CrossRef CAS.
- Y. Ye, J. Wang, Q. Hu, G. M. Hochu, H. Xin, C. Wang and Z. Gu, Synergistic Transcutaneous Immunotherapy Enhances Antitumor Immune Responses through Delivery of Checkpoint Inhibitors, ACS Nano, 2016, 10(9), 8956–8963 CrossRef CAS.
- C. Wang, Y. Ye, G. M. Hochu, H. Sadeghifar and Z. Gu, Enhanced Cancer Immunotherapy by Microneedle Patch-Assisted Delivery of Anti-PD1 Antibody, Nano Lett., 2016, 16(4), 2334–2340 CrossRef CAS.
- J. Xu, B. Xu, J. Tao, Y. Yang, Y. Hu and Y. Huang, Microneedle-Assisted, DC-Targeted Codelivery of pTRP-2 and Adjuvant of Paclitaxel for Transcutaneous Immunotherapy, Small, 2017, 13(28), 1–13 CrossRef.
- X. Lan, J. She, D.-a. Lin, Y. Xu, X. Li, W.-f. Yang, V. W. Y. Lui, L. Jin, X. Xie and Y.-x. Su, Microneedle-Mediated Delivery of Lipid-Coated Cisplatin Nanoparticles for Efficient and Safe Cancer Therapy, ACS Appl. Mater. Interfaces, 2018, 10(39), 33060–33069 CrossRef CAS.
- C. Zhang, M. Jugold, E. C. Woenne, T. Lammers, B. Morgenstern, M. M. Mueller, H. Zentgraf, M. Bock, M. Eisenhut, W. Semmler and F. Kiessling, Specific Targeting of Tumor Angiogenesis by RGD-Conjugated Ultrasmall Superparamagnetic Iron Oxide Particles Using A Clinical 1.5 T Magnetic Resonance Scanner, Cancer Res., 2007, 67(4), 1555–1562 CrossRef CAS.
- A. A. Radwan and F. K. Alanazi, Targeting Cancer Using Cholesterol Conjugates, Saudi Pharm. J., 2014, 22(1), 3–16 CrossRef.
- O. Nakagawa, X. Ming, L. Huang and R. L. Juliano, Targeted intracellular delivery of antisense oligonucleotides via conjugation with small-molecule ligands, J. Am. Chem. Soc., 2010, 132(26), 8848–8849 CrossRef CAS.
- R. Kucharski, J. Maleszka and R. Maleszka, Novel cuticular proteins revealed by the honey bee genome, Insect Biochem. Mol. Biol., 2007, 37(2), 128–134 CrossRef CAS.
- Z. Xiao, Z. Su, S. Han, J. Huang, L. Lin and X. Shuai, Dual pH-sensitive nanodrug blocks PD-1 immune checkpoint and uses T cells to deliver NF-κB inhibitor for antitumor immunotherapy, Sci. Adv., 2020, 6(6), eaay7785 CrossRef.
- D. Pozzi, C. Marchini, F. Cardarelli, F. Salomone, S. Coppola, M. Montani, M. E. Zabaleta, M. A. Digman, E. Gratton, V. Colapicchioni and G. Caracciolo, Mechanistic evaluation of the transfection barriers involved in lipid-mediated gene delivery: interplay between nanostructure and composition, Biochim. Biophys. Acta, 2014, 1838(3), 957–967 CrossRef CAS.
- R. R. S. Thakur, I. A. Tekko, F. Al-Shammari, A. A. Ali, H. McCarthy and R. F. Donnelly, Rapidly dissolving polymeric microneedles for minimally invasive intraocular drug delivery, Drug Delivery Transl. Res., 2016, 6(6), 800–815 CrossRef CAS.
- G. T. Gibney, L. M. Weiner and M. B. Atkins, Predictive Biomarkers for Checkpoint Inhibitor-Based Immunotherapy, Lancet Oncol., 2016, 17(12), e542–e551 CrossRef CAS.
- S. L. Topalian, J. M. Taube, R. A. Anders and D. M. Pardoll, Mechanism-Driven Biomarkers to Guide Immune Checkpoint Blockade in Cancer Therapy, Nat. Rev. Cancer, 2016, 16(5), 275–287 CrossRef CAS.
- R. S. Herbst, J.-C. Soria, M. Kowanetz, G. D. Fine, O. Hamid, M. S. Gordon, J. A. Sosman, D. F. McDermott, J. D. Powderly, S. N. Gettinger, H. E. K. Kohrt, L. Horn, D. P. Lawrence, S. Rost, M. Leabman, Y. Xiao, A. Mokatrin, H. Koeppen, P. S. Hegde, I. Mellman, D. S. Chen and F. S. Hodi, Predictive Correlates of Response to The Anti-Pd-L1 Antibody MPDL3280A in Cancer Patients, Nature, 2014, 515, 563 CrossRef CAS.
- E. P.-Y. Lin, C.-Y. Yang, C.-W. Lin, B.-T. Huang, W.-Y. Lai, Y.-T. Tseng and P.-C. Yang, Priming PD-L1 Expression by Chemotherapeutic Agents in Non-Small Cell Lung Cancers, J. Clin. Oncol., 2017, 35(15_suppl), e20087–e20087 Search PubMed.
- J. Pol, E. Vacchelli, F. Aranda, F. Castoldi, A. Eggermont, I. Cremer, C. Sautes-Fridman, J. Fucikova, J. Galon, R. Spisek, E. Tartour, L. Zitvogel, G. Kroemer and L. Galluzzi, Trial Watch: Immunogenic Cell Death Inducers for Anticancer Chemotherapy, Oncoimmunology, 2015, 4(4), e1008866 CrossRef.
- W. Hugo, J. M. Zaretsky, L. Sun, C. Song, B. H. Moreno, S. Hu-Lieskovan, B. Berent-Maoz, J. Pang, B. Chmielowski, G. Cherry, E. Seja, S. Lomeli, X. Kong, M. C. Kelley, J. A. Sosman, D. B. Johnson, A. Ribas and R. S. Lo, Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma, Cell, 2016, 165(1), 35–44 CrossRef CAS.
- S. P. Sullivan, D. G. Koutsonanos, M. del Pilar Martin, J. W. Lee, V. Zarnitsyn, S.-O. Choi, N. Murthy, R. W. Compans, I. Skountzou and M. R. Prausnitz, Dissolving Polymer Microneedle Patches for Influenza Vaccination, Nat. Med., 2010, 16, 915 CrossRef CAS.
- E. S. Esser, J. A. Pulit-Penaloza, H. Kalluri, D. McAllister, E. V. Vassilieva, E. Q. Littauer, N. Lelutiu, M. R. Prausnitz, R. W. Compans and I. Skountzou, Microneedle Patch Delivery of Influenza Vaccine During Pregnancy Enhances Maternal Immune Responses Promoting Survival and Long-lasting Passive Immunity to Offspring, Sci. Rep., 2017, 7(1), 5705–5705 CrossRef.
- S. Duarte, A. Loubat, D. Momier, M. Topi, H. Faneca, M. C. Pedroso de Lima, G. F. Carle and V. Pierrefite-Carle, Isolation of Head and Neck Squamous Carcinoma Cancer Stem-Like Cells in A Syngeneic Mouse Model and Analysis of Hypoxia Effect, Oncol. Rep., 2012, 28(3), 1057–1062 CrossRef.
- S. Guo, Y. Wang, L. Miao, Z. Xu, C. M. Lin, Y. Zhang and L. Huang, Lipid-coated Cisplatin nanoparticles induce neighboring effect and exhibit enhanced anticancer efficacy, ACS Nano, 2013, 7(11), 9896–9904 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr04213g |
| This journal is © The Royal Society of Chemistry 2020 |