
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Silicon oxycarbide-antimony nanocomposites for high-performance Li-ion battery anodes†
Romain J.-C.
Dubey
 ab,
Pradeep Vallachira Warriam
Sasikumar
c,
Noemi
Cerboni
ab,
Marcel
Aebli
ab,
Frank
Krumeich
a,
Gurdial
Blugan
c,
Kostiantyn V.
Kravchyk
ab,
Thomas
Graule
*c and
Maksym V.
Kovalenko
*ab
ab,
Pradeep Vallachira Warriam
Sasikumar
c,
Noemi
Cerboni
ab,
Marcel
Aebli
ab,
Frank
Krumeich
a,
Gurdial
Blugan
c,
Kostiantyn V.
Kravchyk
ab,
Thomas
Graule
*c and
Maksym V.
Kovalenko
*ab
aLaboratory of Inorganic Chemistry, Department of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 1, CH-8093 Zürich, Switzerland. E-mail: mvkovalenko@ethz.ch
bLaboratory for Thin Films and Photovoltaics, Empa – Swiss Federal Laboratories for Materials Science and Technology, Überlandstrasse 129, CH-8600 Dübendorf, Switzerland
cLaboratory for High-Performance Ceramics, Empa – Swiss Federal Laboratories for Materials Science and Technology, Überlandstrasse 129, CH-8600 Dübendorf, Switzerland. E-mail: Thomas.Graule@empa.ch
First published on 10th June 2020
Abstract
Silicon oxycarbide (SiOC) has recently regained attention in the field of Li-ion batteries, owing to its effectiveness as a host matrix for nanoscale anode materials alloying with Li. The SiOC matrix, itself providing a high Li-ion storage capacity of 600 mA h g−1, assists in buffering volumetric changes upon lithiation and largely suppresses the formation of an unstable solid-electrolyte interface. Herein, we present the synthesis of homogeneously embedded Sb nanoparticles in a SiOC matrix with the size of 5–40 nm via the pyrolysis of a preceramic polymer. The latter is obtained through the Pt-catalyzed gelation reaction of Sb 2-ethylhexanoate and a poly(methylhydrosiloxane)/divinylbenzene mixture. The complete miscibility of these precursors was achieved by the functionalization of poly(methylhydrosiloxane) with apolar divinyl benzene side-chains. We show that anodes composed of SiOC/Sb exhibit a high rate capability, delivering charge storage capacity in the range of 703–549 mA h g−1 at a current density of 74.4–2232 mA g−1. The impact of Sb on the Si–O–C bonding and on free carbon content of SiOC matrix, along with its concomitant influence on Li-ion storage capacity of SiOC was assessed by Raman and 29Si and 7Li solid-state NMR spectroscopies.
Introduction
Harnessing the volume changes associated with the lithiation and delithiation of alloying-type anodes such as Si, Sn and Sb, is of paramount importance in Li-ion battery research.1–4 Owing to recent endeavors, cycling stability of these anodes was substantially advanced.5–9 The detrimental implications of uncontrolled volume changes are mainly of two kinds: active-material particle pulverization and the formation of cracks in the rigid solid-electrolyte interface (SEI).10,11 In recent years, significant progress was accomplished with respect to the mitigation of particle pulverization issues through the synthesis of anode materials in the form of nanoparticles (NPs), nanorods, etc.12–16 To address the problem of the SEI cracking around alloying-type NPs, the utilization of a suitable embedding matrix was determined to be the most effective strategy.10 The matrix should spatially disentangle the alloying reaction and the contact of the NPs with the liquid electrolyte, while efficiently buffering their volume changes upon lithiation. The design of a suitable matrix is highly challenging since it must meet stringent requirements in terms of mechanical stability, Li-ion and electrical conductivity, volumetric, and gravimetric charge storage capacity. The majority of the proposed syntheses of a suitable matrix meet these requirements; however, at the cost of utmost pricey and lengthy synthesis procedures.11,17Silicon oxycarbide (SiOC) is a highly appealing candidate matrix material for stabilization of alloying-type inclusions.18 SiOC is an amorphous ceramic material with a complex tetrahedrally bonded network comprising Si, O, and C, and commonly containing also free carbon (Cfree) nanodomains.19,20 This material is attractive for its ability to serve as an active matrix owing to its highly reversible intrinsic Li-ion storage properties (∼600 mA h g−1)21 originating mainly from the quality of its Cfree domains.22,23 The level of disorder in the Cfree phase is decisive for high Li-ion storage capacity and kinetics.24–26 Furthermore, SiOC is a low-cost material and it undergoes low volumetric expansion upon lithiation (7%).21 Some recent examples of the effectiveness of SiOC matrix for improving the cycling stability of alloying-type anodes include the stabilization of Sn NPs18,27,28 and diverse kinds of Si NPs.29–31
In this work, we were motivated to test SiOC as a matrix for the impregnation of alloying-type Sb nanoinclusions. We note that the synthesis of SiOC/Sb was already demonstrated by Lee et al.32 that involves mixing of silicone oil and Sb acetate (Sb(Ac)3) at 400 °C, followed by pyrolysis at 900 °C. However, due to incompatible polarities of the silicone oil with Sb(Ac)3 resulting in the inhomogeneous mixing within the preceramic polymer before pyrolysis, the Sb NPs had large size variations in the range of 10–100 nm and non-uniform distribution within the SiOC matrix. Herein, we attain a favorable SiOC/Sb architecture by altering the polarity of the side-chains of the polysiloxane, thereby ensuring its intimate blending with the antimony 2-ethylhexanoate (Sb(Oct)3) precursor. The electrochemical characterization of SiOC/Sb revealed that the synthesized active material delivers a high capacity of 703 mA h g−1 at a current density of 74.4 mA g−1. Notably, 78% of this capacity was retained at a high current density of 2232 mA g−1. The mechanism of Li+ ion insertion into SiOC/Sb was assessed using ex situ Raman and 29Si and 7Li solid-state NMR spectroscopies.
Results and discussion
Synthesis and characterization of SiOC/Sb nanocomposites
SiOC/Sb nanocomposite was obtained in three steps that include the functionalization of a polymethylhydrosiloxane (PMHS) polymer with apolar divinyl benzene (DVB) side-chains, its subsequent gelation with Sb(Oct)3 precursor yielding a preceramic SiOC/Sb polymer (pre-SiOC/Sb) and then pyrolysis of pre-SiOC/Sb at 1000 °C (Fig. 1). In short, the polymer with apolar side-chains was synthesized through a hydrosililation reaction between PMHS and 200 wt% of DVB. As follows from the Fourier-transform infrared spectroscopy (FTIR) measurements of pre-SiOC/Sb, the absence of the Si–H absorption band at 2100 cm−1 demonstrates the completeness of the hydrosililation reaction of PMHS with DVB (Fig. 2a). This step was followed by the gelation of the polymer precursor with 40 wt% Sb(Oct)3. Sb(Oct)3 was synthesized in a high yield using an anion-exchange reaction between Sb(Ac)3 and 2-ethylhexanoic acid in xylene at elevated temperatures. The Sb(Oct)3 precursor was chosen because of its complete miscibility with the polysiloxane precursor and the absence of a reaction with the side-chains of polysiloxane. The pyrolysis of pre-SiOC/Sb yielded a black powder (SiOC/Sb). Pure SiOC was synthesized according to our earlier report.18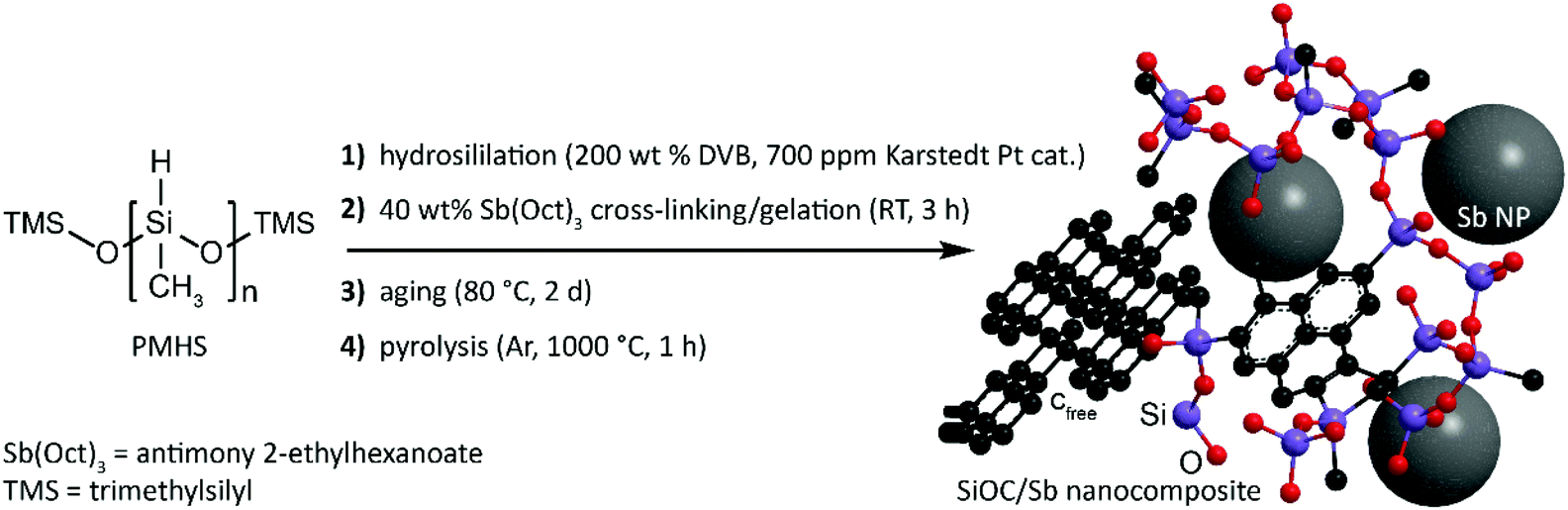 | ||
| Fig. 1 Schematics of the synthetic protocol of SiOC/Sb nanocomposite. | ||
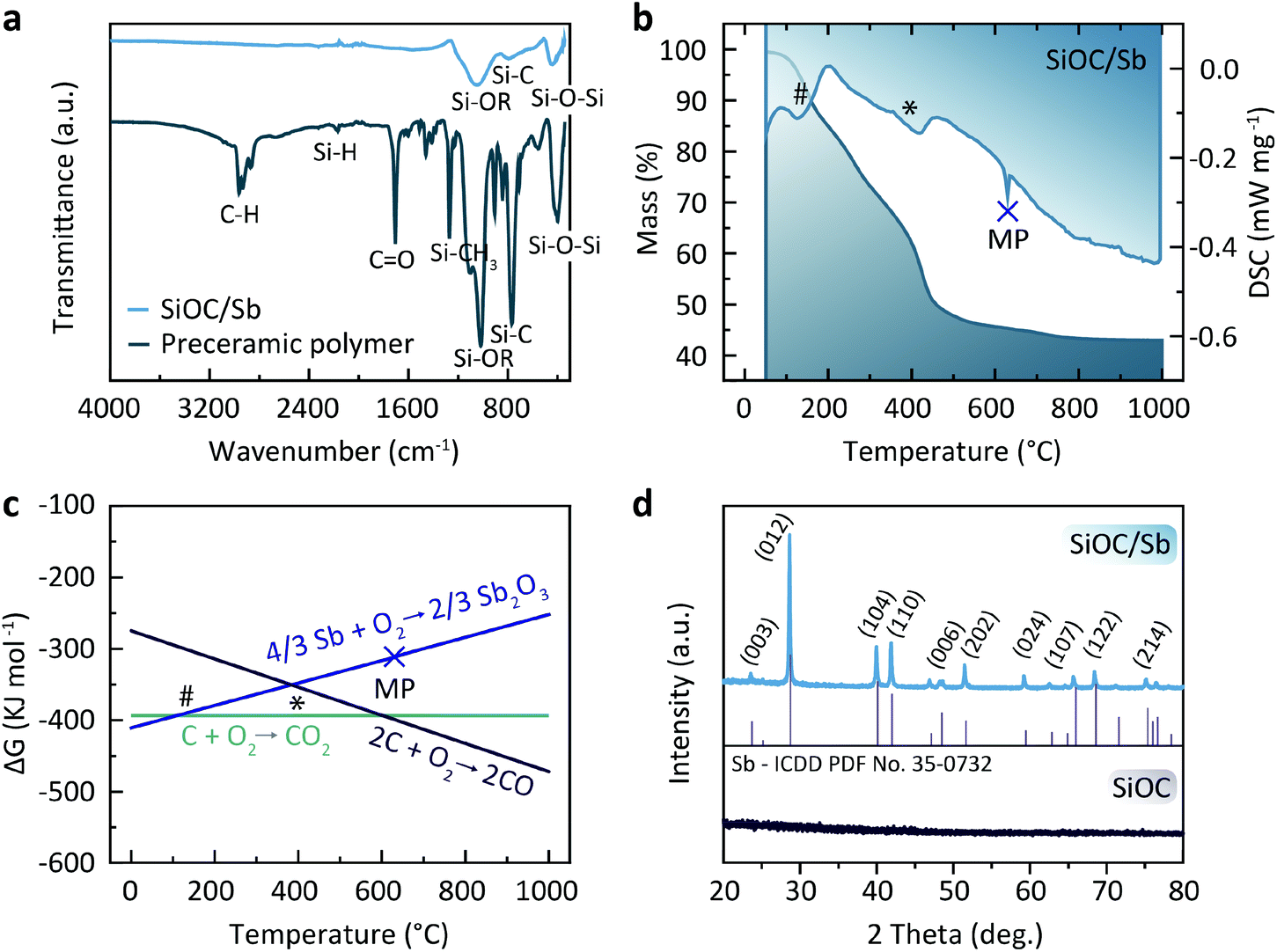 | ||
| Fig. 2 (a) FTIR absorption spectra of pre-SiOC/Sb and SiOC/Sb. (b) TGA/DSC curves of pre-SiOC/Sb. (c) Ellingham diagram for Sb. (d) PXRD patterns of SiOC and SiOC/Sb. | ||
To assess the mechanism of the pre-SiOC/Sb pyrolysis, a thermogravimetric analysis (TGA), coupled with differential scanning calorimetry (DSC) was performed. As follows from Fig. 2b, TGA curve of pre-SiOC/Sb remains relatively stable up to ca. 100 °C. However, at higher temperatures, it drops slowly with the concomitant appearance of a DSC peak at 130 °C, which is attributed to the carbothermal reduction of Sb. We note that these results are in line with the carbothermal reduction temperature of Sb evidenced from Ellingham diagram (Fig. 2c). Upon further heat-treatment, we assume that the Sb reduction proceeds with the concomitant loss of non-crosslinked DVB (b.p. 195 °C) at 200 °C and accelerates around 400 °C. The reduced Sb particles then melt at 630.6 °C, which is evidenced by the appearance of a sharp DSC peak. In the temperature range of 500–1000 °C, the conversion of the pre-SiOC polymer to SiOC ceramics take place with concomitant losses of organics and other volatile moieties. Interestingly, the cooling rate after pyrolysis played a decisive role in the formation of Sb NPs. For instance, the cooling rate of 5 °C min−1 yielded clusters of aggregated Sb NPs, not homogeneously distributed within the SiOC matrix (Fig. 3a). On the contrary, a uniform dispersion of Sb NPs was observed at a cooling rate of 1 °C min−1. Representative high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) and transmission electron microscopy (TEM) images of the SiOC/Sb obtained at optimized cooling rate are shown in Fig. 3b, c, d, e and f respectively. The size of Sb NPs was in the order of 5–40 nm, while the vast majority was ca. 15 nm.
 | ||
| Fig. 3 HAADF STEM images of a SiOC/Sb obtained using a cooling rate of (a) 5 °C min−1 and (b–d) 1 °C min−1, respectively. EDXS analysis of SiOC/Sb indicates that the bright spots on HAADF-STEM images represent Sb NPs (Fig. S1 and S2†) (e and f) TEM images of SiOC/Sb. | ||
Powder X-ray diffraction (PXRD) patterns of SiOC/Sb after pyrolysis confirmed the formation of a rhombohedral Sb structure (a = b = 0.4307 nm, c = 1.1273 nm, space group R![[3 with combining overline]](https://www.rsc.org/images/entities/char_0033_0305.gif) m, ICDD PDF no. 35-0732). As follows from the elemental analysis, the content of Sb within the SiOC matrix was ca. 19.00 wt%. The full compositions of SiOC/Sb and pure SiOC, along with the weight ratios between SiOC and Cfree, are shown in Table 1. Notably, the Cfree content in SiOC/Sb was lower than in SiOC, although the exact same amount of DVB and PMHS was used for hydrosililation reaction in both samples. The difference can be explained by carbothermal reduction that consumes part of the carbon in the SiOC matrix. The impact of the carbothermal reduction was more pronounced with higher amount of Sb(Oct)3 (50 wt%), seen as further decrease of Cfree content down to 18.61 wt% (Table S1†). Notably, at 50 wt% of Sb(Oct)3, a much broader Sb particle size distribution (Fig. S3†), resulting in poor electrochemical performance of SiOC/Sb (Fig. S4†), was observed. The disorder of the Cfree phase in SiOC/Sb and SiOC was estimated by Raman spectroscopy measurements (Fig. 4a), showing D and G carbon bands at ≈1350 and 1600 cm−1. The intensity of the D band for SiOC/Sb is higher than for SiOC. Additionally, the second-order region of the Raman spectrum for SiOC/Sb shows a high intensity of D + G combination mode at 2940 cm−1. Such features can be attributed to a high level of carbon disorder in the SiOC matrix within SiOC/Sb, which might favorably enhance its charge storage capacity.
m, ICDD PDF no. 35-0732). As follows from the elemental analysis, the content of Sb within the SiOC matrix was ca. 19.00 wt%. The full compositions of SiOC/Sb and pure SiOC, along with the weight ratios between SiOC and Cfree, are shown in Table 1. Notably, the Cfree content in SiOC/Sb was lower than in SiOC, although the exact same amount of DVB and PMHS was used for hydrosililation reaction in both samples. The difference can be explained by carbothermal reduction that consumes part of the carbon in the SiOC matrix. The impact of the carbothermal reduction was more pronounced with higher amount of Sb(Oct)3 (50 wt%), seen as further decrease of Cfree content down to 18.61 wt% (Table S1†). Notably, at 50 wt% of Sb(Oct)3, a much broader Sb particle size distribution (Fig. S3†), resulting in poor electrochemical performance of SiOC/Sb (Fig. S4†), was observed. The disorder of the Cfree phase in SiOC/Sb and SiOC was estimated by Raman spectroscopy measurements (Fig. 4a), showing D and G carbon bands at ≈1350 and 1600 cm−1. The intensity of the D band for SiOC/Sb is higher than for SiOC. Additionally, the second-order region of the Raman spectrum for SiOC/Sb shows a high intensity of D + G combination mode at 2940 cm−1. Such features can be attributed to a high level of carbon disorder in the SiOC matrix within SiOC/Sb, which might favorably enhance its charge storage capacity.
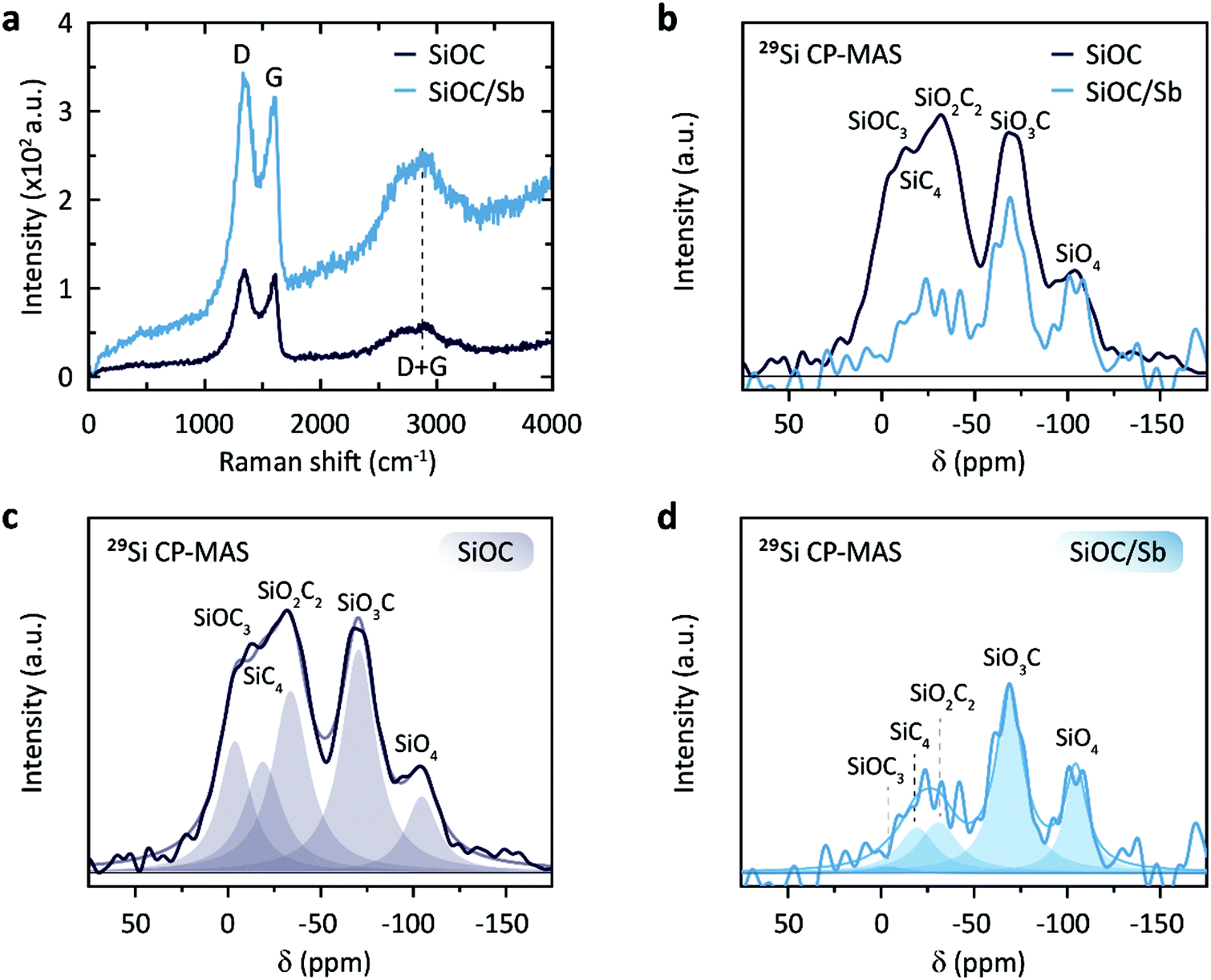 | ||
| Fig. 4 (a) Raman spectra of SiOC and SiOC/Sb. (b) 29Si CP-MAS NMR spectra of SiOC and SiOC/Sb. The intensities are scaled to the SiO4 peak intensity. (c) Fitted 29Si CP-MAS NMR spectrum of SiOC. (d) Fitted 29Si CP-MAS NMR spectrum of SiOC/Sb. | ||
| Sample | Elemental content [wt%] | Formula (normalized) | SiCxO2(1−x) | Cfree | Cfree | SiOC | |||
|---|---|---|---|---|---|---|---|---|---|
| Si | C | O | Sb | [wt%] | |||||
| SiOC | 32.40 | 49.42 | 17.80 | — | SiC3.56O0.96 | SiC0.51O0.96 | 3.05 | 42.37 | 57.63 |
| SiOC/Sb | 27.02 | 29.85 | 23.80 | 19.00 | SiC2.58O1.54Sb0.16 | SiC0.11O1.54 | 2.47 | 27.29 | 53.71 |
Solid-state NMR spectroscopy with magic angle spinning (MAS) is a powerful tool to compare qualitatively the nature of the bonds surrounding the excited nucleus in a solid. To probe the nature of the local bonding in the Si–O–C network, we performed a 29Si solid-state NMR analysis of the synthesized samples (Fig. 4b–d). Due to the low sensitivity of the 29Si nuclei, a transfer of magnetization, i.e. cross polarization (CP) from the proton traces to the 29Si nuclei was effective for achieving suitable signal-to-noise ratios. The obtained chemical shifts are in good agreement with the previously reported values for SiO4, SiO3C, SiO2C2, SiOC3, and SiC4 (Table S2†).33,34 A direct comparison of the spectra of SiOC and SiOC/Sb scaled to the intensity of the SiO4 peak reveals that the carbon content drastically decreases in the SiOC/Sb sample (Fig. 4c and d). Interestingly, the SiOC3 feature in SiOC/Sb is almost absent, while the SiO3C character remained almost unchanged. This observation is in line with the loss of carbon during the carbothermal reduction and the increased oxygen level from the carboxylic acid groups. Furthermore, complementary 13C MAS NMR measurements of SiOC and SiOC/Sb revealed that the carbothermal reduction also decreases the amount of Cfree in SiOC/Sb in comparison with pure SiOC (Fig. S5†).
Electrochemical performance
The electrochemical performance of the SiOC/Sb nanocomposite is summarized in Fig. 5. For electrochemical testing, the SiOC and SiOC/Sb electrodes were prepared by mixing finely ground powders of SiOC and SiOC/Sb with carbon black, carboxymethyl cellulose and water. The obtained slurries were then cast onto a copper foil current collector. Resulting active material loadings were in the range of 2.3–3.1 mg cm−2. All electrochemical measurements presented herein were conducted as half-cells vs. Li+/Li using 1 M LiPF6 in ethylene carbonate/dimethyl carbonate (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by weight) + 3% fluoroethylene carbonate as an electrolyte.
:1 by weight) + 3% fluoroethylene carbonate as an electrolyte.
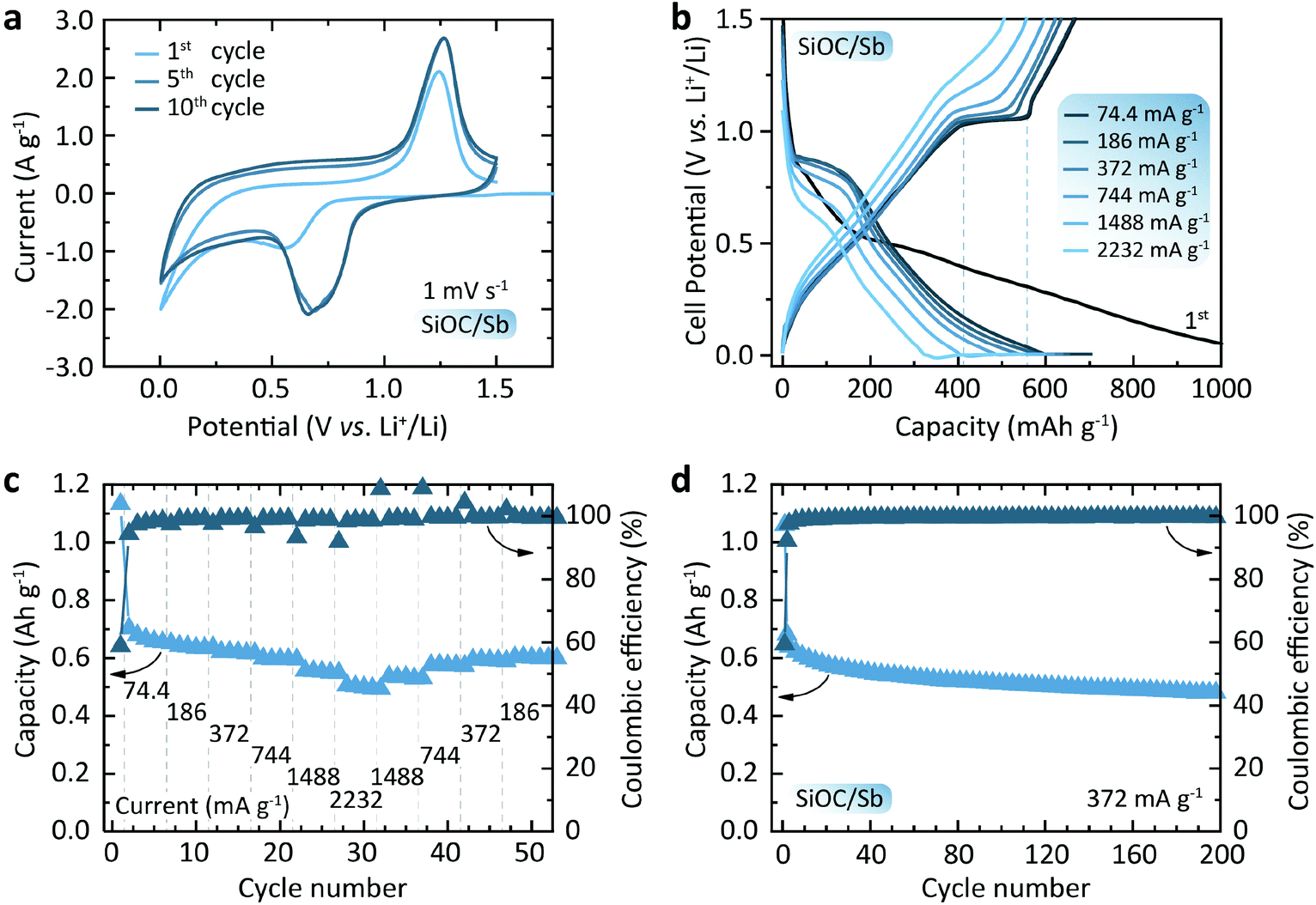 | ||
| Fig. 5 (a) CV curves of SiOC/Sb measured at a scan rate of 1 mV s−1. (b) Galvanostatic charge–discharge curves of SiOC/Sb. The first cycle is measured at a current density of 18.6 mA g−1 and the plotted curves correspond to the 1st, 2nd, 7th, 17th, 22nd, 27th cycles. (c) Discharge capacities measured with a constant voltage step contribution at different current densities ranging from 18.6 to 2232 mA g−1. (d) Cyclic performance of SiOC/Sb tested at a current density of 372 mA g−1. | ||
Fig. 5a shows the cyclic voltametry (CV) curves of the SiOC/Sb electrodes at a scan rate of 1 mV s−1. Upon lithiation and delithiation (cathodic and anodic cycles) of SiOC/Sb, the broad, yet distinct peaks at ca. 0.70 and 1.25 V were attributed to the alloying and dealloying of Sb NP with Li. An additional smooth increase of current in the voltage range of 0.01–1 V vs. Li+/Li can be ascribed to the gradual lithiation/delithiation of the amorphous SiOC matrix. Fig. 5b shows the discharge and charge voltage profiles of the SiOC/Sb measured at different currents of 74.4–2232 mA g−1 and yielding capacities ranging from 703 to 549 mA h g−1 for SiOC/Sb (Fig. 5c). These measurements were performed using a constant current–constant voltage protocol with a constant voltage step at 5 mV (see the Experimental section for details). The shape of the voltage profiles in Fig. 5b has two distinct features. A smooth part can be attributed to the lithiation of the interstitial spaces and edges of graphene within Cfree in SiOC.35 The plateaus at 0.9 and 1.1 V, eventually correspond to the lithiation and delithiation of Sb, respectively. The comparison of discharge and charge capacities of 1133 mA h g−1 and 667 mA h g−1 for the first cycle point to the high irreversible capacity loss of 41%, which can be attributed the formation of the SEI layer as well as irreversible bonding of Li ions to oxygen sites in SiOC mixed bonds.
We note that the overall reversible capacity of SiOC/Sb was higher than for pure SiOC (621 mA h g−1 initially and ca. 595 mA h g−1 at 372 mA g−1 for the subsequent cycles, see Fig. S6†). Additionally, SiOC/Sb was characterized by low polarization upon charge at high currents, resulting in only a slight increase of an average charge voltage of 0.82 and 0.87 V at C/20 and 6C-rates, respectively. On the contrary, a higher increase of the average delithiation voltage was observed for SiOC at higher currents (from 0.71 V and 0.83 V at C/20 and 6C-rate). The cyclic stability tests (Fig. 5d) of half-cells employing SiOC/Sb at a current density of 0.372 mA g−1 (1C for graphite) showed a high delithiation capacity retention of 76% after 200 cycles. Notably, as observed by HAADF-STEM, no evidence of significant Sb NP pulverization or aggregation was found upon lithiation and delithiation of SiOC/Sb (see Fig. S7 and S8†).
Li-ion storage mechanism and particle stability
Following the electrochemical measurements, the detailed lithiation mechanism of SiOC/Sb was assessed by 7Li solid-state NMR spectroscopy. A series of samples were recovered after electrochemical lithiation and delithiation (either fully lithiated or delithiated after a single discharge or charge). To ensure an optimal differentiation between the Li inside the SiOC composites and traces of electrolyte, the recovered materials were washed several times with EC/DMC (1:1 by weight) and then thoroughly dried under inert conditions (see ESI† for details). 7Li MAS NMR experiments showed common Li signals in all spectra between 0 and 20 ppm (Table 2, Fig. 6). In lithiated SiOC, a broad signal at 9.6 ppm is observed (Fig. 6a), corresponding to the reversibly inserted Li in the SiOC matrix. This signal has a full width at half maximum (FWHM) of 1.9 kHz and can be attributed to the lithiation of the graphene layer edges, which are the main sites of Li-ion storage in SiOC ceramics.36 The signal line displays a slight asymmetry, indicating the presence of another species at 5.10 ppm. These species were ascribed to irreversibly inserted Li, visible for delithiated SiOC at 4.40 ppm (Fig. 6b). Upon lithiation of SiOC/Sb (Fig. 6c), five species were identified. The irreversibly inserted Li species within the SiOC matrix are present at 5.44 ppm. A narrow peak at 5.37 ppm with a FWHM of 220 Hz is assigned to the Li3Sb species. A small shoulder at −4.20 ppm may be ascribed to SEI formed around the naked Sb NPs, as it remains after delithiation (Fig. 6d). Two resolvable, broad Li species at 6.82 and 8.89 ppm can be ascribed to the reversibly inserted Li within the SiOC matrix. This is not surprising, as several Li-ion species have already been observed in SiOC with different precursors.36 However, it confirms the different nature of the Cfree and mixed Si–O–C bond character in SiOC in the presence of Sb as already observed from Raman and 29Si CP-MAS NMR spectroscopy measurements. Consequently, the NMR results show that the primary source of charge storage contribution within the SiOC/Sb nanocomposite comes from SiOC/Cfree active sites. In lithiated SiOC/Sb, the matrix accounts for ca. 78.7% of the reversible Li sites, while the Sb contribution is ca. 21.3%.
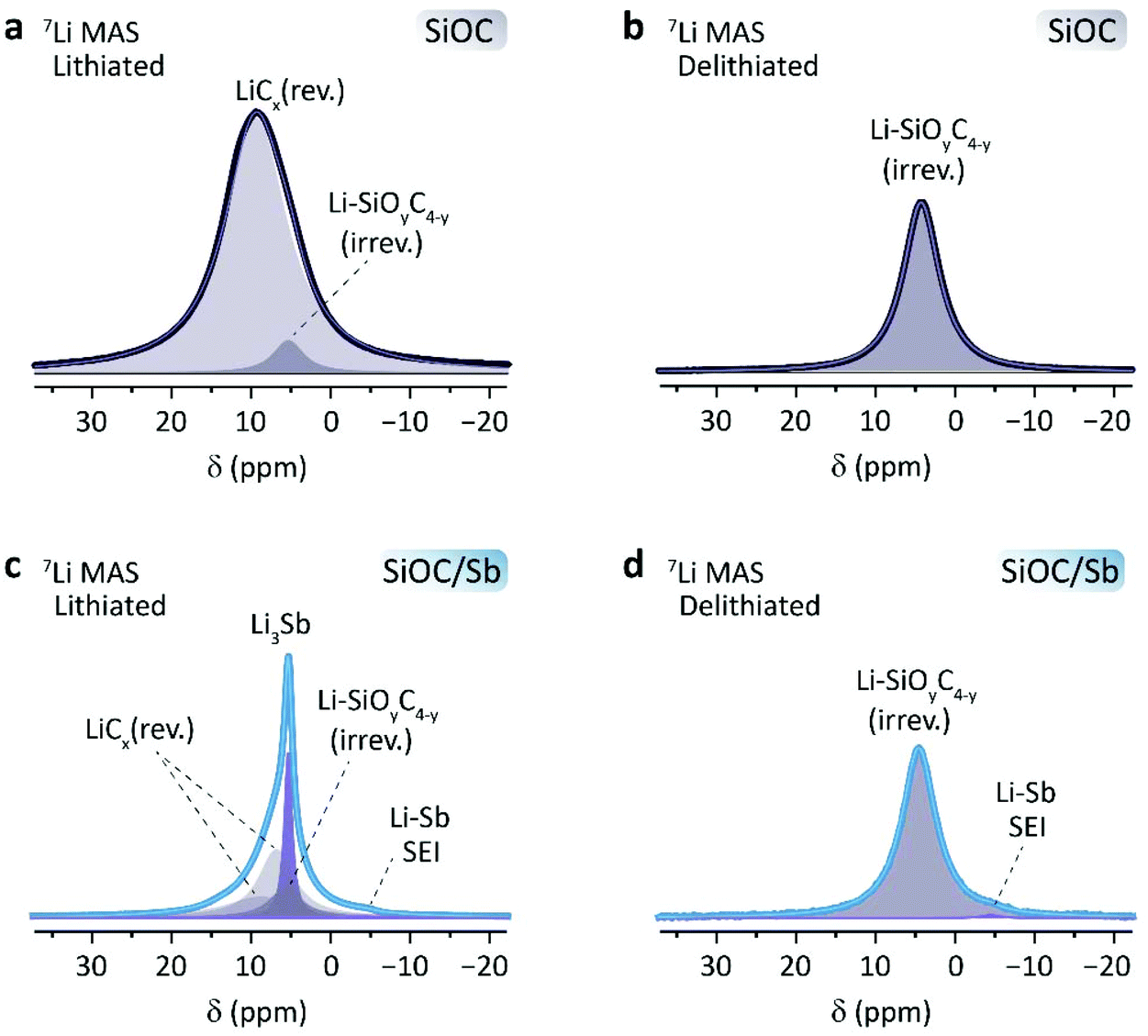 | ||
| Fig. 6 7Li MAS NMR (12 kHz) spectra of (a and c) lithiated and (b and d) delithiated SiOC and SiOC/Sb, respectively. | ||
| Sample | δ (ppm) | Species | Int. (%) | FWHM (Hz) |
|---|---|---|---|---|
| SiOC (lith.) | 9.62 | LiCx/rev. | 96.2 | 1903 |
| 5.1 | SiOC/irrev. | 3.8 | 975 | |
| SiOC (delith.) | 4.4 | SiOC/irrev. | 100 | 1077 |
| SiOC/Sb (lith.) | 8.89 | LiCx/rev. | 25.4 | 2134 |
| 6.82 | LiCx/rev. | 41.4 | 969 | |
| 5.44 | SiOC/irrev. | 14.6 | 883 | |
| 5.37 | Li3Sb | 18.1 | 220 | |
| −4.2 | Sb/SEI | 0.5 | 751 | |
| SiOC/Sb (delith.) | 4.56 | SiOC/irrev. | 99.1 | 1009 |
| −4.48 | Sb/SEI | 0.9 | 796 |
Conclusions
In summary, we report the synthesis of a SiOC/Sb nanocomposite by the pyrolysis of a preceramic single-source precursor, which was prepared through the gelation reaction of Sb(Oct)3 and PMHS with apolar DVB side-chains. The SiOC/Sb nanocomposite displays a uniform distribution of Sb NPs (5–40 nm) within the SiOC matrix. SiOC/Sb showed improved Li-ion storage characteristics as compared to SiOC. In particular, SiOC/Sb exhibits high rate capability, with a charge-storage capacity of 549 mA h g−1 at a current density of 2232 mA g−1 and at high active material loadings of 2.3–3.1 mg cm−2. 29Si and 7Li NMR measurements revealed that the incorporation of Sb into SiOC matrix alters the carbon in the Si–O–C network, yielding different Li-ion storage sites. We suggest that future work should be focused on the improvement of the initial coulombic efficiency of SiOC/Sb, which remains a major hurdle for the commercialization of such composite electrodes.Experimental
Chemicals for the synthesis of SiOC and SiOC/Sb
Polymethylhydrosiloxane (MW ≈ 1900, PMHS, Sigma Aldrich), divinyl benzene (DVB, technical grade, 80%, Sigma Aldrich), Sb acetate (Sb(Ac)3, 99.99%, Sigma Aldrich), 2-ethylhexanoic acid (≥99%, Sigma Aldrich), ortho-xylene (99%, pure, Acros Organics) and platinum Karstedt's catalyst (platinum(0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxane complex solution in xylene, Pt ≈2%, Sigma-Aldrich) were used as received.Battery components
Carboxymethyl cellulose (CMC, SUNROSE MAC 500LC, Nippon Paper Group), poly(vinylidene fluoride) (PVDF, Mw ≈ 534000, Sigma-Aldrich), carbon black (CB, Super P, TIMCAL, Switzerland), ethylene carbonate (EC, battery grade, BASF), LiPF6 (battery grade, Novolyte Technologies), graphite (SLP50, TIMCAL), dimethyl carbonate (DMC, battery grade, BASF), and fluoroethylene carbonate (FEC, >98%, TCI Chemicals) were used as received.
Synthesis of Sb(Oct)3 precursor
Sb(Oct)3 was obtained from Sb(Ac)3 through anion exchange. In a three-necked flask equipped with a distillation apparatus and a magnetic stir bar, Sb(Ac)3 (5.011 g) and 2-ethylhexanoic acid (8.25 mL) were added to o-xylene (20 mL). The white solution was heated up to 120 °C for 15 min, following treatment at 240 °C for 2 h. The mixture was then dried under vacuum at 80 °C for 12 h to form a yellowish liquid product. The yield was ca. 96%.Synthesis of SiOC/Sb nanocomposite
DVB (2.5 g) and a platinum Karstedt's catalyst (5 μL) were combined into a 25 mL flask, and the mixture was stirred at room temperature for 10 min. PMHS (1.25 g) was then added, and the mixture was stirred for another 10 min. Afterwards, Sb(Oct)3 (2.5 g, corresponding to 40 wt% of the total preceramic polymer mixture) was added to the flask and the reaction mixture was stirred at RT for another 15 min following the formation of gel within 3 h. The preceramic polymer gel was then aged via heat-treatment in a drying oven at 80 °C for 48 h. Subsequently, the obtained solid was crushed and pyrolyzed in an alumina crucible under Ar gas at 1000 °C. A heating rate was set to 5 °C min−1, followed by a holding step of 1 h at the dwell temperature. The cooling rate was set at 1 °C or 5 °C min−1.Characterization
Attenuated total reflectance FTIR spectra were recorded using a Bruker Tensor 27 spectrometer (Bruker, USA) equipped with a Golden Gate ATR crystal. Raman spectra of the samples were recorded by a confocal Raman spectrometer (Renishaw, UK) using a laser beam of 488 nm. Scanning transmission electron microscopy (STEM) investigations were performed on an aberration-corrected HD2700CS (Hitachi) operated at an acceleration potential of 200 kV. Images were recorded with a high-angle annular dark field (HAADF) detector resulting in atomic number contrast (Z-contrast). TEM measurements were performed on the Talos F200X (ThermoFisher scientific, FEG, Uacc = 200 kV). Powder X-ray diffraction (XRD) spectra were collected on a STOE STADI P powder X-ray diffractometer.The contents of C, H, O and Sb in the SiOC and SiOC/Sb samples were determined by elemental analysis at the Mikroanalytisches Labor Pascher (Remagen-Bandorf, Germany). The content of Si was estimated as the difference between the total amount of all elements (100 wt%) and the sum of O, C, H and Sb contents (in wt%). The SiCxO2(1−x) and Cfree = Ctotal − Cx formulas were applied for calculations of the ratio between SiOC and Cfree, based on the total amount of Si, O, and C, which was determined from the elemental analysis. 7Li,13C and 29Si solid-state NMR spectroscopy was performed using a Bruker 11.7 T spectrometer equipped with an Avance III console and a double resonance 2.5 mm solid-state probe head. Samples were prepared for NMR as described in the ESI† and filled into a 2.5 mm zirconia rotor in an argon glovebox. All experiments were performed at room temperature while spinning the sample at 12 kHz MAS frequency. The 7Li chemical shifts were referenced to aqueous LiCl solution (0.1 M), the 13C chemical shifts to Si(CH3)4. For 29Si chemical shifts, Q8M8 was used as external reference. The number of transients acquired was 4k for 7Li, 100k for 13C NMR and 16k for 29Si NMR experiments. All 7Li spectra were acquired without decoupling using one-pulse excitation sequences with a 90° pulse length of 11 μs and a recycle delay of 1 s. All 13C and 29Si NMR spectra were acquired with a CP sequence with contact times of 2.6 ms and 10 ms, respectively, and proton decoupling. A recycle delay of 1 s was used for both experiments.
Electrode fabrication
SiOC or SiOC/Sb (550 mg, 85%) and carbon black (48.5 mg, 7.5%) were ball-milled in a planetary ball-mill at 350 rpm for 30 min. CMC [48.5 mg in water (1.94 mL), 7.5%] was added to the dry mass and the mixture was ball-milled in a planetary ball-mill at 350 rpm for 30 min. The obtained slurry was cast onto Cu-foil using a doctor-blading technique. 12 mm electrode disks were cut and dried overnight in a vacuum oven at 80 °C (2 × 10−2 mbar) prior to cell assembly. Electrode active material loadings were in the range of 2.3–3.1 mg cm−2. To determine the gravimetric capacities, the full mass of the SiOC and SiOC/Sb composites (85 wt% of the total electrode material) was used.Cell assembly and testing
Stainless-steel coin-type cells (316 L, Hohsen Corp) were assembled in a glovebox under Ar atmosphere (<0.1 ppm H2O/O2) with a glass microfiber separator, 200 μL of 1 M LiPF6 in EC:DMC (1:1 by weight) with 3 wt% FEC, and a thin disk of Li-metal (16 mm in diameter). The coin cells were electrochemically cycled after a waiting time of 2 h using a multichannel workstation (Astrol BAT-Flex). Cyclic voltammetry was performed using a separate multichannel workstation (MPG-2, Bio-Logic SAS). The capacity values at 74.4 and 2232 mA g−1 correspond to the 2nd and 27th cycle, respectively.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is part of the activities of SCCER HaE, which was financially supported by the Innosuisse – Swiss Innovation Agency. P. V. W. S. and T. G. acknowledge the funding from the EMPAPOSTDOCS-II programme. The EMPAPOSTDOCS-II programme has received funding from the European Union's Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie grant agreement number 754364. We thank the Swiss National Science Foundation for funding of the TGA analytic instrument (SNF 206021_64024). STEM was done at the Scientific Center for Optical and Electron Microscopy (ScopeM, ETH Zürich). We thank Roland Widmer and Samuel Stolz for XPS measurements. We thank Eirini Kakkava (Optics Laboratory, EPFL) for Raman spectroscopy measurements.Notes and references
- L. Y. Beaulieu, K. W. Eberman, R. L. Turner, L. J. Krause and J. R. Dahn, Electrochem. Solid-State Lett., 2001, 4, A137–A140 CrossRef CAS.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359 CrossRef CAS PubMed.
- M. R. Palacín, Chem. Soc. Rev., 2009, 38, 2565–2575 RSC.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- W.-J. Zhang, J. Power Sources, 2011, 196, 13–24 CrossRef CAS.
- W.-J. Zhang, J. Power Sources, 2011, 196, 877–885 CrossRef CAS.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- M. Ashuri, Q. He and L. L. Shaw, Nanoscale, 2016, 8, 74–103 RSC.
- X. Shen, Z. Tian, R. Fan, L. Shao, D. Zhang, G. Cao, L. Kou and Y. Bai, J. Energy Chem., 2018, 27, 1067–1090 CrossRef.
- X. Zuo, J. Zhu, P. Müller-Buschbaum and Y.-J. Cheng, Nano Energy, 2017, 31, 113–143 CrossRef CAS.
- Y. Sun, N. Liu and Y. Cui, Nat. Energy, 2016, 1, 16071 CrossRef CAS.
- C. Jiang, E. Hosono and H. Zhou, Nano Today, 2006, 1, 28–33 CrossRef.
- A. B. Yaroslavtsev, T. L. Kulova and A. M. Skundin, Russ. Chem. Rev., 2015, 84, 826–852 CrossRef CAS.
- N. Mahmood, T. Tang and Y. Hou, Adv. Energy Mater., 2016, 6, 1600374 CrossRef.
- F.-F. Cao, H. Ye and Y.-G. Guo, in Nanostructures and Nanomaterials for Batteries: Principles and Applications, Springer Singapore, Singapore, 2019, pp. 89–158, DOI:10.1007/978-981-13-6233-0_3.
- M. He, K. Kravchyk, M. Walter and M. V. Kovalenko, Nano Lett., 2014, 14, 1255–1262 CrossRef CAS PubMed.
- Y. Jin, B. Zhu, Z. Lu, N. Liu and J. Zhu, Adv. Energy Mater., 2017, 7, 1700715 CrossRef.
- R. J.-C. Dubey, P. V. W. Sasikumar, F. Krumeich, G. Blugan, J. Kuebler, K. V. Kravchyk, T. Graule and M. V. Kovalenko, Adv. Sci., 2019, 6, 1901220 CrossRef CAS PubMed.
- P. Colombo, G. Mera, R. Riedel and G. D. Sorarù, J. Am. Ceram. Soc., 2010, 93, 1805–1837 CAS.
- C. Stabler, E. Ionescu, M. Graczyk-Zajac, I. Gonzalo-Juan and R. Riedel, J. Am. Ceram. Soc., 2018, 101, 4817–4856 CrossRef CAS.
- M. Halim, C. Hudaya, A. Y. Kim and J. K. Lee, J. Mater. Chem. A, 2016, 4, 2651–2656 RSC.
- M. Wilamowska-Zawlocka, P. Puczkarski, Z. Grabowska, J. Kaspar, M. Graczyk-Zajac, R. Riedel and G. D. Sorarù, RSC Adv., 2016, 6, 104597–104607 RSC.
- M. Graczyk-Zajac, L. M. Reinold, J. Kaspar, P. V. W. Sasikumar, G.-D. Soraru and R. Riedel, Nanomaterials, 2015, 5, 233 CrossRef CAS PubMed.
- M. Graczyk-Zajac, D. Vrankovic, P. Waleska, C. Hess, P. V. Sasikumar, S. Lauterbach, H.-J. Kleebe and G. D. Sorarù, J. Mater. Chem. A, 2018, 6, 93–103 RSC.
- A. M. Wilson, G. Zank, K. Eguchi, W. Xing and J. R. Dahn, J. Power Sources, 1997, 68, 195–200 CrossRef CAS.
- P. Vallachira Warriam Sasikumar, E. Zera, M. Graczyk-Zajac, R. Riedel and G. D. Soraru, J. Am. Ceram. Soc., 2016, 99, 2977–2983 CrossRef CAS.
- J. Kaspar, C. Terzioglu, E. Ionescu, M. Graczyk-Zajac, S. Hapis, H.-J. Kleebe and R. Riedel, Adv. Funct. Mater., 2014, 24, 4097–4104 CrossRef CAS.
- A. Tolosa, M. Widmaier, B. Krüner, J. M. Griffin and V. Presser, Sustainable Energy Fuels, 2018, 2, 215–228 RSC.
- D. Vrankovic, M. Graczyk-Zajac, C. Kalcher, J. Rohrer, M. Becker, C. Stabler, G. Trykowski, K. Albe and R. Riedel, ACS Nano, 2017, 11, 11409–11416 CrossRef CAS PubMed.
- J. Kaspar, M. Graczyk-Zajac, S. Lauterbach, H.-J. Kleebe and R. Riedel, J. Power Sources, 2014, 269, 164–172 CrossRef CAS.
- Z. Wu, W. Lv, X. Cheng, J. Gao, Z. Qian, D. Tian, J. Li, W. He and C. Yang, Chem. – Eur. J., 2019, 25, 2604–2609 CAS.
- Y. Lee, K. Y. Lee and W. Choi, Adv. Funct. Mater., 2017, 27, 1702607 CrossRef.
- S. J. Widgeon, S. Sen, G. Mera, E. Ionescu, R. Riedel and A. Navrotsky, Chem. Mater., 2010, 22, 6221–6228 CrossRef CAS.
- C. G. Pantano, A. K. Singh and H. Zhang, J. Sol-Gel Sci. Technol., 1999, 14, 7–25 CrossRef CAS.
- P. Kroll, MRS Online Proc. Libr., 2011, 1313, 1–6 Search PubMed.
- H. Fukui, H. Ohsuka, T. Hino and K. Kanamura, ACS Appl. Mater. Interfaces, 2010, 2, 998–1008 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr02930k |
| This journal is © The Royal Society of Chemistry 2020 |