
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA site-specific self-assembled light-up rotor probe for selective recognition and stabilization of c-MYC G-quadruplex DNA†
Marco
Deiana
 a,
Karam
Chand
b,
Jan
Jamroskovic
a,
Rabindra Nath
Das
b,
Ikenna
Obi
a,
Erik
Chorell
*b and
Nasim
Sabouri
*a
a,
Karam
Chand
b,
Jan
Jamroskovic
a,
Rabindra Nath
Das
b,
Ikenna
Obi
a,
Erik
Chorell
*b and
Nasim
Sabouri
*a
aDepartment of Medical Biochemistry and Biophysics, Umeå University, 90187 Umeå, Sweden. E-mail: nasim.sabouri@umu.se
bDepartment of Chemistry, Umeå University, 90187 Umeå, Sweden. E-mail: erik.chorell@umu.se
First published on 3rd June 2020
Abstract
Direct and unambiguous evidence of the formation of G-quadruplexes (G4s) in human cells have shown their implication in several key biological events and has emphasized their role as important targets for small-molecule cancer therapeutics. Here, we report on the first example of a self-assembled molecular-rotor G4-binder able to discriminate between an extensive panel of G4 and non-G4 structures and to selectively light-up (up to 64-fold), bind (nanomolar range), and stabilize the c-MYC promoter G4 DNA. In particular, association with the c-MYC G4 triggers the disassembly of its supramolecular state (disaggregation-induced emission, DIE) and induces geometrical restrictions (motion-induced change in emission, MICE) leading to a significant enhancement of its emission yield. Moreover, this optical reporter is able to selectively stabilize the c-MYC G4 and inhibit DNA synthesis. Finally, by using confocal laser-scanning microscopy (CLSM) we show the ability of this compound to localize primarily in the subnuclear G4-rich compartments of cancer cells. This work provides a benchmark for the future design and development of a new generation of smart sequence-selective supramolecular G4-binders that combine outstanding sensing and stability properties, to be utilized in anti-cancer therapy.
Introduction
G-quadruplex nucleic acids (G4s) are non-canonical higher-order four-stranded guanine-rich sequences stabilized through Hoogsteen-type hydrogen bonding. G4s have received increasing attention as they have been implicated in biologically important roles such as oncogene transcriptional regulation, DNA replication, and telomere stability.1–3 The prevalence of putative G4 structures in promoters of cancer genes provides a firm basis for the concept of G4s being plausible therapeutic targets in oncology. One such example is the G4 structure in the proto-oncogene, c-MYC.4–6 The c-MYC protein is a transcription factor that regulates cellular proliferation, differentiation, and apoptosis.4–6 The expression of c-MYC is upregulated in 70% of different cancer types, and it is connected to ca. 100.000 deaths world-wide.7–10 The c-MYC protein has proven challenging to target with conventional drug design strategies and it has thus been considered undruggable.8 However, as about 90% of c-MYC expression is regulated by a G4 forming sequence found in the nuclease hypersensitive element III (1) region (NHEIII1) of the c-MYC gene,2,11 G4-binding small molecules able to inhibit c-MYC functions at the gene level rather than the protein level are an alternative anti-cancer therapeutic strategy.1,3,12 Although indisputable progresses have been made over the past years in the use of G4-binders as therapeutic agents, the design of dual-tasking compounds capable of both stabilizing and detecting G4 structures with high selectivity and sensitivity for certain sequences rather than a certain topology is still one of the major challenges in the field.1–3,12Supramolecular fluorescence sensors with distinguishable and controllable readout responses were recently designed as topology-specific G4-binders.13–16 Their G4-interactive binding model relies on the disassembly of the molecular aggregate (disaggregation-induced emission, DIE) in the presence of highly accessible π-surfaces such as those found in parallel G4 topologies. Parallel G4 structures are devoid of either adjacent lateral or diagonal loops, and can therefore provide better π-stacking platforms for the accommodation of the aromatic core of these ligands. Indeed, supramolecular sensors operating via DIE are successful in selectively detecting parallel G4 topologies, however neither of these G4-binders have been sequence/site-specific.13–16 This sensing mechanism strongly differs from the aggregation-caused quenching (ACQ) previously reported for G4- or duplex minor-groove binders based on cyanine dyes.17,18
In this study, we envisioned that a two-step switching mechanism based on (1) recognition-driven disassembly (i.e. DIE)19 of the hydrophobic fluorophore and (2) surrounding-induced motional restriction (motion-induced change in emission, MICE)20 in the monomeric ligand structure would give rise to a specific readout signal only when the optical reporter fits into the binding pocket of the c-MYC G4 DNA.
By synthesizing a self-assembled quinazoline-quinazolinone derivative, hereafter 4b, we were able to overcome this challenge. 4b featured outstanding binding affinity, sensitivity, selectivity, and stabilization for the c-MYC G4 DNA (Fig. 1). Moreover, the association with the c-MYC G4 simultaneously triggered the disassembly of its supramolecular state and induced geometrical restrictions leading to a significant increase in its fluorescence intensity and lifetime decay. The outstanding recognition process of 4b for c-MYC G4 via end-stacking provided a binding constant and a limit of detection in the nanomolar regime awarding it as one of the most sensitive sequence-selective G4-binders currently available.21–24 Moreover, DNA polymerase stop assay clearly demonstrated the ability of 4b to inhibit DNA synthesis of Taq-DNA polymerase by selectively stabilizing the c-MYC G4. Finally, confocal laser scanning microscopy (CLSM) images showed that 4b was primarily enriched in the G4-rich nucleolar regions of human cancer cells. 4b is, to the best of our knowledge, the first prototype of a supramolecular ligand with a drug-like chemical structure that is capable of site-specific detection and stabilization of a particular G4-sequence. The drug-like chemical structure of this compound, featured by its low-molecular weight and absence of charged moieties, along with its chemical versatility and scalability is rare in the G4 field. Thus, this opens up avenues for the development of a brand-new generation of site-specific G4-binders with desirable optical and biological properties.
 | ||
Fig. 1 Schematic illustration of the molecular recognition mechanism operated by 4b in the presence of G4 and non-G4 structures. (Middle) 4b self-assembles in water forming a non-emissive supramolecular aggregate. (Right) The high binding affinity for c-MYC Pu22 displaces 4b from its aggregated state (DIE) forming a rigid 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex (MICE) that induce up to 64-fold fluorescence enhancement. (Left) ssDNA, dsDNA and various G4s with different topologies (parallel, antiparallel or hybrid structures) can neither efficiently induce disassembly and/nor molecular restrictions on the 4b scaffold leading to poor affinities and negligible light-up responses. :1 complex (MICE) that induce up to 64-fold fluorescence enhancement. (Left) ssDNA, dsDNA and various G4s with different topologies (parallel, antiparallel or hybrid structures) can neither efficiently induce disassembly and/nor molecular restrictions on the 4b scaffold leading to poor affinities and negligible light-up responses. | ||
Results and discussion
Molecular design and self-assembly process
The synthesis of the desired quinazoline-quinazolinones 4a–d was achieved in three to four steps from commercially available anilines (1a–b) or 4-carboxybenzyl (Cbz) protected 1,4-benzenediamine (1c).25 The anilines were first converted to 1,2-dihydroquinoline derivative (2a–c) via a Skraup's synthesis and next reacted with 2-cynoguanidine in acidic conditions to give the N-(4-methyl-quinazolin-2-yl)-guanidine intermediates (3a–c). Condensation of 3a–c with isatoic anhydride under basic conditions gave the quinazoline-quinazolinones (4a–c) and 4d was obtained by subsequent hydrolysis of the Cbz group in 4c (Fig. 2). | ||
| Fig. 2 Synthetic route of quinazoline-quinazolinone (4a–d) hybrids. Reagents and conditions: (i)dry acetone, I2, t-butylcatechol, MgSO4, reflux, 24 h, (49–68%); (ii)2-cynogunidine, 2 M HCl, 100 °C, 0.5 h, (43–57%); (iii)DMF, isatoic anhydride, N,N-diisopropylethylamine, 100 °C, 12 h, (51–63%); (iv)HBr–acetic acid (3:7), reflux, 12 h, 66%. | ||
The quinazoline-quinazolinone 4b was the main focus of this investigation and fluorescence studies showed that 4b was sensitive to polarity changes and exhibited positive solvatochromism in organic solvents (Fig. S1 and S2†). Temperature-dependent UV/Vis studies of 4b in aqueous buffered solution at varying temperatures from 25 to 95 °C showed that the absorption maxima were hypsochromically shifted with a gain of vibronic fine structures and an overall narrowing of the full width at half-maximum (FWHM) of the high-energy transition band of ∼33% (Fig. S3 and S4†). The absorption behavior corresponded well with the strongly quenched blue-shifted emission band observed at 25 °C compared with that obtained at 95 °C (Fig. S4†). The above spectral features suggest the formation of higher-order aggregated structures of 4b.26 Concentration-dependent UV/Vis experiments provided the aggregation constant (Kagg) = 7.3 × 105 M−1 (Fig. S5†). The aggregated state of 4b was favored by increasing the ionic strength in the solution, which could be detected by the hypochromically reduced absorption maxima (ca. 60% reduction at potassium chloride concentrations above 5 mM) (Fig. S6†).15 On the contrary, DMSO is a highly disaggregating solvent and upon increasing the DMSO volume fraction χ(DMSO), 4b was present in the monomeric form (Fig. 3 and S7†).15 Finally, the monomer spectrum of 4b could also be observed in buffered water solution in the presence of sodium dodecyl sulfate (SDS) micelles (Fig. S8†).14,16
 | ||
| Fig. 3 UV/Vis absorption and emission (inset) spectra of 4b in water/DMSO solutions at increasing DMSO volume fractions, χ(DMSO) at 25 °C. In the upper-right panel, the change of 4b from non-fluorescent (100% water) to fluorescent (100% DMSO) is shown. (c4b = 15 and 2 μM for absorption and fluorescence studies, respectively). | ||
G4-interactive binding studies
The DMSO-disaggregation condition of 4b induced a colorimetric change of 4b and a high fluorescence response (Fig. 3). The observed optical changes in disaggregation-favouring solutions, prompted us to study the recognition process of 4b towards a large panel of biologically relevant synthetic and natural G4 structures with different topologies to examine their ability to disaggregate 4b (see Table S1 and Fig. S9† for their morphological characterization). 4b exhibited the highest fluorescence response (∼64-fold) in the presence of c-MYC Pu22 G4 DNA, a commonly used and well-characterized parallel G4 with exceptional thermal stability (Fig. 4).27–30 | ||
| Fig. 4 Fluorimetric binding isotherms of 4b in the presence of G4 and non-G4 structures (c4b = 2.5 μM, Tris buffer (10 mM), pH = 7.5, KCl (100 mM), T = 25 °C, λexc = 320 nm). The black dashed curves result from a global fitting by using either a 1:1 (4b-c-MYC Pu24T) or 2:1 (4b-c-MYC Pu22/Pu22 mut) binding model. | ||
The truncated c-MYC Pu22 sequence (c-MYC Pu22, 5′-TGAG3TG3TAG3TG3TTA-3′) form the major G4 conformation representing the wild-type G4 found in the NHEIII1 region of the c-MYC gene and provides a great molecular system for G4-interactive binding studies.31 To investigate if the emission enhancement of 4b in the presence of c-MYC Pu22 was specific to only this ligand, we examined the G4 light-up binding response of all the synthesized compounds (3a–4d) in the presence of c-MYC Pu22. As depicted in Fig. 5, the highest fluorescence response was detected for the 4b-c-MYC Pu22 system supporting its outstanding selectivity for this particular G4 template.
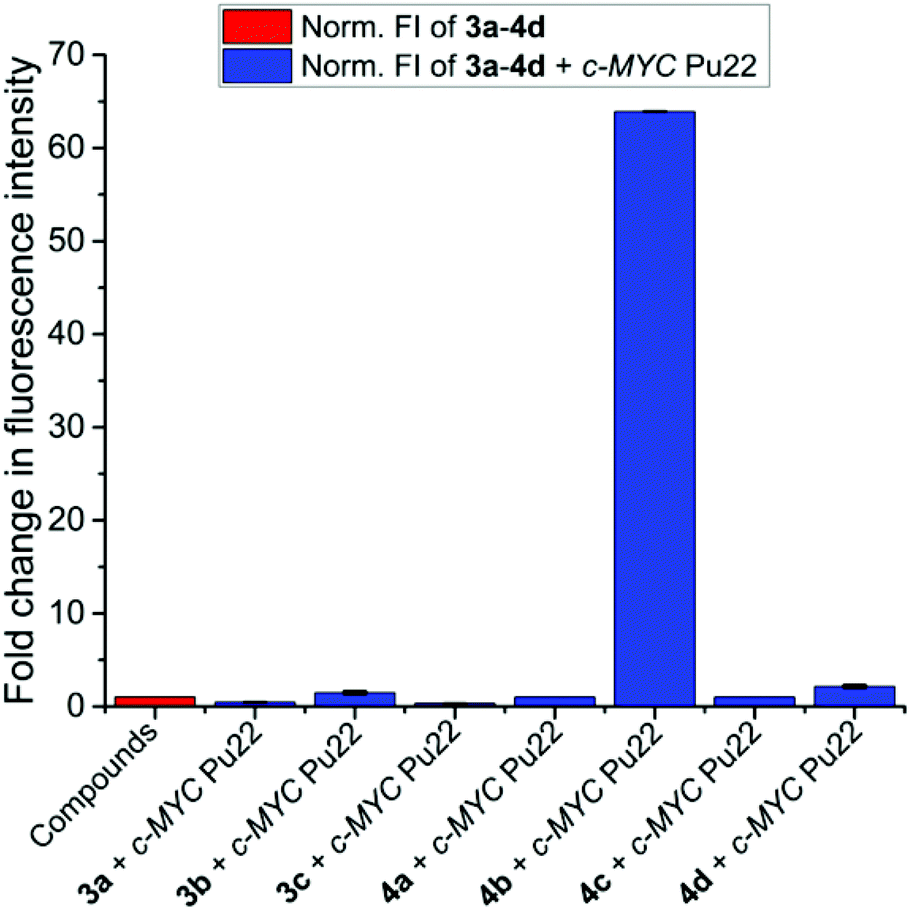 | ||
| Fig. 5 Fluorescence light-up response of all synthesized compounds (3a–4d) in the presence of c-MYC Pu22 (c3a−4d = 2.5 μM, cc-MYC Pu22 = 2.5 μM, Tris buffer (10 mM), pH = 7.5, KCl (100 mM), T = 25 °C, λexc = 320 nm). | ||
Importantly, the presence of long genomic dsDNA did not induce any significant light-up response from 4b even at an excess of 140 eq. However, addition of 0.5 eq. of c-MYC Pu22 to the same dsDNA-containing solution enhanced the fluorescence intensity from 4b with over 50 folds (Fig. S10†). This outstanding selectivity to recognize and light-up c-MYC Pu22 was further supported by 4b-stained non-denaturing polyacrylamide gels loaded with different G4 and non-G4 structures (Fig. 6).
 | ||
| Fig. 6 Non-denaturing PAGE images of G4 and non-G4 structures stained with 4b (left panel). The same gel was later co-stained with Thiazole Orange (TO), (right panel) to show the specificity of binding and to detect loading of DNA in all lanes. c4b = 5 μM, cTO = 5 μM and coligos = 10 μM, λexc = 457 nm. | ||
Next, we performed in-depth biophysical analysis on the interaction between 4b and c-MYC Pu22. The absorption spectrum of 4b in aqueous buffered solution in the absence of c-MYC Pu22 featured high and low energy transitions centered at 334 nm and 445 nm, respectively (Fig. 7). Upon addition of c-MYC Pu22, the absorption maxima of 4b showed a concomitant monotonic hypochromism and hyperchromism of the low and high energy transitions, respectively, along with a marked hypsochromic shift resulting in a well-defined isosbestic point centered at 355 nm. In line with the absorption changes, the steady-state emission of 4b is almost fully quenched in its unbound state (photoluminescence quantum yield (PLQY), ΦF(4b) = 0.14%) and light-up in the presence of c-MYC Pu22 (ΦF(4b-c-MYC Pu22) = 5.5%) inducing a clear colour change in the solution, and resulting in a large Stokes shift of 226 nm (i.e. ca. 12000 cm−1) indicative of excited-state charge transfer (Fig. 7).
 | ||
| Fig. 7 Spectrophotometric (solid lines) and fluorimetric (dashed lines) titration of 4b (c4b = 15 or 2.5 μM for absorption and emission, respectively, in 10 mM Tris buffer and 100 mM KCl at pH = 7.5, λexc = 320 nm) upon addition of c-MYC Pu22 at 25 °C (cc-MYC Pu22 = 0.0, 0.9, 1.8, 2.6, 3.5, 4.4, 5.3, 7.0, 8.8, 10.6, 13.2 and 17.6 μM and cc-MYC Pu22 = 0.0, 0.3, 0.6, 0.9, 1.2, 1.5, 2.1, 2.7, 3.3, 4.2, 5.1, 6.6, 8.1, 10.5, 12.9, 16.5 and 20.1 μM in the absorption and fluorescence titrations, respectively). The blue and red lines correspond to the spectra at 0.0 eq. and 1.17 eq. or 8.04 eq. in the absorption and emission spectra, respectively. Superimposed black dotted lines resulted from a global fitting with a 2:1 binding model. The inset shows the change in fluorescence derived by the addition of c-MYC Pu22 to 4b. | ||
These results suggest that the c-MYC Pu22 structure triggers disassembly of the aggregated 4b state into a stable c-MYC Pu22-4b complex, which results in a significant increase in its emission quantum yield, likely through geometrical (vibrational and rotational) and/or structural (molecular conformation) changes.
To examine this hypothesis, we compared 4b with its analogous, 4a and 4d, that lacked the diethyl-amino substituent in position C-6 (Fig. 2 and 5). The diethyl-amino substituent is prone to favour the formation of a twisted intramolecular charge transfer (TICT) process in the excited state. 4a had a hydrogen in the C-6 position and was therefore, expected to be non-emissive in solution, while 4d carried an electron-donating primary amine group in the same position, and was expected to be fluorescent but unable to form TICT processes. Indeed, 4a was neither fluorescent in the presence nor absence of c-MYC Pu22, highlighting the pivotal role played by the electron-donating C-6 substituent in conferring the emissive properties of the compound (Fig. 5 and S11†). On the other hand, 4d displayed an emission band with maximum at 548 nm. The addition of c-MYC Pu22 to 4d did not largely affect its light-up ability (∼2-fold) but blue-shifted its emission maximum resulting in a clear colour change of the solution (Fig. S12†). Together, these results confirmed the direct involvement of the diethyl-amino moiety into the complexation process with the c-MYC Pu22 structure and its role as an external flexible turn-on signal reporting motif.
Next, the association mechanism of the 4b-c-MYC Pu22 complex was quantitatively determined by performing the mole ratio method, Job's plot titration assay, and a global fitting procedure (Fig. 7 and S13–S15†). Binding data analysis indicated the formation of a 4b-c-MYC Pu22 complex with a 2:1 (guest/host) stoichiometry and nanomolar association constant (Table 1). A similar 2:1 ligand:c-MYC Pu22 complex was earlier reported for quindoline and indenoisoquinolines compounds.31,32
| G4s | K 11/M−1 | K 21/M−1 | ΔΔG/kj mol−1 | Φ F/% | τ/ns |
|---|---|---|---|---|---|
| Data fitting with 1:1 (4b-c-MYC Pu24T) or 2:1 (4b-c-MYC Pu22/Pu22 mut) binding models was performed with Bindfit by using multiple global fitting methods (Nelder-Mead method) and are provided as the average values of both fluorimetric and spectrophotometric titrations.33,34 The microscopic stepwise free energy changes are defined as: ΔΔG = ΔG21 − ΔG11 considering that in case of 2:1 binding ΔG11 = −RT ln(K11/2) and ΔG21 = −RT ln(K21 × 2) while, in case of 1:1 binding ΔΔG = −RT lnK11. R is the gas constant (8.314 J mol−1 K−1) and T is the temperature at 25 °C expressed in K (298 K). 5-(Dimethylamino)-1-naphthalenesulfonamide is used as reference standard (ΦF = 5%, in water). The average lifetime is derived by using a tri-exponential decay fitting. |
|||||
| c-MYC Pu22 mut | 1.1 × 102 | 4.3 × 109 | −46.8 | 5.9 | 4.9 |
| c-MYC Pu22 | 4.9 × 102 | 3.4 × 108 | −36.8 | 5.5 | 4.4 |
| c-MYC Pu24T | 3.4 × 106 | — | −37.3 | — | 3.7 |
The association constant calculated for 4b-c-MYC Pu22 complex is among the highest values reported in literature so far.21–24 Moreover, a limit of detection (LOD) of 49 nM was obtained which is 2-fold lower than that reported for N-methyl mesoporphyrin IX (NMM), a renowned selective parallel G4 binder, LOD = 104 nM (Fig. S16†).15 Kinetic studies on the mechanism of interaction between 4b and c-MYC Pu22 showed that the G4 structure actively binds 4b which catalyses the dissociation of the aggregated form (Fig. S17†). To further characterize and confirm 4b binding, we performed NMR titrations of the c-MYC Pu22 G4 structure with 0, 0.1, 0.2, 0.5, 1, and 2 equivalents of 4b (Fig. S18†). This showed a clear line-broadening of the signals in the imino-region of c-MYC Pu22 already at 0.1 equivalents of 4b. Increasing concentrations of 4b resulted in an almost complete disappearance of NMR signals from the imino-protons which confirm that 4b is strongly binding the c-MYC Pu22 G4 structure. To confirm that the observed line-broadening is an effect of exchange processes caused by binding rather than a compound-induced unfolding of the G4 structure, we next performed electronic circular dichroism (ECD) measurements (Fig. S19†). This showed that the native parallel morphology of c-MYC Pu22 was fully conserved upon addition of four equivalents of 4b, without any sign of induced CD (ICD) peaks.
The binding mode of 4b to the c-MYC Pu22 template was investigated by fluorescence displacement assay using the well-known G4 end-stackers, Phen-DC3 and Pyridostatin (PDS, Fig. 8 and S20†).35–37 As expected, both Phen-DC3 and PDS were able to fully displace 4b from the G-tetrad ends, highlighting the ability of 4b to target the G4 template via an end-stacking binding mode. We observed a slightly different colour change in cuvette III (compare cuvettes marked as I with III, Fig. 8) upon Phen-DC3-mediated 4b displacement from the G4 structure. This may be due to a small fraction of the unbound 4b molecules that remained free in solution without reforming molecular aggregates.
 | ||
| Fig. 8 (A) Schematic illustration of the fluorescence displacement assay performed by using the G4 end-stacker Phen-DC3. 4b is non-emissive in its free state (I). Complexation with c-MYC Pu22 induced a clear colour change of the solution (II). Replacement of 4b from the G4 template by Phen-DC3 caused the fluorescence quenching of 4b (III). (B and C) Fluorescence emission changes on the 4b-c-MYC Pu22 system upon increasing concentration of Phen-DC3. The dashed white line corresponds to the emission of 4b alone. The blue and red lines correspond to 4b-cMYC Pu22 and 4b-c-MYC Pu22-Phen-DC3 systems, respectively (4b = 2.5 μM, c-MYC Pu22 = 1.25 μM and Phen-DC3 ranged from 0 to 1.5 μM). | ||
In addition to the core G-quartets of the c-MYC Pu22, this oligonucleotide also includes 5′ TGA and 3′ TAA flanking residues (Table S1†). These flanking residues form together with the G-tetrad ends two suitable binding pockets for ligand accommodation through a sandwiched-type binding mode.31,32 The importance of these flanking sequences in the interactions with 4b is apparent based on the fluorimetric titration of 4b in the presence of c-MYC sG4, a mutated c-MYC Pu22 sequence (c-MYC sG4, 5′-G3TG3TAG3TG3-3′) that still features the high accessible parallel G-quartets, but lacks the flanking sequences (Fig. 4). These titrations showed that the fluorescence enhancement signal of 4b with c-MYC sG4 was dramatically reduced compared with the c-MYC Pu22 sequence (Fig. 4). Furthermore, the direct fluorescence titration of 4b to the c-MYC Pu22 sequence where the 3′ end is mutated to TGA instead of TAA (c-MYC Pu22 mut, 5′-TGAG3TG3TAG3TG3TGA-3′) showed an equally strong fluorescence enhancement and an increased binding constant compared to the unmutated c-MYC Pu22 sequence (Fig. 4 and Table 1), suggesting that the flanking sequences are important for disrupting the aggregated form of 4b to promote the formation of a stable and highly emissive 4b-c-MYC G4 complex. We also tested the G4-interactive binding properties of 4b with the c-MYC Pu24T (c-MYC Pu24T, 5′-TGAG3TG2TGAG3TG4AAG2-3′) sequence that features the same 5′-G-tetrad/flanking residue of c-MYC Pu22 but considerably different 3′-terminal G-tetrad end and flanking sequence. Association of 4b with c-MYC Pu24T showed a limited fluorescence light-up response (Fig. 4). These fluorimetric data could be well-fitted by using a 1:1 binding model indicating the 5′-end as the major binding site involved in the complexation process between 4b and the c-MYC Pu24T template. Importantly, the sequence specificity was strongly connected to the G4 structure, as the fluorimetric titrations of 4b to a single-stranded DNA sequence (TGA-ssDNA, 5′-G2ATGTGAGTGTGAGTGTGAG2-3′) that features three TGA motifs but cannot fold into a G4 structure, provided minimal fluorescence changes (Fig. 4). Next, we performed ligand-induced fluorescence quenching assays to probe the direct involvement of the terminal G-tetrads and flanking residues in the complexation mechanism.38 We either labeled the 5′ or 3′-end of the c-MYC Pu22 with the Texas Red fluorophore (5′-Txred c-MYC Pu22 or 3′-Txred c-MYC Pu22), and hypothesized that binding of 4b to either of the G-tetrads/flanking ends would mediate proximal ligand-binding quenching. Indeed, the addition of 4b to either the 5′-Txred c-MYC Pu22 or 3′-Txred c-MYC Pu22 induced a dose-dependent loss of fluorescence intensity (Fig. S21†). The 5′-TGA end gave the highest loss of fluorescence, suggesting that the 5′-end is the main site of 4b binding. Together, these data show that the synergetic interplay between the hydrophobic surface of the terminal G-tetrads with the presence of specific flanking sequences is the major driving force for the selective association of 4b toward the c-MYC Pu22 scaffold.
In order to investigate the photophysical properties of the excited-state, time-correlated single photon counting (TCSPC) measurements of 4b bound to different c-MYC Pu22 analogues were performed (Fig. 9 and Table S2†).
 | ||
| Fig. 9 Singlet excited state decay traces of 4b (λexc = 365 nm) in the absence and presence of different c-MYC G4 oligonucleotides. [DNA] = 20 μM and [4b] = 2 μM. | ||
In its unbound state, 4b was non-emissive and its time-dependent decay overlapped with the Instrumental Response Function (IRF) signal of our time-resolved setup (0.055 ns) showing the formation of radiationless pathways of the aggregated form. However, the lifetime of 4b dramatically increased in the presence of c-MYC Pu22. The average singlet state lifetime obtained by fitting the time-resolved fluorescence decay of the 4b:c-MYC Pu22 complex with a triexponential function was ∼4.4 ns (Table 1 and S2†).
G4-selective stabilization properties
To determine if 4b can stabilize G4 structures, we performed the Taq-DNA polymerase stop assay.39 Using this assay, we showed that 4b inhibited replication by the Taq-DNA polymerase by selectively stabilizing the c-MYC G4 just before the first G-tract in the DNA template (Fig. 10A). This process resulted in enhanced replication pausing and decreased amounts of full-length products (Fig. 10B). No enhanced pausing effects were observed in the presence of 4b when using the human telomeric G4 template or a non-G4 forming sequence, showing the selectivity of 4b for stabilizing the c-MYC G4 (Fig. 10). | ||
| Fig. 10 (A) Taq-DNA polymerase stop assay with 4b using non-G4 DNA, c-MYC Pu24T G4 DNA and Tel-22 G4 DNA as a template. The G4 sequences of the templates are shown on the side of the gels. The Gs forming the G4 structures are indicated in bold. Concentration of 4b is indicated above each line. Dark arrows represent start of the reaction – primer; grey arrows represent full-length products; asterisks represent pausing site. (B) Relative quantification of full-length products normalized to DMSO treated sample. | ||
Next, the ability of 4b to induce G4 stabilization was investigated by CD melting analysis (Fig. S22†). As shown in Table 2, 4b induced a high level of thermal stabilization (ΔTm) on the c-MYC Pu22 template that reached 10 °C at 5 equivalents. In contrast, under the same experimental conditions, 4b destabilized the Tel-22 G4 structure with 6 °C. Together these data show a selective G4 stabilization effect by 4b.
| System | T m (G4) | T m (4b-G4) | ΔTm |
|---|---|---|---|
| 4b-c-MYC Pu22 | 59.7 | 69.7 | +10.0 |
| 4b-Tel-22 | 37.9 | 31.8 | −6.1 |
Cellular imaging and associated G4-nucleolar binding
Confocal laser scanning microscopy (CLSM) was used to evaluate the cellular emission fingerprint of 4b (Fig. 11).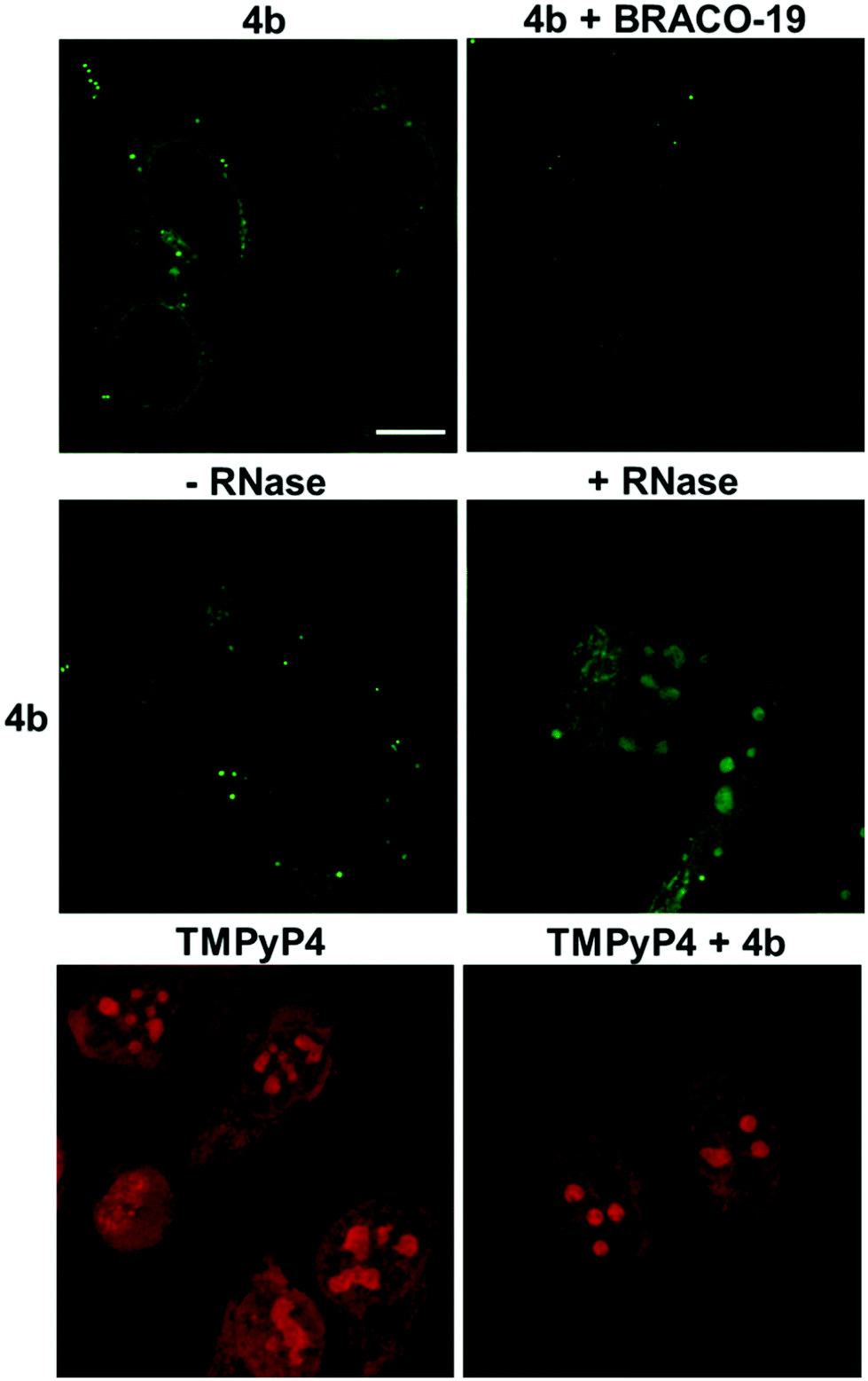 | ||
| Fig. 11 Confocal fluorescence images of fixed HeLa cells. Upper panel: HeLa cells stained with 4b (20 μM) in the absence or presence of BRACO-19 (20 μM). Middle panel: Fluorescence images of HeLa cells stained with 4b (20 μM) with or without RNase treatment. Lower panel: TMPyP4 (10 μM) in the absence and presence of 4b (60 μM). 4b alone does not emit upon TMPyP4 excitation. Scale bar = 10 μm. Diode 405 nm laser was used for 4b excitation λexc = 405 nm, λem 470–700 nm; Argon laser λexc = 514 nm, λem = 630–780 nm was used for TMPyP4. | ||
HeLa cells treated with 4b revealed an intense fluorescence signal in the cytoplasmic/mitochondrial, and nuclear networks, with clear peaks in the subnuclear compartments, whose appearance was compatible with that of G4-rich nucleolar regions (Fig. 11).16,40–44 To confirm the nature of the main binding target of 4b, we treated cells with RNase (Fig. 11). RNase-treatment did not greatly modify the staining pattern of the cells, supporting the ability of the compound to mainly target G4 DNA sites. To validate the G4-binding ability of 4b in the nucleolar compartments we carried out a competition assay by using the well-known G4-binder, BRACO-19.45 The 4b-associated emission in the nucleolar regions decreased significantly in the presence of BRACO-19 confirming the specific staining of 4b towards DNA G4s. Finally, TMPyP4 was used as a non-selective G4/duplex porphyrin-based binder displaying a red emission in the cytoplasmic, nuclear, and sub-nuclear regions (Fig. 11).41 The staining pattern of the nucleus by TMPyP4 was more intense and spread out compared to the staining by 4b, suggesting that TMPyP4 stain the nuclear DNA differently, perhaps due to its non-selective G4/duplex binding affinity. Incubation of TMPyP4 with 4b did not affect the fluorescence signal of TMPyP4, suggesting that 4b do not compete with the same DNA sites as TMPyP4.
Conclusions
In summary, we have reported on the first example of a supramolecular fluorescent rotor with outstanding binding strength, sensitivity and selectivity for the c-MYC G4 DNA. In particular, the compound displayed a two-step switching mechanism based on recognition-driven disassembly and surrounding induced motional restriction on the ligand's chromophoric structure combined with the ability to discriminate between an extensive panel of G4 and non-G4 structures and selectively light-up the c-MYC promoter G4 DNA. In-depth biophysical studies highlighted the role exerted by the terminal flanking sequences of the c-MYC Pu22 template in these effects and identified the localization of the ligand's binding site. Beside its enticing quadruplex interacting/optical properties, our fluorescent sensor showed an outstanding ability to selectively stabilize the c-MYC G4 structure. Finally, our probe preferentially targeted the nucleolar G4-rich compartments of cancer cells. This is the first prototype of a small molecule that address the quest toward sequence/site-specific G4-binders and open new directions for the development of self-assembled fluorescent molecular rotors for tracking and controlling non-canonical DNA structures in a cellular context.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Work in the Sabouri lab was supported by Knut and Alice Wallenberg Foundation (KAW2015-0189) and the Swedish Research Council (VR-MH 2018-02651). MD was supported by a fellowship from the MIMS Excellence by Choice Postdoctoral Programme. Work in the Chorell lab was supported by the Kempe foundations (SMK-1632) and the Swedish Research Council (VR-NT 2017-05235). We thank Dr. Irene Martinez Carrasco and the Biochemical Imaging Center at Umeå University and the National Microscopy Infrastructure (VR-RFI 2016-00968) for providing assistance in confocal laser scanning microscopy. We also thank Dr. Mattias Hedenström at the Knut and Alice Wallenberg foundation program, NMR for Life (http://www.nmrforlife.se), for NMR spectroscopy support.Notes and references
- S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261–275 CrossRef CAS PubMed.
- R. Hänsel-Hertsch, M. Di Antonio and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2017, 18, 279–284 CrossRef PubMed.
- S. Neidle, Nat. Rev. Chem., 2017, 1, 0041 CrossRef CAS.
- L. M. Boxer and C. V. Dang, Oncogene, 2001, 20, 5595–5610 CrossRef CAS PubMed.
- B. Amati, S. R. Frank, D. Donjerkovic and S. Taubert, Biochim. Biophys. Acta, Rev. Cancer, 2001, 1471, M135–M145 CrossRef CAS.
- E. B. Thompson, Annu. Rev. Physiol., 1998, 60, 575–600 CrossRef CAS PubMed.
- M. A. Gregory and S. R. Hann, Mol. Cell Biol., 2000, 20, 2423–2435 CrossRef CAS PubMed.
- J. R. Whitfield, M. E. Beaulieu and L. Soucek, Front. Cell Dev. Biol., 2017, 5, 10 Search PubMed.
- C. V. Dang, Cell, 2012, 149, 22–35 CrossRef CAS PubMed.
- L. Zhang, Y. Hou, H. Ashktorab, L. Gao, Y. Xu, K. Wu, J. Zhai and L. Zhang, Mol. Cell. Biochem., 2010, 344, 125–135 CrossRef CAS PubMed.
- A. Siddiqui-Jain, C. L. Grand, D. J. Bearss and L. H. Hurley, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11593–11598 CrossRef CAS PubMed.
- S. Neidle, J. Med. Chem., 2016, 59, 5987–6011 CrossRef CAS PubMed.
- V. Grande, F. Doria, M. Freccero and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 7520–7524 CrossRef CAS PubMed.
- V. Grande, C.-A. Shen, M. Deiana, M. Dudek, J. Olesiak-Banska, K. Matczyszyn and F. Würthner, Chem. Sci., 2018, 9, 8375–8381 RSC.
- M. Zuffo, A. Guédin, E.-D. Leriche, F. Doria, V. Pirota, V. Gabelica, J.-L. Mergny and M. Freccero, Nucleic Acids Res., 2018, 46, e115 CrossRef PubMed.
- M. Deiana, K. Chand, J. Jamroskovic, I. Obi, E. Chorell and N. Sabouri, Angew. Chem., Int. Ed., 2020, 59, 896–902 CrossRef CAS PubMed.
- B.-L. Wang and C. Jiang, Anal. Chem., 2019, 91, 1541–1547 CrossRef CAS PubMed.
- J. L. Seifert, R. E. Connor, S. A. Kushon, M. Wang and B. A. Armitage, J. Am. Chem. Soc., 1999, 121, 2987–2995 CrossRef CAS.
- D. Zhai, W. Xu, L. Zhang and Y.-T. Chang, Chem. Soc. Rev., 2014, 43, 2402–2411 RSC.
- D. Su, C. L. Teoh, L. Wang, X. Liu and Y.-T. Chang, Chem. Soc. Rev., 2017, 46, 4833–4844 RSC.
- D. R. Calabrese, X. Chen, E. C. Leon, S. M. Gaikwad, Z. Phyo, W. M. Hewitt, S. Alden, T. A. Hilimire, F. He, A. M. Michalowski, J. K. Simmons, L. B. Saunders, S. Zhang, D. Connors, K. J. Walters, A. Beverly, B. A. Mock and J. S. Schneekloth Jr., Nat. Commun., 2018, 9, 4429 CrossRef PubMed.
- K. M. Felsenstein, L. B. Saunders, J. K. Simmons, E. Leon, D. R. Calabrese, S. Zhang, A. Michalowski, P. Gareiss, B. A. Mock and J. S. Schneekloth Jr., ACS Chem. Biol., 2016, 11, 139–148 CrossRef CAS PubMed.
- Q. Zhai, C. Gao, J. Ding, Y. Zhang, B. Islam, W. Lan, H. Hou, H. Deng, J. Li, Z. Hu, H. I. Mohamed, S. Xu, C. Cao, S. M. Haider and D. Wei, Nucleic Acids Res., 2019, 47, 2190–2204 CrossRef CAS PubMed.
- M.-H. Hu, J. Zhou, W.-H. Luo, S.-B. Chen, Z.-S. Huang, R. Wu and J.-H. Tan, Anal. Chem., 2019, 91, 2480–2487 CrossRef CAS PubMed.
- O. I. Afanasyev, A. A. Tsygankov, D. L. Usanov and D. Chusov, Org. Lett., 2016, 18, 5968–5970 CrossRef CAS PubMed.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- R. D. Gray, J. O. Trent, S. Arumugam and J. B. Chaires, J. Phys. Chem. Lett., 2019, 10, 1146–1151 CrossRef CAS PubMed.
- A. T. Phan, Y. S. Modi and D. J. Patel, J. Am. Chem. Soc., 2004, 126, 8710–8716 CrossRef CAS PubMed.
- E. Hatzakis, K. Okamoto and D. Yang, Biochemistry, 2010, 49, 9152–9160 CrossRef CAS PubMed.
- D. Yang and L. H. Hurley, Nucleosides, Nucleotides Nucleic Acids, 2006, 25, 951–968 CrossRef CAS.
- K.-B. Wang, M. S. A. Elsayed, G. Wu, N. Deng, M. Cushman and D. Yang, J. Am. Chem. Soc., 2019, 141, 11059–11070 CrossRef CAS PubMed.
- J. Dai, M. Carver, L. H. Hurley and D. Yang, J. Am. Chem. Soc., 2011, 133, 17673–17680 CrossRef CAS PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- L. K. S. von Krbek, C. A. Schalley and P. Thordarson, Chem. Soc. Rev., 2017, 46, 2622–2637 RSC.
- W. J. Chung, B. Heddi, F. Hamon, M.-P. Teulade-Fichou and A. T. Phan, Angew. Chem., Int. Ed., 2014, 53, 999–1002 CrossRef CAS PubMed.
- A. De Cian, E. DeLemos, J.-L. Mergny, M.-P. Teulade-Fichou and D. Monchaud, J. Am. Chem. Soc., 2007, 129, 1856–1857 CrossRef CAS PubMed.
- M. I. Umar, D. Ji, C.-Y. Chan and C. K. Kwok, Molecules, 2019, 24, 2416 CrossRef CAS PubMed.
- D. D. Le, M. Di Antonio, L. K. M. Chan and S. Balasubramanian, Chem. Commun., 2015, 51, 8048–8050 RSC.
- J. Jamroskovic, M. Livendahl, J. Eriksson, E. Chorell and N. Sabouri, Chem. – Eur. J., 2016, 22, 18932–18943 CrossRef CAS.
- J. Li, X. Yin, B. Li, X. Li, Y. Pan, J. Li and Y. Guo, Anal. Chem., 2019, 91, 5354–5361 CrossRef CAS PubMed.
- O. Domarco, C. Kieler, C. Pirker, C. Dinhof, B. Englinger, J. M. Reisecker, G. Timelthaler, M. D. Garcia, C. Peinador, B. K. Keppler, W. Berger and A. Terenzi, Angew. Chem., Int. Ed., 2019, 58, 8007–8012 CrossRef CAS PubMed.
- F. Doria, M. Nadai, M. Zuffo, R. Perrone, M. Freccero and S. N. Richter, Chem. Commun., 2017, 53, 2268–2271 RSC.
- J. Lefebvre, C. Guetta, F. Poyer, F. Mahuteau-Betzer and M.-P. Teulade-Fichou, Angew. Chem., Int. Ed., 2017, 56, 11365–11369 CrossRef CAS PubMed.
- J. Jamroskovic, M. Doimo, K. Chand, I. Obi, R. Kumar, K. Brännström, M. Hedenström, R. N. Das, A. Akhunzianov, M. Deiana, K. Kasho, S. S. Sato, P. L. Pourbozorgi, J. E. Mason, P. Medini, D. öhlund, S. Wanrooij, E. Chorell and N. Sabouri, J. Am. Chem. Soc., 2020, 142, 2876–2888 CrossRef CAS PubMed.
- E. Ruggiero and S. N. Richter, Nucleic Acids Res., 2018, 46, 3270–3283 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, optical studies, G4 characterization, G4-binding studies and chemical synthesis of the compounds. See DOI: 10.1039/d0nr03404e |
| This journal is © The Royal Society of Chemistry 2020 |