
Protein corona fingerprinting to differentiate sepsis from non-infectious systemic inflammation†

Abstract
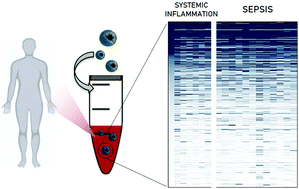
Rapid and accurate diagnosis of sepsis remains clinically challenging. The lack of specific biomarkers that can differentiate sepsis from non-infectious systemic inflammatory diseases often leads to excessive antibiotic treatment. Novel diagnostic tests are urgently needed to rapidly and accurately diagnose sepsis and enable effective treatment. Despite investment in cutting-edge technologies available today, the discovery of disease-specific biomarkers in blood remains extremely difficult. The highly dynamic environment of plasma restricts access to vital diagnostic information that can be obtained by proteomic analysis. Here, we employed clinically used lipid-based nanoparticles (AmBisome®) as an enrichment platform to analyze the human plasma proteome in the setting of sepsis. We exploited the spontaneous interaction of plasma proteins with nanoparticles (NPs) once in contact, called the ‘protein corona’, to discover previously unknown disease-specific biomarkers for sepsis diagnosis. Plasma samples obtained from non-infectious acute systemic inflammation controls and sepsis patients were incubated ex vivo with AmBisome® liposomes, and the resultant protein coronas were thoroughly characterised and compared by mass spectrometry (MS)-based proteomics. Our results demonstrate that the proposed nanoparticle enrichment technology enabled the discovery of 67 potential biomarker proteins that could reproducibly differentiate non-infectious acute systemic inflammation from sepsis. This study provides proof-of-concept evidence that nanoscale-based ‘omics’ enrichment technologies have the potential to substantially improve plasma proteomics analysis and to uncover novel biomarkers in a challenging clinical setting.
- This article is part of the themed collections: Editor’s Choice: Recent breakthroughs in nanobiotechnology, Nanoscale Most Popular 2020 Articles and Advisory Board research selection
 Please wait while we load your content...
Please wait while we load your content...

