
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterogeneous catalysis by ultra-small bimetallic nanoparticles surpassing homogeneous catalysis for carbon–carbon bond forming reactions†
Nazgol
Norouzi‡
a,
Mrinmoy K.
Das‡
 a,
Alexander J.
Richard
a,
Amr A.
Ibrahim
b,
Hani M.
El-Kaderi
*a and
M. Samy
El-Shall
*a
a,
Alexander J.
Richard
a,
Amr A.
Ibrahim
b,
Hani M.
El-Kaderi
*a and
M. Samy
El-Shall
*a
aDepartment of Chemistry, Virginia Commonwealth University, Richmond, Virginia 23284-2006, USA. E-mail: helkaderi@vcu.edu; mselshal@vcu.edu; Fax: +1 (804) 828-8599; Tel: +1 (804) 828-7505 Tel: +1 (804) 828-2753
bDepartment of Chemistry, Faculty of Science, Mansoura University, Al-Mansoura 35516, Egypt
First published on 19th August 2020
Abstract
Palladium catalyzed cross-coupling reactions represent a significant advancement in contemporary organic synthesis as these reactions are of strategic importance in the area of pharmaceutical drug discovery and development. Supported palladium-based catalysts are highly sought-after in carbon–carbon bond forming catalytic processes to ensure catalyst recovery and reuse while preventing product contamination. This paper reports the development of heterogeneous Pd-based bimetallic catalysts supported on fumed silica that have high activity and selectivity matching those of homogeneous catalysts, eliminating the catalyst's leaching and sintering and allowing efficient recycling of the catalysts. Palladium and base metal (Cu, Ni or Co) contents of less than 1.0 wt% loading are deposited on a mesoporous fumed silica support (surface area SABET = 350 m2 g−1) using strong electrostatic adsorption (SEA) yielding homogeneously alloyed nanoparticles with an average size of 1.3 nm. All bimetallic catalysts were found to be highly active toward Suzuki cross-coupling (SCC) reactions with superior activity and stability for the CuPd/SiO2 catalyst. A low CuPd/SiO2 loading (Pd: 0.3 mol%) completes the conversion of bromobenzene and phenylboronic acid to biphenyl in 30 minutes under ambient conditions in water/ethanol solvent. In contrast, monometallic Pd/SiO2 (Pd: 0.3 mol%) completes the same reaction in three hours under the same conditions. The combination of Pd with the base metals helps in retaining the Pd0 status by charge donation from the base metals to Pd, thus lowering the activation energy of the aryl halide oxidative addition step. Along with its exceptional activity, CuPd/SiO2 exhibits excellent recycling performance with a turnover frequency (TOF) of 280![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 under microwave reaction conditions at 60 °C. Our study demonstrates that SEA is an excellent synthetic strategy for depositing ultra-small Pd-based bimetallic nanoparticles on porous silica for SCC. This avenue not only provides highly active and sintering-resistant catalysts but also significantly lowers Pd contents in the catalysts without compromising catalytic activity, making the catalysts very practical for large-scale applications.
000 h−1 under microwave reaction conditions at 60 °C. Our study demonstrates that SEA is an excellent synthetic strategy for depositing ultra-small Pd-based bimetallic nanoparticles on porous silica for SCC. This avenue not only provides highly active and sintering-resistant catalysts but also significantly lowers Pd contents in the catalysts without compromising catalytic activity, making the catalysts very practical for large-scale applications.
1. Introduction
Catalytic processes that lead to carbon–carbon bond formation are vital for advancements in contemporary organic chemistry as these reactions are of strategic importance in the synthesis of complex organics and the assembly of highly functionalized molecules.1–6 This area of catalysis has increased the accessibility for molecules of greater chemical complexity, particularly in the field of pharmaceutical drug discovery and development.7–9 Many reactions such as Suzuki, Miyaura, Heck, Negishi, Sonogashira, and others are extensively used in the assembly of active pharmaceutical ingredients (APIs) and are typically performed under palladium catalyzed homogeneous catalysis utilizing ligands and complex solvent systems to enhance activity and selectivity.1–9In spite of their high catalytic activity, homogeneous palladium catalysts still face several challenges that hinder their wide and effective use in industrial processes.10 Among these challenges are high solubility which precludes catalyst recovery and reuse, product contamination, and high cost.4,10 One of the major issues, particularly in pharmaceutical applications where this chemistry is extensively used, is residual metal contamination in the reaction products since palladium compounds can be highly toxic.9,11 Moreover, the difficulty associated with both metal and ligand recycling makes API applications very costly.11
To address these challenges, there has been great interest in designing cost-effective heterogeneous catalysts to allow for catalyst isolation and recycling without compromising catalytic activity.9,12,13 Given the indispensable role of the Suzuki cross-coupling (SCC) reaction in organic synthesis and its simplicity and effectiveness, intense efforts have been directed toward the development of cost-effective heterogeneous catalysts.3 However, the heterogeneous catalysis mechanism is still a matter of debate.14 It has been reported that catalytic events can take place at the metal surface in specific defect/edge sites of Pd nanoparticles or catalytically competent Pd species leached into the solution. The leached species can later precipitate over the support, and this process is affected by various factors such as temperature, nature of the support, and nature of the base.15–17 The precipitation problem can result in larger colloidal particles and extensively restructure the nanoparticles.18 The dissolution mechanism by itself can proceed through small Pd (0) clusters17 or surface detachment induced by the oxidative addition of aryl halide. The latter usually results in Pd(II) complexes, which further undergo an in situ reduction forming catalytically active Pd(0) species.19,20
One of the most pursued approaches has been the synthesis of highly dispersed and supported metal nanoparticles (NPs) with controlled size, morphology, and composition.21 By controlling these parameters and careful selection of the support, such catalysts have the potential to rival their homogeneous counterparts in terms of activity and stability.22–24 Although considerable advances have been made in this area, only very few catalysts have activity comparable to established homogeneous catalysts.6,25–27 Thus, there is an urgent need for highly active heterogeneous catalysts for SCC reactions. The requirements for a superior catalyst are: the high activity of a ligated homogeneous catalyst and the recyclability of a heterogeneous catalyst.
Palladium is the most active metal utilized at both the laboratory and industrial scales for cross-coupling reactions with ligated Pd complexes being the major player in homogeneous catalytic processes. Such complexes rely on in situ reduction of Pd(II) to Pd(0) to initiate the catalytic cycle via oxidative addition of aryl halides in SCC.4,28,29 Although the use of the supported Pd metal in cross-coupling catalysis dates back to the 1970s, often elevated temperatures and pressure are required for these reactions.30,31 To alleviate these undesired reaction conditions, a variety of synthetic strategies such as decreasing Pd NP size, use of unique supports, and electronic modification of Pd NPs by other metals were targeted. Although base metals, mainly nickel, copper and iron, have been reported to catalyze various steps of the cross-coupling reaction, these catalysts are deemed inferior to Pd NPs.32–35 Very interestingly, alloying these metals with Pd affords bimetallic nanoparticles with superior catalytic properties compared to their monometallic counterparts because of the ability of base metals to donate charge to the Pd centers.36–38 It is accepted that base metals increase the electron density on Pd in bimetallic alloys, thereby lowering the activation energy of the oxidative addition step of SCC. Alloying with base metals not only enhances the catalytic activity of Pd catalysts but also makes them more cost-effective because the Pd content is significantly reduced. To further advance these bimetallic catalysts and make them viable, precise control over NP size and dispersity, tunable chemical composition, and strong interaction with the support to prevent metal leaching and NP sintering are needed. In this regard, strong electrostatic adsorption (SEA) is uniquely suited for achieving all these desirable features.39–41 Catalyst supports are also very important in stabilizing metal nanoparticles against agglomeration and several supports were tested in SCC such as activated carbon,42 graphene oxide,43 reduced and partially reduced graphene oxide,36,43–47 carbon nanotubes,48 polymers,49 metal organic frameworks,50 and silica.51–53 The latter is commercially available, has high porosity, high physicochemical stability, and a well-established surface chemistry which make it an excellent candidate for SCC catalyst design and scale up.
Herein, we demonstrate the effective use of co-SEA in the preparation of ultra-small bimetallic nanoparticles from Pd and base metals Cu, Ni and Co and report their remarkable catalytic activity in SCC reactions. The catalytic activity of the catalysts and the effect of alloying base metals with Pd toward SCC were investigated and it was shown that the CuPd/SiO2 catalyst is superior to Pd/SiO2 and the other bimetallic nanoparticle catalysts achieving full bromobenzene conversion to biphenyl using benzeneboronic acid in 30 minutes at room temperature as opposed to three hours under the same conditions for the monometallic Pd/SiO2 catalyst. Studying the recyclability and activity toward different derivatives confirm the outstanding catalytic performance and stability of the bimetallic CuPd/SiO2 catalyst. High turnover frequencies for all the catalysts are comparable to or surpass those of homogeneous catalysts, especially the CuPd/SiO2 catalyst with a turnover frequency of 280000 h−1, attesting to the superior activity of the newly developed heterogeneous catalysts.
2. Experimental section
2.1. Materials and preparation of silica supported bimetallic nanoparticles
Amorphous fumed silicon(IV) oxide (Alfa Aesar, surface area 350 m2 g−1) was used without any further modification. Copper(II) nitrate trihydrate (Sigma Aldrich, 98.0–103%), nickel(II) nitrate hexahydrate (Alfa Aesar, 98%), cobalt(III) nitrate hexahydrate (J.T. Baker, 99.1%), and tetraamminepalladium(II) nitrate solution in 10 wt% H2O (Sigma Aldrich) were used for metal loading. 2000 ppm stock solutions of base metal salts were prepared using 5 N NH3 (aq). The total volume of the solution used for the SEA studies was calculated to yield a surface loading of 1000 m2 l−1 (eqn (S1), ESI†). The intended weight loading of the metal stock solution was pipetted into the support solution and stirred for 30 min to allow the adsorption of the cationic metal precursor on the support. The solution containing deposited metal salts on the surface of fumed silica is centrifuged in water and then in EtOH to replenish the basic solution. All samples were air-dried overnight under ambient conditions followed by drying in an oven for four hours at 120 °C. After drying, the samples were reduced under a flow of H2 (5% in Ar) at 400 °C in a tube furnace for one hour.2.2. Instrumentation
Inductively coupled plasma optical emission spectrometry (ICP-OES) was used to measure the bulk composition of the metals. Temperature programmed reduction (TPR) profiles were measured using a Micromeritics AutoChem II instrument coupled with a thermal conductivity detector (TCD) in a quartz u-tube to examine the reduction temperatures of the metals. Powder X-ray diffraction (PXRD) was examined with X-ray diffraction patterns collected at room temperature using a PANalytical X'Pert PRO Multipurpose Diffractometer (MPD). The samples were mounted on a zero-background sample holder measured in transmission mode using Ni-filtered Cu Kα radiation (operated at 40 kV, 45 mA; λ = 0.15418 nm), and the diffraction angle ranging from 10° to 80° with a step size of 0.07878° and a rate of 4.67° per min. X-ray photoelectron spectroscopy (XPS) analysis was performed using a PHI VersaProbe III Scanning XPS Microprobe employing a monochromatic, micro-focused, scanning X-ray source equipped with a hemispherical analyzer and an argon gun. The samples for XPS measurements were prepared by pressing the specimen into a piece of double-sided tape, which was then mounted onto the sample holder. During XPS analysis, a combination of a low-energy electron flood gun and an Ar ion flood gun was utilized for charge compensation. For some samples, the Ar gun was also used to sputter ∼10 nm from the top of the samples with a rate of 2 nm min−1 before performing XPS analysis. The binding energy scale was calibrated by setting the Si 2p peak at 103.5 eV. The XPS results were analyzed with the CasaXPS software (v4.84). High Resolution Transmission Electron Microscopy (HR-TEM) was performed using a FEI Titan TEM 300 kV equipped with a Gatan 794 Multi-Scan Camera, a HAADF-STEM detector. The samples were dropcast on carbon-coated grids (Ted Pella, Inc.) and left to dry over night at room temperature. The captured images were processed using ImageJ software. A gas chromatography – flame ionization detector (GC-FID) has been used to analyze the SCC reactants and products. An Agilent 6890N GC system equipped with an Agilent J&W GC HP-5MS capillary column was used in conjunction with an FID detector.2.3. Suzuki cross-coupling reactions under ambient conditions
The Pd-supported catalyst (Pd: 0.96 μmol, 0.3 mol%) was added to and dispersed in a glass vial with 2 ml of ethanol and 2 ml of deionized (DI) water. Bromobenzene (50 mg, 0.32 mmol, 1 equiv.) along with benzeneboronic acid (47 mg, 0.38 mmol, 1.2 equiv.), and potassium carbonate (133 mg, 0.96 mmol, 3 equiv.) were added to the solution. The reaction was performed under room temperature and atmospheric pressure conditions with continuous stirring until completion or after 3 hours. For the duration of the reaction, small aliquots were collected at regular intervals to monitor product formation. The biphenyl product was extracted with ethyl acetate via centrifugation and the progression of the reaction was monitored using a GC-FID. Product yields (%) were determined by GC-MS analysis using the formula: [(product peak area)/(bromo-reactant peak area + product peak area)] × 100.2.4. Suzuki cross-coupling reactions under microwave reaction conditions
The Pd-supported catalysts (Pd: 0.96 μmol, 0.3 mol%) were added to and dispersed in a microwave reaction vessel with 2 ml of ethanol and 2 ml of DI water. Bromobenzene (50 mg, 0.32 mmol, 1 equiv.) along with benzeneboronic acid (47 mg, 0.38 mmol, 1.2 equiv.), and potassium carbonate (133 mg, 0.96 mmol, 3 equiv.) were added to the solution. The vessel was sealed and heated at 60 °C for 5 min under MWI (CEM Discover Microwave, 300 W, 2.45 MHz). During the reaction, aliquots were collected and the reaction yield was monitored as described above. SCC reactions for different derivatives of the reactants and the recyclability of the catalyst were studied under MWI reaction conditions. For catalyst recycling, after the reaction was completed, the catalyst was collected by centrifugation, washed thoroughly three times with DI water and three times with ethanol, and then dried at 80 °C overnight. The dried catalyst was used for the consecutive run as described above. A small aliquot of the sample was collected after each run for analysis with a GC-FID to monitor the conversion. After each run, a small amount of the spent catalyst was used for ICP, XRD and XPS analyses.2.5. Suzuki cross-coupling reactions in an oil bath at 60 °C
The CuPd/SiO2 catalyst (Pd: 0.96 μmol, 0.3 mol%) was dispersed in a glass vial with 2 ml of ethanol and 2 ml of deionized (DI) water and the vial was then placed in the 60 °C oil bath. After the solution temperature reaches 60 °C, bromobenzene (50 mg, 0.32 mmol, 1 equiv.) was added to the solution along with benzeneboronic acid (47 mg, 0.38 mmol, 1.2 equiv.) and stirred for 30 seconds to ensure a homogeneous mixture. Finally, potassium carbonate (133 mg, 0.96 mmol, 3 equiv.) was added to the solution to start the reaction. During the reaction, small aliquots were collected every minute and the biphenyl product was extracted using ethyl acetate and centrifugation and analyzed using GC-FID and GC-MS.2.6. NMR analysis of the reaction products
Upon completion of the Suzuki cross coupling reactions under MW reaction conditions, the reaction mixture was mixed with 75 ml of CH2Cl2 in a 125 ml separatory funnel. Then, the organic layers were combined and dried in a vacuum at 80 °C to obtain the solid product. The products were then washed with hexane by centrifugation. The solid products were kept under vacuum for further characterization studies. NMR analysis was performed by mixing ∼50 mg of the solid to 750 μl of CDCl3. 1H NMR spectra were recorded using a Bruker NanoBay AVANCE III 400 MHz NMR spectrometer.3. Results and discussion
3.1. Preparation and characterization of the catalysts
Given the effectiveness and versatility of co-SEA in the preparation of bimetallic nanoparticles,39–41 we have used this method to prepare silica-supported Pd bimetallic catalysts containing Cu, Ni and Co. The SEA and co-SEA reaction schematics are shown in Scheme 1 below, and the process is illustrated schematically in Fig. S1 (ESI†). The fumed silica used in this study has a point of zero charge (PZC) of 3.6 in line with other silica supports reported in the literature.39,54,55 The SEA process was initiated by dispersing the silica support in DI water and then the pH of the solution was adjusted to 11.8 using 5 N NH3 (aq) to optimize electrostatic adsorption of metal complexes.39 As the pH increases, the native hydroxyl groups of the surface become deprotonated. It is worth mentioning that addition of an excess amount of base results in lower adsorption because metal precursors are shielded from the surface charge due to high ionic strength which according to kinetic studies diminishes the adsorption equilibrium constant and results in a phenomenon known as the double-layer screening effect.54,56 High pH values above 12 are also known to disintegrate silica supports. While the silica solution is continuously stirred, the intended weight loading of the metal stock solution is pipetted into the support solution, allowing the cationic metal precursor to adsorb, and then the solution is further stirred for 30 minutes. Following the deposition, the solution is centrifuged to replenish the basic solution, the sample is air-dried overnight, oven dried at 120 °C for four hours and then reduced under a flow of 5% H2 (in Ar) at 400 °C for one hour using a tube furnace. Four monometallic catalysts were prepared by SEA, namely Cu/SiO2, Co/SiO2, Ni/SiO2, and Pd/SiO2. The co-SEA was used to synthesize bimetallic catalysts, where positively charged precursor ions of the two metals are introduced into the solution simultaneously. These ions participate in competitive physical adsorption on the silica surface. To investigate the impact of base metal alloying on catalytic activity, three bimetallic catalysts were synthesized by co-SEA: CuPd/SiO2, NiPd/SiO2 and CoPd/SiO2. Furthermore, the most active catalyst CuPd/SiO2 was optimized by varying the Pd content in the Cu matrix: 0.5Cu1Pd/SiO2, 1Cu0.5Pd/SiO2, and 2Cu1Pd/SiO2. Although the latter catalyst has a similar Cu/Pd ratio to 1Cu0.5Pd/SiO2, it was prepared with the intention to maximize catalyst loading by saturating the surface of the support (complete monolayer coverage) by adsorption of [Cu(NH3)4]2+ and [Pd(NH3)4]2+ complexes. As such, this comparison could help in identifying the minimum amount of Pd needed for optimum catalytic activity. | ||
| Scheme 1 Schematic illustration of the SEA and Co-SEA reaction. | ||
3.2. Characterization
For mono and bimetallic catalysts, the comparison between targeted weight loading and the final bulk composition of each metal in wt% was measured by ICP-OES. The close agreement between intended wt% and bulk composition for the bimetallic catalysts suggests that the simultaneous adsorption of metal ions during co-SEA is not affected by their different competitive adsorption at a low weight loading of 1 wt% (Table 1 and Table S1, ESI†). The amount of metal adsorbed on the surface of the support by the SEA method depends on its surface area known as adsorption density (μmol m−2).56 This defines the maximum ability of metal adsorption (μmol) on the support surface area (m2). Other studies have also concluded that in similar bimetallic complexes, for a complete monolayer coverage, a maximum adsorption of 1.0 to 1.3 μmol m−2 can be achieved.39 According to ICP analysis, it is clear that for all the catalysts more than 85% of the metals were adsorbed on the support surface. In comparison with the monometallic catalysts, metal adsorption in bimetallic catalysts also maintains a close correlation between the targeted and the actual weight loading according to the ICP data.| Catalyst | Targeted weight loading (wt%) | Adsorbed metal (%) | Actual weight loading (wt%) | Catalyst denomination | |||
|---|---|---|---|---|---|---|---|
| BM | Pd | BM | Pd | BM | Pd | ||
| Cu/SiO2 | 1 | — | 99 | — | 0.99 | — | 0.99 Cu |
| Co/SiO2 | 1 | — | 86 | — | 0.86 | — | 0.86 Co |
| Ni/SiO2 | 1 | — | 99 | — | 0.99 | — | 0.99 Ni |
| Pd/SiO2 | — | 1 | — | 94 | — | 0.94 | 0.94 Pd |
| CuPd/SiO2 | 1 | 1 | 97 | 97 | 0.97 | 0.97 | 0.97 Cu 0.97 Pd |
| CoPd/SiO2 | 1 | 1 | 88 | 87 | 0.88 | 0.87 | 0.88 Co 0.87 Pd |
| NiPd/SiO2 | 1 | 1 | 85 | 86 | 0.85 | 0.86 | 0.85 Ni 0.86 Pd |
TPR was performed and the bimetallic samples were compared with their monometallic counterparts. The TPR profile of monometallic Pd/SiO2 shows a reduction peak at 190 °C (Fig. 1a) and Cu/SiO2 shows a peak at 250 °C with a broad shoulder starting at 480 °C (Fig. 1a). It is expected that Cu2+ was reduced to metallic Cu0 directly and there is no transient stage. The presence of complexes with higher reduction temperatures attests to their stronger interaction with the support.39 For the bimetallic catalyst CuPd/SiO2, a sharp peak at 190 °C and a smaller peak at 365 °C are present. The first peak can be assigned to the reduction of CuPd alloyed NPs, and the second, a much smaller peak, corresponds to the metallic Cu reduction shoulder with an ∼100 °C shift toward lower reduction temperatures. This shift is due to the hydrogen spillover from Pd species to the remaining unreduced Cu confirming the close proximity of Pd and Cu atoms on the support surface (Fig. 1a).39 The TPR profile of Ni/SiO2 shows a broad peak centered at 510 °C corresponding to the reduction of the Ni complex (Fig. 1b). In contrast, NiPd/SiO2 shows two sharp peaks at 170 °C and 315 °C corresponding to the reduction of bimetallic alloyed NiPd complexes and the shifted monometallic Ni complexes, respectively (Fig. 1b). The TPR profile of CoPd/SiO2 (Fig. 1c) also shows two peaks, a sharp peak at 178 °C and a small peak at 328 °C, while the monometallic catalyst Co/SiO2 also has two peaks but at much higher temperatures of 463 °C and 775 °C (Fig. 1c These peaks can be assigned to the reduction of Co3+ to Co2+ and another due to the reduction of Co2+ to metallic Co0.57 Low metal loading for Co causes these two peaks to merge. The reduction temperatures for the bimetallic catalyst CoPd/SiO2 follows the same pattern as that of the other bimetallic samples (Fig. 1c). Overall, the TPR profiles of bimetallic nanoparticle catalysts exhibit temperature shifts induced by the presence of Pd in close proximity to the base metal as well as the effect of hydrogen spillover on the reduction temperatures which validate the intimacy of the alloyed nanoparticles.
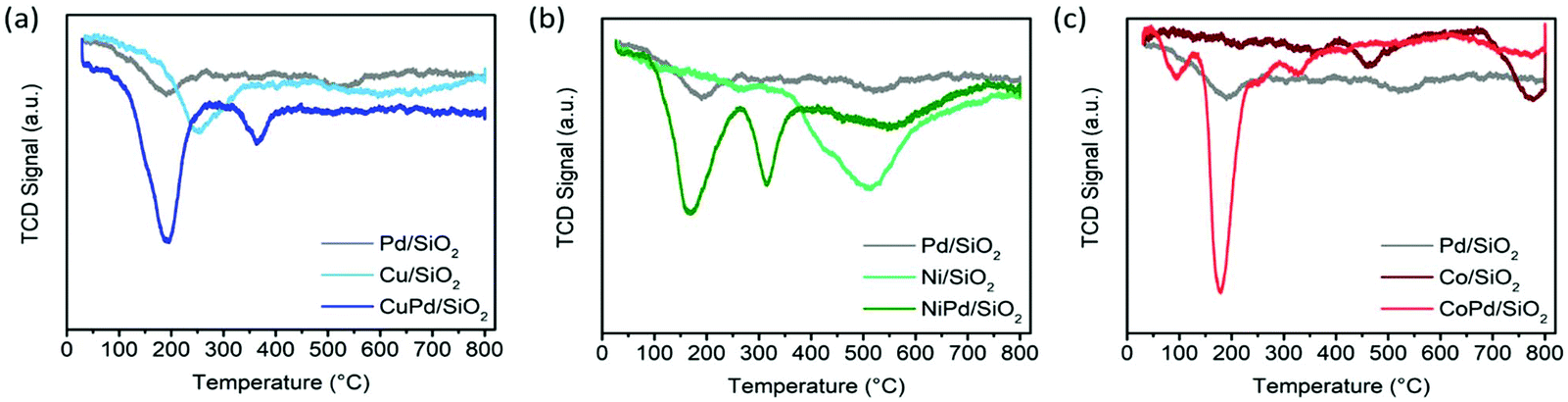 | ||
| Fig. 1 TPR patterns of (a) Pd/SiO2, Cu/SiO2, and CuPd/SiO2, (b) Pd/SiO2, Ni/SiO2, and NiPd/SiO2, and (c) Pd/SiO2, Co/SiO2, and CoPd/SiO2. | ||
The XRD patterns of the mono and bimetallic catalysts are shown in Fig. 2a. There are no distinguishable metal characteristic peaks present for any of the catalysts. The hump present at ∼22° for all the samples is characteristic of the amorphous structure of SiO2.55,58 The absence of any other peaks indicates the absence of crystalline materials and the formation of ultra-small particles with no signs of particle aggregation as typically XRD has a 2–2.5 nm size limit.59 XRD analysis was also performed on recycled CuPd/SiO2 as shown in Fig. 2b. The SiO2 amorphous hump at ∼22° is prominent for all the recycled samples, and no other distinguishable peaks are noticed; a very small hump starts to appear at ∼40° from the 3rd run of the recycled catalyst. This hump is correlated to the Pd (111) plane.60–62 The small and broad nature of the peak instead of a sharp peak may suggest that while the metallic nanoparticles start to aggregate in the recycled catalyst, the size of the particle is still smaller than 2.5 nm. The particle size is further discussed with the TEM and STEM results. For the CuPd/SiO2 catalyst, the absence of the Pd XRD small hump until after the 3rd recycling implies that CuPd alloying is playing a role in preventing Pd from aggregating.
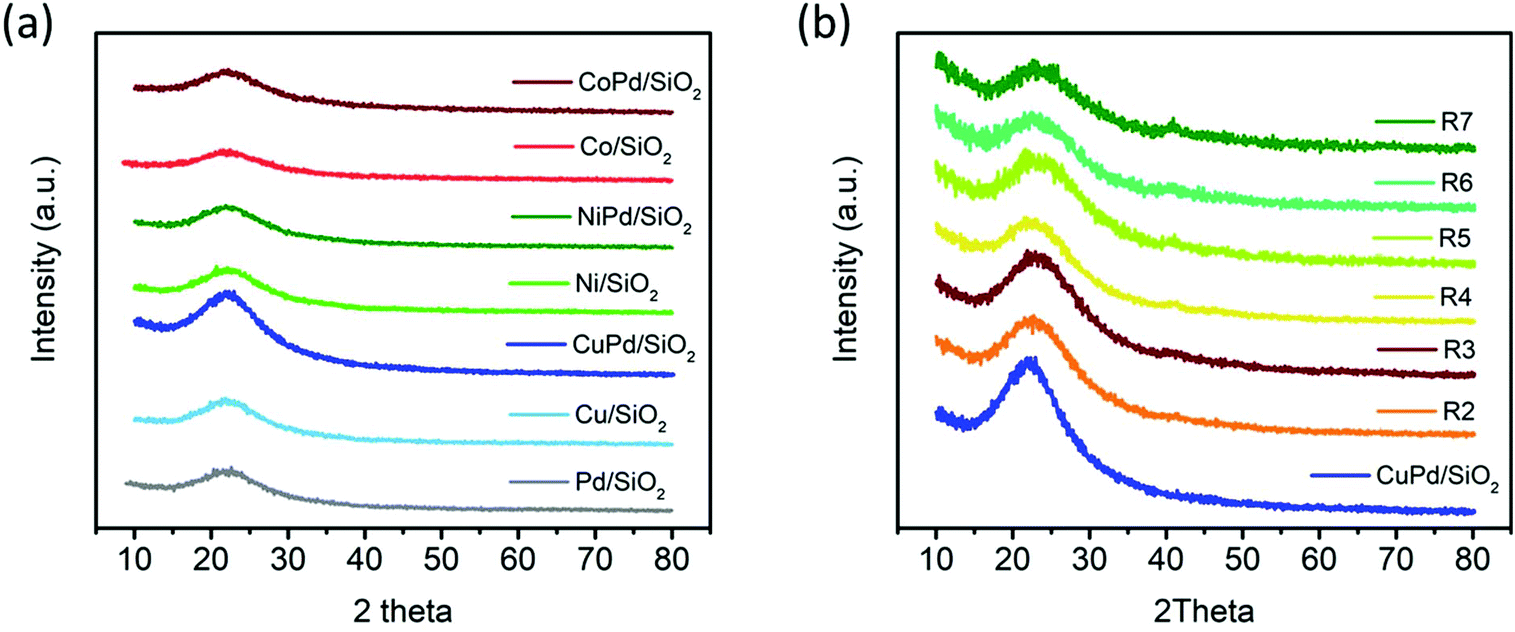 | ||
| Fig. 2 (a) XRD patterns of all mono and bimetallic catalysts. (b) XRD patterns of pristine CuPd/SiO2 (blue, bottom) and consecutive recycling test catalysts (R2–R7). | ||
XPS has also been performed on all of the catalysts. The surface composition of silicon and oxygen is reported in Table S2 (ESI†). The silica support shows a single peak at 103.5 eV for the Si 2p spectra (Fig. S2b†), and a single peak at 532.9 for O 1s spectra (Fig. S2c†).63 The fitted/assigned peaks for Si and O and their corresponding elemental composition (at%), reported in Fig. S2,† suggest that exposure to the 5 N ammonium hydroxide solution does not affect the chemical composition of the surface of silica. The deconvolution of the Cu 2p3/2 spectra shows a single peak at 933.1 eV for the monometallic sample, Cu/SiO2, and a single peak at 933.2 eV for the bimetallic catalyst CuPd/SiO2. This 0.1 eV shift for the bimetallic sample corresponds to the alloying of the two metal species (Fig. S2d†). It is also known that any small shift from metallic copper with a binding energy of 932.864,65 may also be induced by surface oxidation. Copper(I) oxide and copper(II) oxide have a binding energy of 933.0 and 933.5 eV respectively, making the differentiation between the metallic species and the Cu+ challenging.66 XPS is considered to be a surface-sensitive method, consequently for all samples, low contents of the metals and their ultra-small particle size result in low intensities. In the case of Co 2p3/2 spectra, the monometallic catalyst Co/SiO2 has a very weak peak at 783.0 eV, which has the closest correlation to Co2+.67,68 No peaks in the Co 2p spectra for the bimetallic sample can be detected (Fig. S2f†). Deconvolution of Ni 2p3/2 spectra shows a peak at 856.2 eV and a satellite hump at 861.8 eV for the monometallic Ni/SiO2 (Fig. S2e†). The bimetallic catalyst, NiPd/SiO2, shows a peak at 856.5 eV and a satellite peak at 861.7 eV can also be fitted (Fig. S2e†).69 The 0.3 eV shift for the bimetallic sample in comparison with Ni/SiO2 indicates a minor shift in the electron cloud distribution for the NiPd alloy, suggesting that Ni atoms do not contribute significantly to the desired Pd0 state essential for catalytic activity. As the comparison between the transition metal species in bimetallic samples suggests, the deconvoluted Cu spectra for those samples more closely correlates with metallic Cu, which can be play a significant role in the catalytic activity of the cross-coupling reactions. The deconvolution of Pd 3d spectra in Fig. S2g† characterized by spin-orbital splitting (Pd 3d5/2 and Pd 3d3/2) shows two peaks at 335.6 eV and 340.3 eV for the bimetallic CuPd/SiO2 catalyst which can both be assigned to the Pd0 state;70 336.1 eV and 341.9 eV are the corresponding peaks for CoPd/SiO2, and 336.1 eV and 341.2 eV are the corresponding peaks for NiPd/SiO2. Interestingly, the Pd atoms in the CuPd/SiO2 catalyst (Fig. S2g†) have average binding energies closer to the metallic Pd at 334.8–335.4 eV and 340.1–340.7 eV.50,70,71 Chemical shifts in the range of 0.4–0.7 can also be traced back to the strong interaction between the Pd 3p core level and the base metal.70 It has also been reported that Cu may strongly modify the Pd valence shell through electrons being injected into the sp orbital.70 On the other hand, the binding energy of Pd atoms alloyed with Co and Ni correlates better with the Pd2+ species with a binding energy of 336.7 eV, which can be traced back to possible surface oxidation or shifts in electron cloud distribution; either would affect the catalytic activity of these catalysts.72 In the case of monometallic catalyst Pd/SiO2, a peak at 336.3 eV and a peak at 341.6 eV can be deconvoluted (Fig. S2g†). The positive shift toward higher energies and a closer correlation with Pd2+ binding energy highlights the important role base metals play in keeping the catalytically active Pd at the zero oxidation state upon introduction into the reaction solution and during catalyst handling after reduction with hydrogen.
To study the effects of each cross-coupling experiment on CuPd/SiO2, the catalysts after the 3rd and 6th run were chosen to investigate the changes in the intimacy of the alloy and their binding energies. Deconvolution of Cu 2p spectra, for both the 3rd and 6th experiment, shows a single peak at 933.2 eV (Fig. S2d†). This finding indicates that copper atoms maintain their integrity without going through any surface changes. In the case of the Pd 3d spectra (Fig. S2g†), the 3rd run peaks at 336.0 eV and 341.1 eV are deconvoluted, and the 6th run peaks at 336.7 eV and 341.6 eV indicate a shift towards higher binding energies suggesting that the Pd atoms are becoming more positively charged (Fig. S2g†). The formation of Pd(II) complexes is traced back to the surface detachment induced by the oxidative addition of aryl halide, which further undergo an in situ reduction forming the catalytically active Pd(0) species.19,20 Well-alloyed CuPd catalysts can enhance the in situ reduction and further improve the ability to achieve full conversion in the cross-coupling reaction even after the 6th recycling run.
HRTEM and STEM images were analyzed to determine the metal nanoparticle size and distribution. It is clear from the particle size analysis that all catalysts have active metal particle sizes in the 0.7–2.3 nm range (Fig. 3). The particle size distribution (Fig. S3, ESI†) is narrow for all the catalysts. Of particular interest is that the addition of a transition metal along with Pd did not change the particle size significantly. This control over nanoparticle's size stems from the fact that SEA limits the surface coverage of the support to a monolayer of metal ions and upon reduction of the physically adsorbed precursors, large nanoparticles are more difficult and less likely to form. This finding is in agreement with the absence of any peaks in the XRD patterns as even small particles can result in broad peaks with very low intensity and that only becomes apparent after a few consecutive recycling experiments. Smaller particles also have a higher exposed surface area per particle, which should result in a higher catalytic activity compared to bigger particles.
 | ||
| Fig. 3 HRTEM, STEM, and the corresponding lattice spacing of (a) Pd/SiO2, (b) CuPd/SiO2, (c) NiPd/SiO2 and (d) CoPd/SiO2. | ||
STEM analysis of the CuPd/SiO2 catalyst studied for recyclability was performed on the 3rd and 6th recycling runs to examine the integrity of the catalyst and any changes that might take place on average particle size (Fig. S4, ESI†). The particle size analysis shows that there is almost no change in the average particle size during the recycling runs which indicates a strong catalyst–support interaction and the superior sustainability of a well alloyed CuPd catalyst. The strong interaction with the silica support achieved by the co-SEA method protects the active metal particles from sintering.
3.3. Suzuki cross-coupling catalytic performance
The developed monometallic and bimetallic catalysts were studied for SCC reactions at room temperature (RT) as depicted in Fig. 4. Among all catalysts, CuPd/SiO2 exhibits superior activity reaching a bromobenzene conversion of 35% after one minute and a full conversion in only 30 min under ambient conditions (Fig. 4b). This catalyst surpasses all the other bimetallic catalysts and the monometallic Pd catalyst, which reaches full conversion after three hours. The second-best catalyst, NiPd/SiO2, reaches a conversion of 18% after one minute and a full conversion in 90 min (Fig. 4b). On the other hand, dilution of Pd in a Co matrix (CoPd/SiO2) does not seem to enhance the catalytic activity when compared to the other bimetallic catalysts and has a similar activity to that of the Pd/SiO2 catalyst where full conversion to the biphenyl product is reached in three hours (Fig. 4b). The catalysts composed of the monometallic base metals (Ni/SiO2, Co/SiO2 and Cu/SiO2) were found to be inactive towards the SCC reaction at RT (Fig. 4b and c).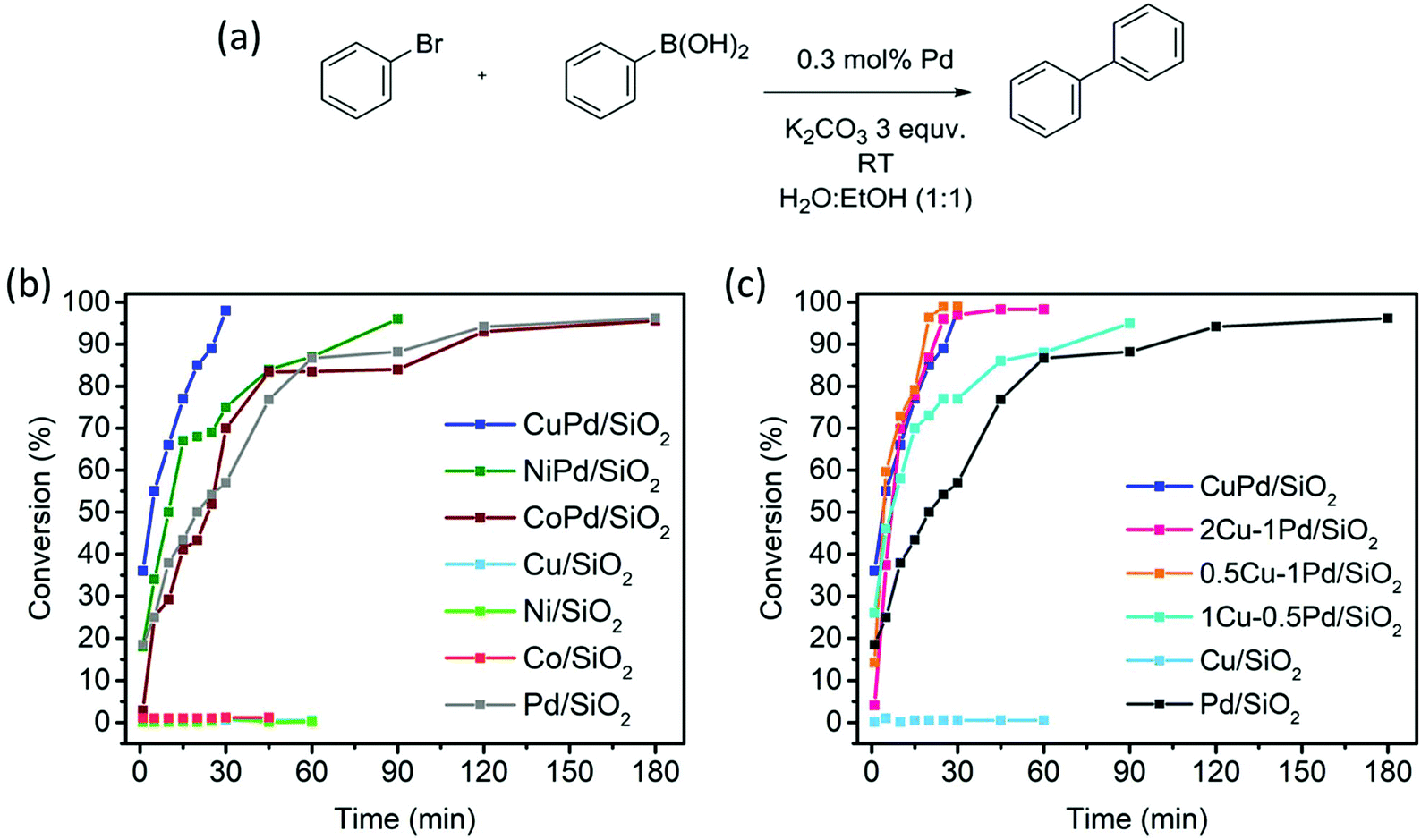 | ||
| Fig. 4 (a) Cross-coupling reactions between bromobenzene and benzeneboronic acid. (b) Bromobenzene conversion results by the mono and bimetallic nanoparticle catalysts. (c) Bromobenzene conversion results by different Pd contents in Cu nanoparticle catalysts. | ||
The superior activity of the CuPd alloyed catalyst encouraged us to investigate the impact of the Cu:Pd ratio on catalytic activity. These catalysts defined using the targeted metal loading and the actual loading determined from the ICP measurements are listed in Table S1 (ESI†). To study the effect of Pd dilution in the Cu matrix, 0.5Cu–1Pd/SiO2, 1Cu–0.5Pd/SiO2 and 2Cu–1Pd/SiO2 catalysts were synthesized. Even though 2Cu–1Pd/SiO2 has a similar Cu/Pd ratio to 1Cu–0.5Pd/SiO2, this catalyst was prepared by saturating the support surface with metal ions for complete monolayer coverage formation.39 The bromobenzene conversion yields from the SCC reaction show that the dilution of 1 wt% Pd in various Cu loadings of 0.5, 1 to 2 wt%, in the 0.5Cu–1Pd/SiO2, CuPd/SiO2 and 2Cu–1Pd/SiO2 catalysts, respectively, has almost no effect on the overall catalytic activities, with all the catalysts achieving 100% conversion in 30 min (Fig. 4c). Lowering the loading of Pd in the 1Cu–0.5Pd/SiO2 catalyst results in full conversion in almost 90 min as compared to 30 min for the CuPd/SiO2 catalyst. A control test was also performed in which homo-coupling of bromobenzene by Pd/SiO2 and CuPd/SiO2 catalysts were investigated. Both catalysts yielded zero conversion of the substrate to biphenyl, which attests to the excellent chemoselectivity of the catalysts.
3.4. Catalyst recycling under microwave assisted reactions
Recyclability of the catalyst is a very important criterion for large-scale applications. The ability to recycle the best performing catalyst, CuPd/SiO2, was investigated under MWI at 60 °C for five minutes and compared with the monometallic catalyst Pd/SiO2. The CuPd/SiO2 catalyst shows an excellent recyclability with a bromobenzene conversion of 100% achieved for seven consecutive runs (Table 2). On the other hand, the recycled Pd/SiO2 catalyst under similar conditions exhibits a small steady drop-in activity upon cycling, where the conversion drops to 93% and 89% by the 3rd and 6th runs, respectively (Table S2, ESI†).| Run | Conversion (%) | Run | Conversion (%) |
|---|---|---|---|
| Conditions: 0.3 mol% Pd, 0.32 mmol bromobenzene, 0.38 mmol phenylboronic acid, 1 mmol K2CO3. Microwave irradiation: 300 W, 60 °C, 5 min reaction time. | |||
| 1 | 100 | 5 | 100 |
| 2 | 100 | 6 | 100 |
| 3 | 100 | 7 | 100 |
| 4 | 100 | ||
In order to confirm that the activity of the CuPd/SiO2 catalyst is not influenced by MWI, the Suzuki cross coupling reaction was also performed in an oil bath at 60 °C for comparison with the MWI reaction at 60 °C. As shown in Fig. S5 (ESI†), the CuPd/SiO2 catalyst shows a full conversion of bromobenzene to the biphenyl product in 4 min, thus confirming that microwave irradiation has no effect on the superior activity of the catalyst (except for the heating effect).
The excellent catalytic activity of the current catalysts can be traced back to nanoparticles’ size, narrow size distribution, homogeneous particle distribution on the support, and base metal alloying which appears to be crucial in inhibiting the oxidation of Pd and providing catalyst stability during consecutive recyclability runs. The superior catalytic activity of CuPd/SiO2 even after successive runs suggests that the catalyst maintains remarkable activity indicating high stability against sintering and deactivation. The stability of the catalyst was probed by studying the surface morphology, particle size and chemical composition of the recycled catalyst by using XRD, HRTEM and XPS, respectively. The XRD patterns for the recycled catalyst after each run are almost identical to those for the pristine catalyst with a new very broad peak at ∼40° correlated to the Pd(111) plane which appears after the 3rd recycling run of the CuPd/SiO2 catalyst (Fig. 2b). STEM analysis of the particle size (Fig. S4, ESI†) shows no significant change in the average particle size during the recycling runs indicating a strong catalyst–support interaction. XPS measurements after the 3rd and 6th recycling runs suggest that the chemical nature of the CuPd alloy is maintained after the runs (Fig. S2 and Table S3 ESI†). ICP measurements on the Pd/SiO2 and CuPd/SiO2 catalysts after each run of the recycling experiments (Tables S4 and S5, ESI†) show that for the Pd/SiO2 catalyst only 3 wt% of Pd has leached away throughout the entire five recycling runs (Table S4†). Also, the CuPd/SiO2 catalyst does not lose much Pd (4 wt%) throughout the first four recycling runs (Table S5†). The amount of leached Pd increases to 11% and 26% by the end of the 5th and 7th recycling runs, respectively (Table S5†). In contrast, 19% of Cu is leached from the surface after the first run, and then during the following consecutive runs, the amount of Cu does not change significantly (Table S5†). The loss of Cu during the first run may be explained by the leaching of some weakly adsorbed Cu particles on the surface of the support resulting in a sudden decrease in the Cu concentration from 0.97 wt% to 0.78 wt% after the first run. However, as the Cu remains consistent throughout the rest of the recyclability runs (about 0.76 wt%), it can be assumed that the rest of the Cu was adsorbed strongly and thus the amount of Cu does not change much during the consecutive runs. The observation of 100% reaction conversion by the CuPd/SiO2 catalyst in spite of the 26 wt% and 23 wt% leaching of Pd and Cu, respectively after the seven consecutive runs suggests that the actual active site of the bimetallic catalyst has an optimum composition of ∼0.75Cu–0.75Pd and the excess Cu and Pd particles are probably weakly adsorbed on the silica surface and therefore do not contribute significantly to the catalytic activity. On the other hand, the negligible amount of Pd leaching from the Pd/SiO2 catalyst (3 wt% after the 5th run, Table S4†) rules out leaching as the primary cause for small catalyst deactivation (91% reaction conversion after the 5th run, Table S2†).
In order to check the leaching of active catalyst species into solution, the Cu and Pd concentrations in the solution were determined by ICP-MS after the removal of the spent CuPd/SiO2 catalyst by hot filtration from the reaction solution. The ICP-MS data (Table S6, ESI†) clearly show no leaching of Pd or Cu throughout the seven consecutive recyclability runs each resulting in 100% conversion of bromobenzene to the biphenyl product (Table 2). The very small Pd and Cu concentrations (less than 8 ppb, Table S6†) in the reaction solution after the consecutive reaction runs support the complete heterogeneity of the CuPd/SiO2 catalyst. Further evidence for the heterogeneous catalytic mechanism is the failure to observe reactivity after the removal of the CuPd/SiO2 catalyst from the reaction medium.
The superior catalytic activity of the CuPd/SiO2 catalyst in comparison with NiPd/SiO2 and CoPd/SiO2 catalysts can be explained by the electronic properties of each system and the expected synergistic effects of the metals involved in the bimetallic catalysts. According to the XPS studies, Cu in the Cu–Pd alloy helps maintain Pd in the unoxidized state (Pd0), whereas deconvoluted Pd spectra of Ni–Pd and Co–Pd alloys reveal that binding energies shifted toward Pd2+ (Fig. S2, ESI†). It is worth noting that DFT calculations of the bimetallic catalysts suggest the presence of an emerging enhanced electronic structure factor upon base metal doping which increases electron density on Pd, thereby facilitating the oxidative addition of the aryl halide which is the rate limiting step in SCC.28,29
For all of the SCC reactions performed under RT and MWI conditions, the Pd content is normalized to 0.3 mol% Pd. With this catalyst concentration, all the CuPd/SiO2, NiPd/SiO2, CoPd/SiO2, and Pd/SiO2 catalysts show 100% reaction conversion within five minutes of MWI at 60 °C. Further reduction in the catalyst's concentration provides insight into the reactivity of these catalysts (Fig. S6, ESI†). Lowering Pd content from 0.3 mol% to 0.03 mol% results in bromobenzene conversions of 100%, 97%, 99%, and 97% within five minutes of MWI at 60 °C for the CuPd/SiO2, NiPd/SiO2, CoPd/SiO2, and Pd/SiO2 catalysts, respectively. At 0.01 mol% (0.003 mol%) under the same reaction conditions, bromobenzene conversions are 97% (70%), 76% (70%), 90% (61%), and 80% (52%) for the CuPd/SiO2, NiPd/SiO2, CoPd/SiO2, and Pd/SiO2 catalysts, respectively (Fig. S6 and Table S7, ESI†). These results demonstrate the remarkable catalytic activity of the CuPd/SiO2, NiPd/SiO2, CoPd/SiO2, and Pd/SiO2 catalysts at the lowest catalyst concentration ever used in a SCC reaction (0.003 Pd mol%) which result in turnover frequencies (TOFs) of 280000 h−1, 248000 h−1, 240000 h−1, and 208000 h−1, respectively. To our knowledge, these are the highest TOFs observed in a microwave-assisted Suzuki cross coupling reaction by the supported Pd nanoparticle catalysts.13
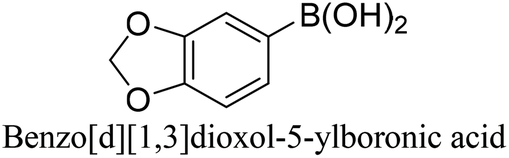
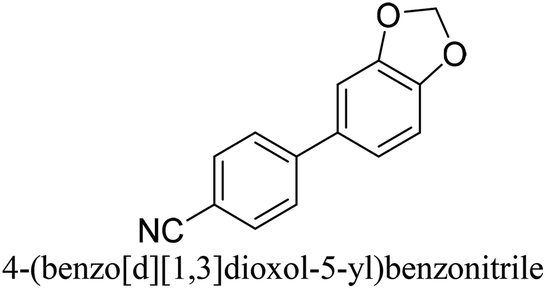
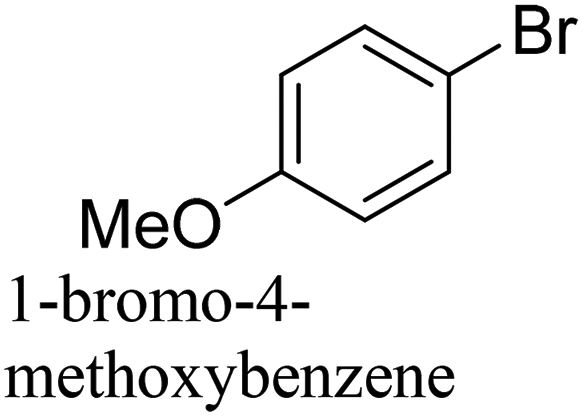
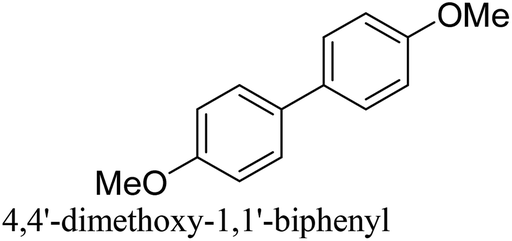
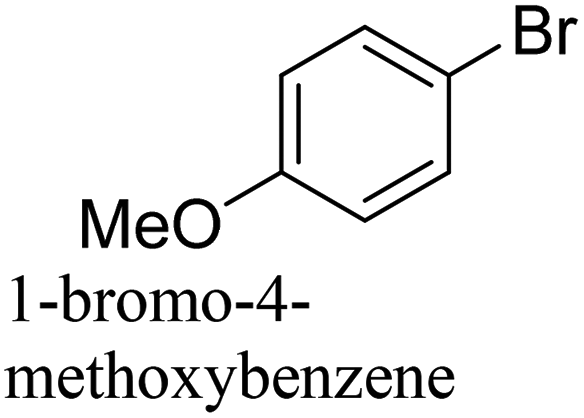
To generalize the above results, the range of catalytic utility in SCC reactions for the preparation of other biphenyl products containing a broader range of functionalities was investigated. For these experiments, we chose to use the CuPd/SiO2 catalyst exclusively due to its superior catalytic activity demonstrated in our prior studies. As illustrated in Table 3, the reaction was carried out in the presence of 0.3 Pd mol%, and potassium carbonate (3 equiv.) using H2O:EtOH (1:1) as environmentally benign solvents. The reactions are heated under microwave irradiation at 60 °C for five minutes. As shown in Table 3, a broad range of aryl bromide containing electron donating and electron withdrawing groups (nitrile, aldehyde and nitro) can be effectively incorporated into the coupling products. In addition, phenyl boronic acids bearing useful functionality such as 4-dimethylamino also led to a good yield of SCC products. Finally, even a more difficult substrate such as aryl chloride 1-chloro-4-nitrobenzene was able to undergo Suzuki coupling with 4-carbamoylphenyl boronic acid although in only 34% yield.
| Aryl bromide | Aryl boronic acid | Product | Yield (%) |
|---|---|---|---|
| Aryl bromide (0.32 mmol, 1 equiv.), arylboronic acid (0.38 mmol, 1.2 equiv.), potassium carbonate (133 mg, 0.96 mmol, 3 equiv.), and CuPd (10.5 mg, 0.3 mol% Pd) in 4 mL (1 v:1 v H2O–EtOH) was heated at 60 °C (MWI) for 5 min. Product yield (%) was determined by GC-MS analysis. All products were characterized by 1H NMR spectra as shown in Table S8 (ESI†). |
|||
|
|
|
96% |
|
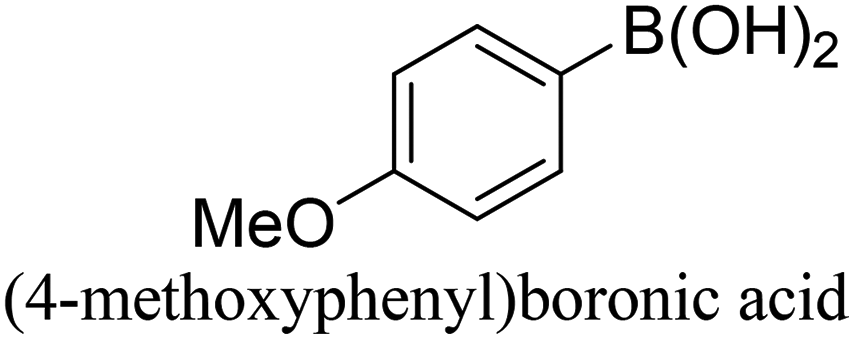
|
|
80% |
|
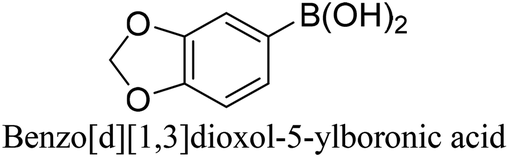
|
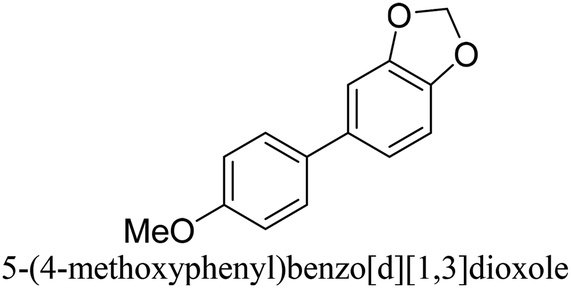
|
72% |
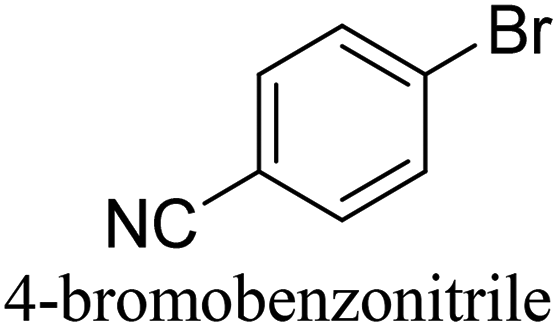
|
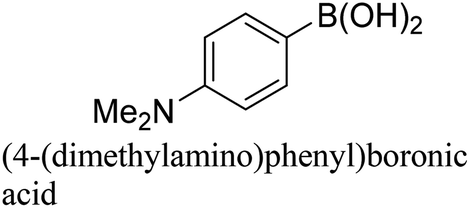
|
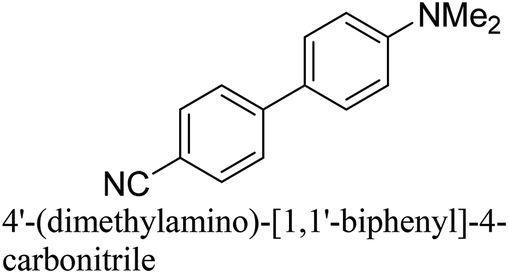
|
91% |
|
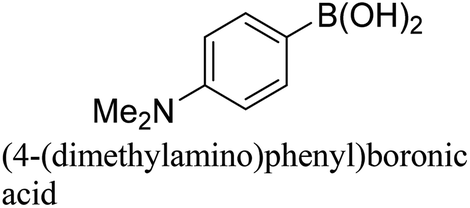
|
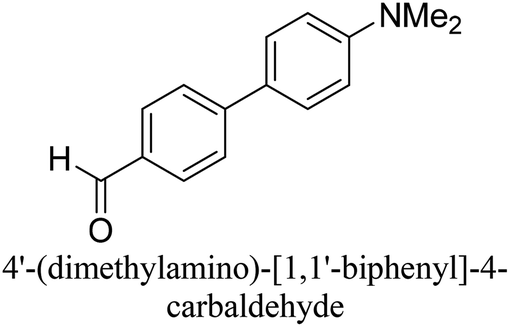
|
94% |
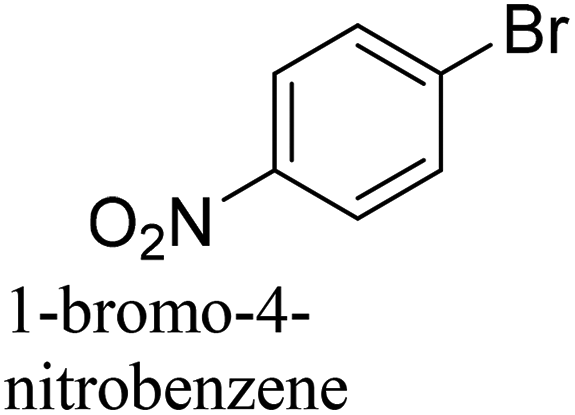
|
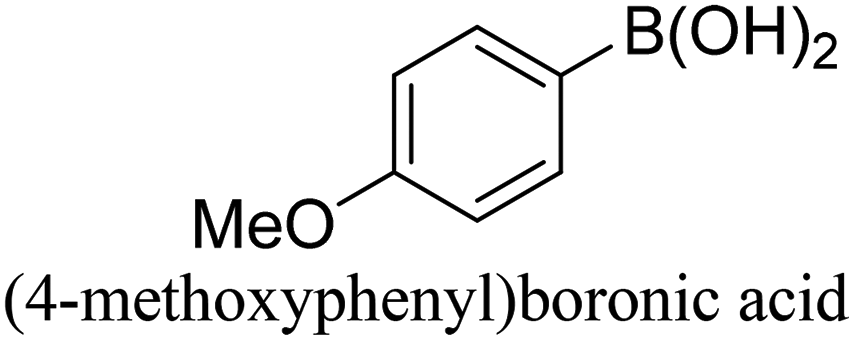
|
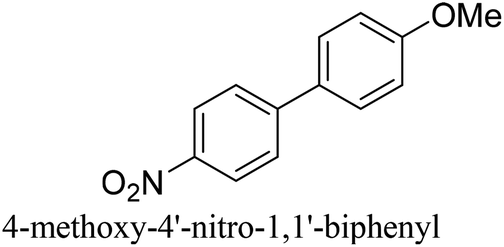
|
94% |
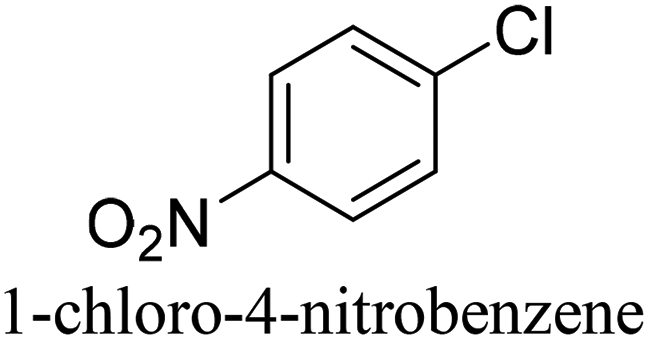
|
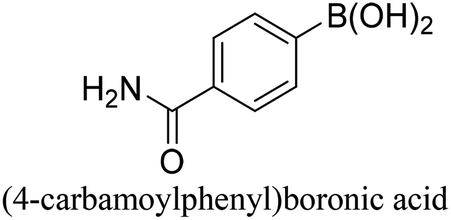
|
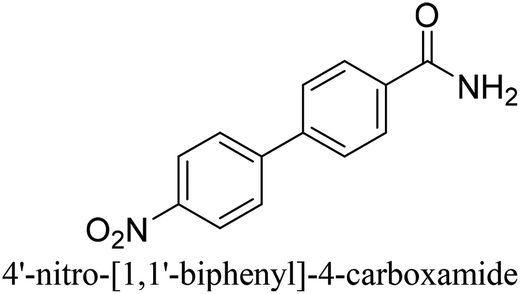
|
34% |
4. Conclusions
A facile, general, reproducible and highly applicable synthetic technique based on strong electrostatic adsorption has been developed for the synthesis of ultra-small bimetallic alloy nanoparticle catalysts supported on high surface area fumed silica for Suzuki cross-coupling reactions. Through this approach, a very low content of the precious metal Pd is alloyed with low content inexpensive base metals (Cu, Ni and Co). TPR patterns of the bimetallic catalysts and their monometallic counterparts confirm the formation of supported alloyed nanoparticles. The absence of characteristic metal peaks in the XRD patterns taken together with the HRTEM images show an average particle size of 1.3 nm. The formation of this ultra-small nanoparticle increases the efficiency of Pd atoms for catalytic activities. Among the three bimetallic catalysts (CuPd/SiO2, NiPd/SiO2 and CoPd/SiO2), the CuPd/SiO2 catalyst exhibits superior activity and unprecedented recyclability. Under room temperature and atmospheric pressure conditions, this catalyst reaches complete bromobenzene conversion to the biphenyl product in 30 min, while the NiPd/SiO2 catalyst performs the same reaction under similar conditions in 90 min. The monometallic Pd/SiO2 and the bimetallic CoPd/SiO2 catalysts reach full conversion to the biphenyl product under similar reaction conditions after three hours. The performance of the CuPd/SiO2 catalyst for the cross-coupling of bromobenzene and phenylboronic acid derivatives also yields high conversion values. The CuPd/SiO2 catalyst is highly recyclable, while maintaining the highest catalytic activity (100% conversion). The catalyst exhibits an unprecedented turnover frequency of 280000 h−1 in the microwave assisted Suzuki reaction at 60 °C that surpasses the performance of other supported Pd catalysts. The superior performance of the CuPd/SiO2 catalyst is attributed to the CuPd alloying which helps in maintaining Pd at the desired catalytic status of Pd0 better than the other bimetallic catalysts. We believe that the facile formation of these ultra-small alloy nanoparticles and their superior catalytic activity would give rise to a new generation of highly efficient heterogeneous catalysts that are comparable to the best homogeneous Pd catalysts for the Suzuki cross-coupling reactions.
Author contributions
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the National Science Foundation (CHE-1900094) for the support of this work. This work was also partially supported by the Center for Rational Catalysis Synthesis (CeRCaS), an Industry/University Cooperative Research Center funded in part by the National Science Foundation [Industry/University Collaborative Research Center grant IIP1464595].References
- Á. Molnár, Chem. Rev., 2011, 111, 2251 CrossRef.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722 CrossRef CAS.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef CAS.
- A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS.
- H. Li, C. C. C. Johansson Seechurn and T. J. Colacot, ACS Catal., 2012, 2, 1147 CrossRef CAS.
- P. Das and W. Linert, Coord. Chem. Rev., 2016, 311, 1–23 CrossRef CAS.
- A. Fihri, M. Bouhrara, B. Nekoueishahraki, J.-M. Basset and V. Polshettiwar, Chem. Soc. Rev., 2011, 40, 5181 RSC.
- A. Zapf and M. Beller, Top. Catal., 2002, 19, 101–109 CrossRef CAS.
- M. Pagliaro, V. Pandarus, R. Ciriminna, F. Béland and P. Demma Carà, Chem. Soc. Rev., 2012, 4, 432–445 CAS.
- Cross Coupling Reactions in Organic Synthesis Themed Issue, Chem. Soc. Rev., 2011, 40, 4877.
- S. R. Chemler, D. Trauner and S. J. Danishefsky, Angew. Chem., Int. Ed., 2001, 40, 4544–4568 CrossRef CAS.
- Y. Li, Y. Dong, Y. Wei, J. Jv, Y. Chen, J. Ma, J. Yao and Y. Dong, Organometallics, 2018, 37, 1645–1648 CrossRef CAS.
- Z. Chen, E. Vorobyeva, S. Mitchell, E. Fako, M. A. Ortuño, N. López, S. M. Collins, P. A. Midgley, S. Richard, G. Vilé and J. Pérez-Ramírez, Nat. Nanotechnol., 2018, 13, 702–707 CrossRef CAS.
- D. B. Eremin and V. P. Ananikov, Coord. Chem. Rev., 2017, 346, 2 CrossRef CAS.
- F. Zhao, B. M. Bhanage, M. Shirai and M. Arai, Chem. – Eur. J., 2000, 6, 843 CrossRef CAS.
- F. Zhao, K. Murakami, M. Shirai and M. Arai, J. Catal., 2000, 194, 479 CrossRef CAS.
- F. Zhao, M. Shirai and M. Arai, J. Mol. Catal. A: Chem., 2000, 154, 39 CrossRef CAS.
- K. Köhler, R. G. Heidenreich, J. G. E. Krauter and J. Pietsch, Chem. – Eur. J., 2002, 8, 622 CrossRef.
- A. Biffis, M. Zecca and M. Basato, Eur. J. Inorg. Chem., 2001, 2001, 1131 CrossRef.
- M. T. Reetz and E. Westermann, Angew. Chem., Int. Ed., 2000, 39, 165–1678 CrossRef CAS.
- R. K. Rai, K. Gupta, D. Tyagi, A. Mahata, S. Behrens, X. Yang, Q. Xu, B. Pathak and S. K. Singh, Catal. Sci. Technol., 2016, 6, 5567–5579 RSC.
- A. M. Trzeciak and A. W. Augustyniak, Coord. Chem. Rev., 2019, 384, 1–20 CrossRef CAS.
- F. Rafiee, P. Khavari, Z. Payami and N. Ansari, J. Organomet. Chem., 2019, 883, 78–85 CrossRef CAS.
- G. Bao, J. Bai, C. Li and D. Yu, Catal. Lett., 2018, 148, 3389–3401 CrossRef CAS.
- K. Lamei, H. Eshghi, M. Bakavoli, S. A. Rounaghi and E. Esmaeili, Catal. Commun., 2017, 92, 40–45 CrossRef CAS.
- I. Hussain, J. Capricho and M. A. Yawer, Adv. Synth. Catal., 2016, 385, 3320–3349 CrossRef.
- J. Yang, Y. Wu, X. Wu, W. Liu, Y. Wang and J. Wang, Green Chem., 2019, 21, 5267–5273 RSC.
- T. Sperger, C. M. Le, M. Lautens and F. Schoenebeck, Chem. Sci., 2017, 8, 2914–2922 RSC.
- E. Alvaro and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 7858–7868 CrossRef CAS.
- M. Julia and M. Duteil, Bull. Soc. Chim. Fr., 1973, 2790, 2790 Search PubMed.
- M. Julia, M. Duteil, C. K. E. Grard and E. Kunz, Bull. Soc. Chim. Fr., 1973, 2791–2794 CAS.
- S. Handa, B. Jin, P. P. Bora, Y. Wang, X. Zhang, F. Gallou, J. Reilly and B. H. Lipshutz, ACS Catal., 2019, 9, 2423–2431 CrossRef CAS.
- K. Pan, H. Ming, H. Yu, H. Huang, Y. Liu and Z. Kang, Dalton Trans., 2012, 41, 2564–2566 RSC.
- C. A. Malapit, J. R. Bour, C. E. Brigham and M. S. Sanford, Nature, 2018, 563, 100–104 CrossRef CAS.
- L. Jiao and J. R. Regalbuto, J. Catal., 2008, 260, 329–341 CrossRef CAS.
- Y. Yang, A. C. Reber, S. E. Gilliland, C. E. Castano, B. F. Gupton and S. N. Khanna, J. Phys. Chem. C, 2018, 122, 25396–25403 CrossRef CAS.
- S. E. Smith, A. R. Siamaki, B. F. Gupton and E. E. Carpenter, RSC Adv., 2016, 6, 91541–91545 RSC.
- R. K. Rai, K. Gupta, S. Behrens, J. Li, Q. Xu and S. K. Singh, ChemCatChem, 2015, 7, 1806–1812 CrossRef CAS.
- A. Wong, Q. Liu, S. Griffin, A. Nicholls and J. R. Regalbuto, Science, 2017, 358, 1427–1430 CrossRef CAS.
- M. Schreier and J. R. Regalbuto, J. Catal., 2004, 225, 190–202 CrossRef CAS.
- K. Ding, D. A. Cullen, L. Zhang, Z. Cao, A. D. Roy, I. N. Ivanov and D. Cao, Science, 2018, 362, 560–564 CrossRef CAS.
- A. A. Kurokhtina, E. V. Larina, A. F. Schmidt, A. Malaika, B. Krzyżyńska, P. Rechnia and M. Kozłowski, J. Mol. Catal. A: Chem., 2013, 379, 327 CrossRef CAS.
- S. Diyarbakir, H. Can and Ö. Metin, ACS Appl. Mater. Interfaces, 2015, 7, 3199–3206 CrossRef CAS.
- A. R. Siamaki, A. E. R. S. Khder, V. Abdelsayed, M. S. El-Shall and B. F. Gupton, J. Catal., 2011, 279, 1–11 CrossRef CAS.
- S. Moussa, A. R. Siamaki, B. F. Gupton and M. S. El-Shall, ACS Catal., 2012, 2, 145–154 CrossRef CAS.
- H. A. Elazab, A. R. Siamaki, S. Moussa, B. F. Gupton and M. S. El-Shall, Appl. Catal., A, 2015, 491, 58–69 CrossRef CAS.
- H. A. Elazab, S. Moussa, A. R. Siamaki, B. F. Gupton and M. S. El-Shall, Catal. Lett., 2017, 147, 1510–1522 CrossRef CAS.
- A. R. Siamaki, Y. Lin, K. Woodberry, J. W. Connell and B. F. Gupton, J. Mater. Chem. A, 2013, 1, 12909–12918 RSC.
- Y. M. A. Yamada, S. M. Sarkar and Y. Uozumi, J. Am. Chem. Soc., 2012, 134, 3190–3198 CrossRef CAS.
- B. Yuan, Y. Pan, Y. Li, B. Yin and H. Jiang, Angew. Chem., Int. Ed., 2010, 49, 4054–4058 CrossRef CAS.
- S. Jana, B. Dutta, R. Bera and S. Koner, Inorg. Chem., 2008, 47, 5512 CrossRef CAS.
- Y. Wan, H. Wang, Q. Zhao, M. Klingstedt, O. Terasaki and D. Zhao, J. Am. Chem. Soc., 2009, 131, 4541 CrossRef CAS.
- B. S. Soled, Science, 2015, 350, 1171–1172 CrossRef.
- H. R. Cho and J. R. Regalbuto, Catal. Today, 2015, 246, 143–153 CrossRef CAS.
- R. Van Den Berg, C. F. Elkjaer, C. J. Gommes, I. Chorkendorff, J. Sehested, P. E. De Jongh, K. P. De Jong and S. Helveg, J. Am. Chem. Soc., 2016, 138, 3433–3442 CrossRef CAS.
- W. A. Spieker and J. R. Regalbuto, Chem. Eng. Sci., 2001, 56, 3491–3504 CrossRef CAS.
- M. L. Carmen, F. Oropeza, J. Alvarez, M. Goldwasser, L. Francisco and M. P. Josefina, J. Mol. Catal. A: Chem., 2008, 281, 146–153 CrossRef.
- E. A. Kyriakidou, O. S. Alexeev, A. P. Wong, C. Papadimitriou, M. D. Amiridis and J. R. Regalbuto, J. Catal., 2016, 344, 749–756 CrossRef CAS.
- K. O'Connell and J. R. Regalbuto, Catal. Lett., 2015, 145, 777–783 CrossRef.
- M. Wang, D. J. Guo and H. L. Li, J. Solid State Chem., 2005, 178, 1996–2000 CrossRef CAS.
- X. Li, Z. Wang, Z. Zhang, G. Yang, M. Jin, Q. Chen and Y. Yin, Mater. Horiz., 2017, 4, 584–590 RSC.
- B. Jamwal, M. Kaur, H. Sharma, C. Khajuria, S. Paul and J. H. Clark, New J. Chem., 2019, 43, 4919–4928 RSC.
- M. S. Hegde, R. Caracciolo, K. S. Hatton and J. B. Wachtman, Appl. Surf. Sci., 1989, 37, 16–24 CrossRef CAS.
- P. Mondal, A. Sinha, N. Salam, A. S. Roy, N. R. Jana and S. M. Islam, RSC Adv., 2013, 3, 5615–5623 RSC.
- C. Gonzalez-Arellano, A. M. Balu, R. Luque and D. J. MacQuarrie, Green Chem., 2010, 12, 1995–2002 RSC.
- S. Natesakhawat, J. W. Lekse, J. P. Baltrus, P. R. Ohodnicki, B. H. Howard, X. Deng and C. Matranga, ACS Catal., 2012, 2, 1667–1676 CrossRef CAS.
- M. Liu, J. Liu, Z. Li and F. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 7052–7060 CrossRef CAS.
- S. Rana and S. Ram, Mater. Sci. Technol., 2005, 21, 243–249 CrossRef CAS.
- N. Mahata, A. F. Cunha, J. J. M. Órfão and J. L. Figueiredo, Appl. Catal., A, 2008, 351, 204–209 CrossRef CAS.
- Z. Yin, W. Zhou, Y. Gao, D. Ma, C. J. Kiely and X. Bao, Chem. – Eur. J., 2012, 18, 4887–4893 CrossRef CAS.
- K. Sawai, R. Tatumi, T. Nakahodo and H. Fujihara, Angew. Chem., Int. Ed., 2008, 47, 6917–6919 CrossRef CAS.
- H. Yang, X. Han, G. Li and Y. Wang, Green Chem., 2009, 11, 1184–1193 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S6 and Tables S1–S8. See DOI: 10.1039/d0nr04105j |
| ‡ Nazgol Norouzi and Mrinmoy K. Das contributed equally as the co-first authors of the paper. |
| This journal is © The Royal Society of Chemistry 2020 |