
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStable, concentrated, biocompatible, and defect-free graphene dispersions with positive charge†
Yuyoung
Shin‡
 a,
Sandra
Vranic‡
bc,
Xavier
Just-Baringo‡
a,
Sai Manoj
Gali
d,
Thomas
Kisby
bc,
Yingxian
Chen
bc,
Alexandra
Gkoutzidou
e,
Eric
Prestat
ef,
David
Beljonne
d,
Igor
Larrosa
*a,
Kostas
Kostarelos
*bd and
Cinzia
Casiraghi
*a
a,
Sandra
Vranic‡
bc,
Xavier
Just-Baringo‡
a,
Sai Manoj
Gali
d,
Thomas
Kisby
bc,
Yingxian
Chen
bc,
Alexandra
Gkoutzidou
e,
Eric
Prestat
ef,
David
Beljonne
d,
Igor
Larrosa
*a,
Kostas
Kostarelos
*bd and
Cinzia
Casiraghi
*a
aDepartment of Chemistry, University of Manchester, Oxford Road, Manchester, UK. E-mail: cinzia.casiraghi@manchester.ac.uk
bNanomedicine Lab, National Graphene Institute and Faculty of Biology, Medicine & Health, The University of Manchester, Manchester M13 9PT, United Kingdom. E-mail: kostas.kostarelos@manchester.ac.uk
cNational Graphene Institute, University of Manchester, Booth Street East, Manchester, UK
dCatalan Institute of Nanoscience and Nanotechnology (ICN2), UAB Campus Bellaterra, Barcelona, Spain
eDepartment of Materials, University of Manchester, Oxford Road, Manchester, UK
fSuperSTEM Laboratory, SciTech Daresbury Campus, Daresbury, UK
First published on 25th May 2020
Abstract
The outstanding properties of graphene offer high potential for biomedical applications. In this framework, positively charged nanomaterials show better interactions with the biological environment, hence there is strong interest in the production of positively charged graphene nanosheets. Currently, production of cationic graphene is either time consuming or producing dispersions with poor stability, which strongly limit their use in the biomedical field. In this study, we made a family of new cationic pyrenes, and have used them to successfully produce water-based, highly concentrated, stable, and defect-free graphene dispersions with positive charge. The use of different pyrene derivatives as well as molecular dynamics simulations allowed us to get insights on the nanoscale interactions required to achieve efficient exfoliation and stabilisation. The cationic graphene dispersions show outstanding biocompatibility and cellular uptake as well as exceptional colloidal stability in the biological medium, making this material extremely attractive for biomedical applications.
1. Introduction
Liquid-phase exfoliation (LPE)1 allows for mass-scalable, cost-effective and versatile production of graphene formulations suitable for a wide range of applications, from composites to biological applications.2–5 Organic solvents have been shown to provide efficient exfoliation.1,6,7 On the other side, LPE in water can also be successfully achieved through covalent functionalisation,8,9 or non-covalent functionalisation with stabilisers.10,11 Different types of stabilisers, including surfactants,10,12–15 polymers16–18 and aromatic molecules,19–24 have been investigated. In particular, pyrene derivatives have shown to be more effective exfoliating agents than traditional surfactants,21 and have been successfully used to make biocompatible and inkjet-printable graphene inks.25 Despite the success of pyrene derivatives for graphene production, mainly negatively charged or neutral functional groups have been studied in detail, leading to production of anionic or neutral graphene.21–23 Only a few studies have reported exfoliation with cationic pyrene molecules, resulting in poorly concentrated and/or unstable graphene dispersions, hindering their use in practical applications.21,26,27 Amphoteric pyrene molecules were also recently used to produce graphene dispersions,28 but their reduced colloidal stability at neutral pH makes this material unsuitable for most applications. One of the major bottlenecks of LPE in water assisted by pyrene derivatives is related to the poor understanding of the nanoscale mechanisms giving rise to exfoliation and stabilisation, i.e. we still do not know how to design the pyrene derivative that can provide the highest exfoliation efficiency.In this work, we have designed a new family of cationic pyrene derivatives that was used to systematically investigate the exfoliation efficiency and quality of the material produced as a function of the charge, type of functional group and length of the linker between the pyrene core and the functional group. Our results show that the most efficient pyrene derivatives are those with the polar group far from the pyrene base as this configuration minimises interference between the adsorption of pyrene on graphene and the interaction of the functional group with the water molecules, in agreement with Molecular Dynamics (MD) simulations. We demonstrate that stable, highly concentrated, defect-free dispersions, containing mostly single and few-layers with average lateral size of about 200 nm, can be obtained using very small amount of stabiliser (0.4 mg mL−1). These cationic graphene dispersions also show exceptional biocompatibility, intracellular uptake profile and stability in the biological medium, even with protein serum, making them a very attractive material for nanomedicine applications.
2. Experimental
2.1. Materials
Natural graphite flakes were provided by Graphexel Ltd. The details of the cationic pyrene synthesis routes and all characterisation methods used for identification of the structure and purity of the compounds are discussed in the ESI (section S7†). 1-Pyrenesulfonic acid sodium salt (PS1) (≥97.0%) was purchased from Sigma-Aldrich. Silicon oxide wafers with 290 nm double side coating were purchased from Inseto Ltd (UK).2.2. Preparation of graphene dispersions
Graphene dispersions were prepared via LPE in water following the protocol developed in previous works.22,25,28 In detail, 30 mg of graphite was added to 10 mL of de-ionised (DI) water, previously mixed with 4 mg of the cationic pyrenes. The mixture was then sonicated at 300 W for 7 days using a Hilsonic bath sonicator. Afterwards, un-exfoliated graphite was removed by 2-steps centrifugation (Sigma 1-14k refrigerated centrifuge) at 3500 rpm (903g) for 20 minutes. After each centrifugation step, the supernatant containing graphene and the pyrene derivatives in water was collected. Graphene dispersions with PS1 were prepared using the same protocol for direct comparison. Graphene dispersions used for biological study are prepared in larger volume (100 mL) using a 600 W Hilsonic bath sonicator. The excess pyrene molecules are removed by 2-steps centrifugation (Sigma 1-14k refrigerated centrifuge) at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm (16600g) for 60 minutes. After each centrifugation step, the supernatant was removed and the sediment was re-dispersed in DI water.
000 rpm (16600g) for 60 minutes. After each centrifugation step, the supernatant was removed and the sediment was re-dispersed in DI water.
2.3. Characterisation
The Raman analysis was performed using a qualitative protocol for analysing LPE graphene, which has been described previously.31–35 In detail, the shape of the 2D peak was used for determination of the thickness distribution. The 2D peak was fitted with a single Lorentzian lineshape, which was used to distinguish between single-layer graphene (SLG), few-layer sheets (FLG) and graphitic material (>10 layers with AB stacking) by evaluating the fit residual, R2. SLG showed a single symmetric peak with R2 > 0.987, FLG a single asymmetric peak with R2 < 0.987, and graphitic material was distinguished by its characteristic peak shape with shoulder.
2.4. Molecular dynamics simulations
The adsorption process of pyrene derivatives on the surface of graphene layer, in the presence of explicit water molecules, was simulated using MD simulations, performed with the NAMD36 software. All pyrene derivatives were described with a customised general AMBER force field37 where in the atomic charges and the torsional potentials were calculated at the M02/6-311G(d) level of theory and used in the force field following the scheme described by ref. 38. Water molecules were represented by the well-known and reliable “modified TIP3P” force field.39 A simulation box of size 63.90 × 51.65 × 68.00 Å3, containing an infinite sheet of SLG at the bottom of the simulation box, was used for each pyrene derivative. Each derivative was positioned at a perpendicular distance of 2.5 Å from the plane of the SLG and the box was filled with 9000 water molecules. In the first step, MD equilibration was performed for 20 ns under constant pressure (1 bar) and temperature (298 K) with the NPT ensemble, and the simulations were extended for an additional 40 ns of production cycle in the NPT ensemble. Subsequently, free energy of interaction of each pyrene derivative with SLG was evaluated by computing the potential of mean force (PMF) of the adsorption process employing the Adaptive Biasing Force (ABF) method.40 Three different PMF profiles were calculated by performing three different/independent ABF simulations for 75 ns in the NPT ensemble by taking the (i) total center of mass of the pyrene derivative (COM), (ii) isolated center of mass of the pyrene unit of the molecule (PYR) and (iii) isolated center of mass of the functional group of the molecule (FUN). For all the ABF simulations, the lower and upper bounds were set to 2 Å and 20 Å, respectively and no specific constraints were employed on the internal degrees of freedom for all the derivatives, meaning that the derivatives are free to rotate along the center of mass and to translate along the ‘xy’ plane (plane parallel to the graphene layer). The three independent PMF profiles were independently adjusted with respect to the free energy at the upper boundary of 20 Å.2.5. Biological studies
000 cells were analysed on a BD FACSVerse™ flow cytometer using 488 nm excitation and 515 and 615 nm band-pass filters for annexin V and PI detection, respectively. The experiment was repeated three times.
000 rpm. The supernatants were discarded, and the graphene-containing pellets were resuspended in Milli-Q water. This washing step was repeated once in order to remove excess unattached biomolecules and electrolytes, enabling the characterisation of graphene by DLS and zeta potential measurements. The experiment was repeated two times.
3. Results and discussion
3.1. Preparation of cationic pyrenes
A family of cationic amphiphilic pyrenes was prepared. The molecules are composed of three parts (Fig. 1A): (i) a pyrene aromatic ring, which allows adsorption to graphene; (ii) a carbon chain linker of variable length (n = 1–4, where n is the number of methylene groups) between the pyrene ring and the polar group; (iii) a cationic group. We have investigated the following functional groups: a trimethylammonium group (TMA family), as example of tetraalkylammonium substitution; a 3-methylimidazolium ring (IMI family) to introduce an electron-rich positively-charged aromatic; and a pyridinium ring (PRD family) to showcase the effect of an electron-poor aromatic cation. The complete family is shown in Fig. 1B. For clarity, the pyrene derivatives produced are indicated with the acronyms: TMAn, IMIn, and PRDn.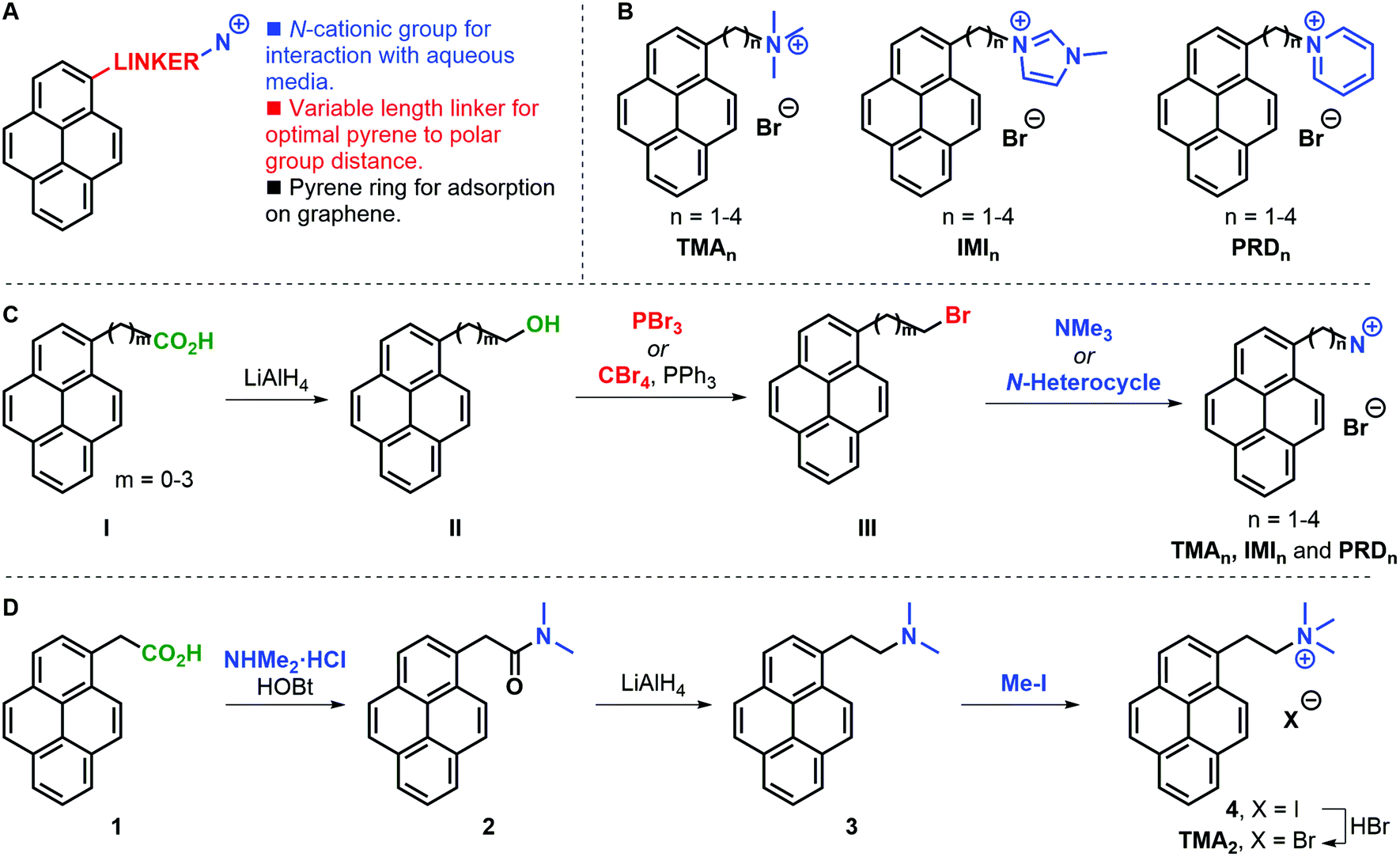 | ||
| Fig. 1 (A) Design of cationic pyrenes. (B) Family of cationic pyrenes designed for this study. (C) General synthetic strategy for the synthesis of cationic pyrenes (NC = nitrogen-based cations). (D) Alternative route towards TMA2 synthesis. | ||
In a general synthetic approach, 1-pyrenyl alcohols II were obtained from a supplier or were prepared by reduction of the parent carboxylic acid I (Fig. 1C). With all alcohols II in hand, the corresponding bromides III were synthesised to serve as electrophiles in the condensation with trimethylamine or an N-heterocycle (i.e. 1-methylimidazole or pyridine). The present strategy gave access to the desired pyrene cations in good overall yields and allowed access to gram amounts of products. Only in the case of TMA2 an alternative approach was required due to the competing elimination reaction leading to large amounts of 1-vinylpyrene being formed. In this case, 1-pyrenylacetic acid 1 was coupled with dimethylammonium chloride to form dimethylamide 2, which was subsequently reduced to amine 3 and alkylated with methyl iodide to yield iodide salt 4 (Fig. 1D). The desired TMA2 bromide salt was obtained after anion exchange to have a consistent family of cationic pyrenes with bromide counter ions.
The solubility of the molecules is reported in Fig. S1, ESI.†
3.2. Liquid-phase exfoliation of graphene: experimental and modelling
The obtained graphene dispersions for each cationic pyrene molecules are shown in Fig. 2A. To better illustrate the concentration difference, the dispersions were diluted by a factor 10. Fig. 2B shows the graphene concentrations obtained with the different cationic pyrene molecules. For comparison, the plot also reports graphene concentration obtained with 1-pyrenesulfonic acid sodium salt (PS1), which is one of the most used anionic pyrene derivative for LPE.22,23Fig. 2B shows that dispersions made with IMI1 and PRD1 have similar or higher concentration than those produced with PS1, while the TMA1 stabiliser is not efficient at exfoliating graphite. However, the concentrations drastically change with increasing chain length: the concentration of graphene prepared by TMA strongly increases with increasing n, becoming comparable, if not even higher than the concentration obtained by IMI and PRD families (Fig. 2B). Note that this behavior cannot be explained by the different solubility of the molecules, as there is no apparent correlation between solubility (Fig. S1†) and graphene concentration (Fig. 2B). The increase in efficiency with increasing distance of the polar group from the hydrophobic region of the molecules, seen for the TMA family, is observed in traditional ionic surfactants, which are more effective at lowering surface tension for increasing chain length.41 This result can be tentatively explained by taking into account that as the charge gets further away from the hydrophobic core, the interaction between the polar group and the water molecules and the π–π interaction between the pyrene and the graphene surface are better separated, i.e. they do not interfere with each other, by enhancing the adsorption of the hydrophobic pyrene on the surface of graphene as well as providing better hydration of the polar group. When the functional group is too close to the pyrene base, as in the case of TMA1, the bulky functional group interferes with the adsorption of the aromatic planar surface of the pyrene and graphene surface. As the distance between the polar functional group and the hydrophobic base increases, steric hindrance is significantly reduced, while the interaction of the localised charge on the functional group and the water molecules is enhanced, therefore improving the stabilisation of graphene flakes in aqueous media. On the other hand, in case of IMI and PRD families, a more complex pattern is observed: relatively high exfoliation efficiency was achieved at n = 1, followed by a dramatic decrease at n = 2, and then a slow increase of exfoliation was observed with increasing n. For n = 1, the aromatic functional groups as well as the pyrene base show strong interaction with the graphene surface, possibly facilitating the adsorption of the stabilisers on the graphene surface, hence the high exfoliation efficiency.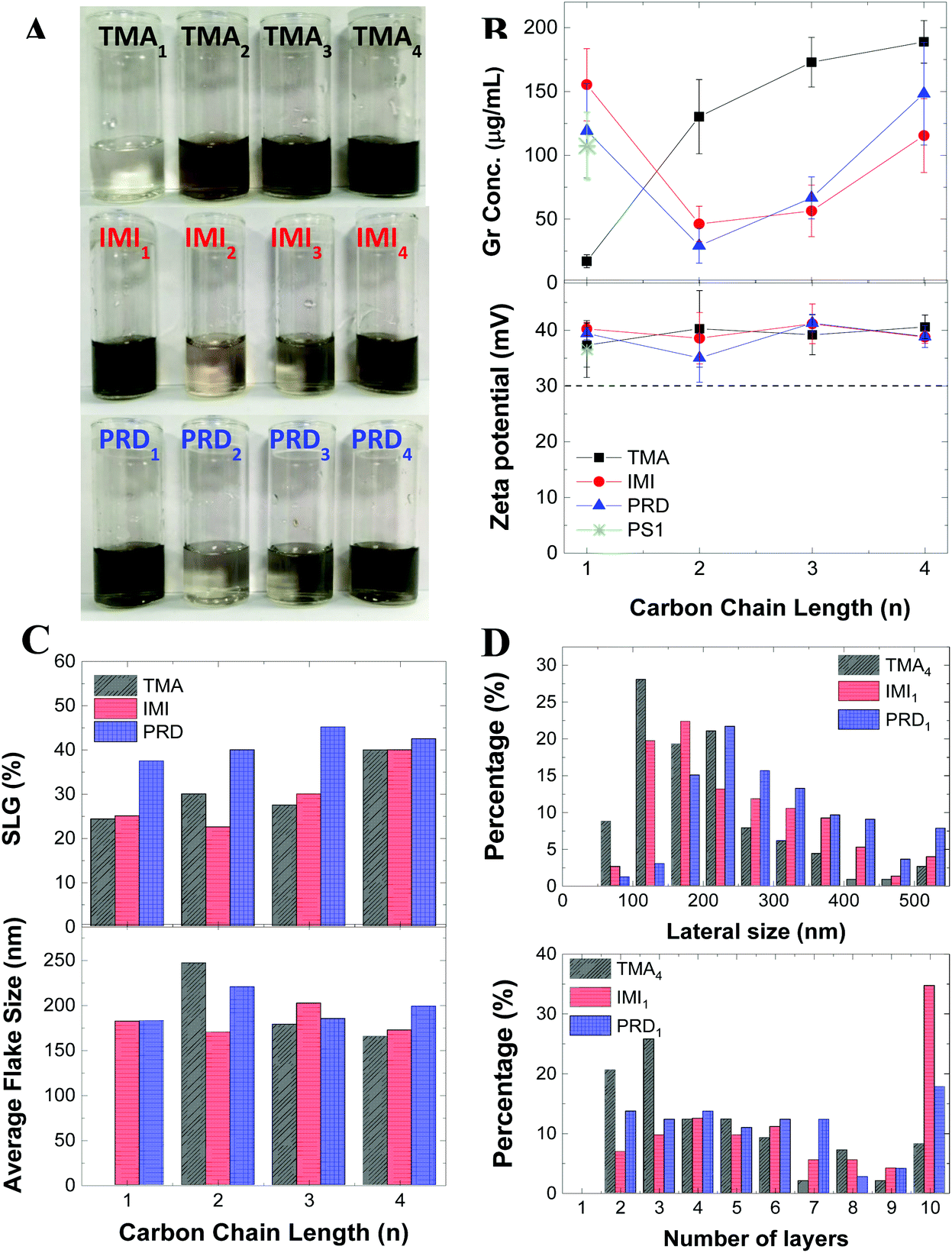 | ||
| Fig. 2 (A) Photographs of graphene dispersions prepared with the cationic pyrenes (diluted by ×10), and (B) evolution of the final graphene concentration with carbon chain length and zeta potential measurements for the graphene dispersions (for PS1, |ζ| is shown). (C) Single layer graphene percentage determined by Raman spectroscopy and mean flake size estimated from AFM measurement, (D) lateral size and number of layers counted from TEM measurement for three samples, TMA4, IMI1 and PRD1. | ||
Note that linear alkyl chains are known to form stable layers on graphene.31,33 However, the length of the chain in our pyrene derivatives is very short, hence this effect is unlikely to be responsible for the higher exfoliation efficiency. Indeed, PRD1 and PRD4 shows similar graphene concentration. Getting insight into the molecular interactions during exfoliation is of fundamental importance because it enables design of the pyrene derivatives providing the highest exfoliation efficiency. Thus, the adsorption process of each cationic pyrene molecule on graphene surface in aqueous medium was assessed by employing MD simulations.23 Each cationic pyrene was placed in a box filled with 9000 water molecules at a starting distance of 2.5 Å from the basis plane consisting of a single layer graphene sheet. The potential of mean force (PMF) of the adsorption process was calculated using adaptive biasing force (ABF) simulations (see Experimental section for more details). To assess the different factors influencing the interaction of graphene with cationic pyrene molecules in the presence of water, three independent PMF profiles were obtained by performing different ABF simulations considering: (1) the center of mass of the whole pyrene molecules (COM), (2) the pyrene base (PYR) and (3) the functional group (FUN).
Fig. 3A shows the PMF curves obtained in the three cases for the TMAn family. The largest changes observed with increasing n are seen for the functional group (right panel): with the increase in n, the PMF profile changes from exhibiting a global minimum (located at the same energy, independent of n) to that of exhibiting an additional broadened local minimum, allowing the charged functional group to be farther away from the graphene surface and to freely interact with water molecules. Similarly, the PMF curves of the pyrene core show one local minimum at ∼3.5 Å, independent from n, as shown in Fig. 3A, middle.
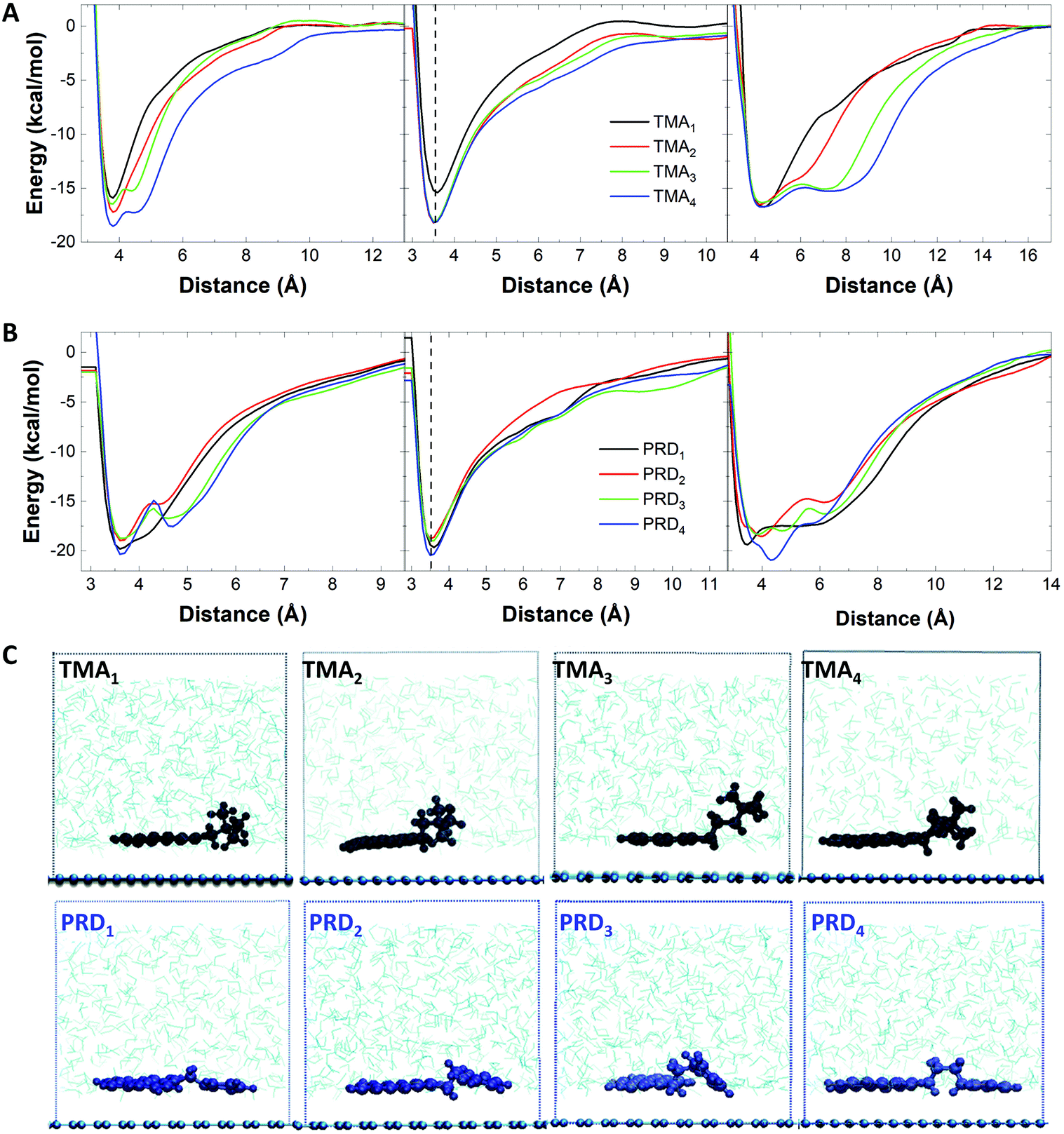 | ||
| Fig. 3 PMF profiles of pyrene derivatives with (A) TMA and (B) PRD functional groups for (top) the center of mass, (middle) pyrene base and (bottom) the functional group. (C) MD snapshots of pyrene derivatives with (left) TMA and (right) PRD functional groups. | ||
However, when n = 1, the energy minimum is much higher (−16.9 kcal mol−1) compared to that observed with n > 1 (−18.2 kcal mol−1), indicating a better interaction of the pyrene base with the graphene surface when n > 1. Fig. 3A, left panel shows the combined effect of pyrene adsorption (Fig. 3A, middle) and hydration of the functional group (Fig. 3A, right), with increasing stabilisation energy for increasing n, in agreement with the experimental results.
In the case of the PRDn family, the PMF profiles of the functional group show a variation in the free energy minima, depending on n (Fig. 3B, right): the global minimum of PRD1 and PRD4 is lower in energy than that of PRD2 and PRD3. As shown in Fig. 3B (left panel), the free energy minima for PRD2 (−18.9 kcal mol−1) and PRD3 (−18.7 kcal mol−1) are higher in value compared to that of PRD1 (−19.3 kcal mol−1) and PRD4 (−20.3 kcal mol−1).
A similar trend is also observed for the PMF profiles of the pyrene base (Fig. 3B, middle panel). These observations indicate stronger interaction of PRD with graphene for n = 1 and 4, driven by the interaction of both the functional group and the pyrene base with the graphene surface. This explains the different exfoliation trends observed with increasing n for TMA: the interaction with the graphene surface is driven mainly by the pyrene base for TMA family and the TMA functional group does not show any variation in the free energy minimum but prefers to interact with water molecules, in comparison to the PRD functional group. This has been confirmed by visualising the relative orientation of TMA and PRD derivatives on the graphene layer. The MD snapshots, taken from the production run of 40 ns in NPT ensemble with the atomic positions of the pyrene derivatives averaged over 20 equally separated time frames, are shown in Fig. 3C. This figure shows that the interaction of the TMA functional group with the surrounding water molecules tends to improve with increasing n, due to the increase in the relative stability of the localised positive charge on the nitrogen interacting with water molecules. For PRD, on the other hand, both the pyrene base and the functional group of the molecules are strongly interacting with the graphene layer. As shown in Fig. 3C (bottom), for PRD1 and PRD4, the plane of the pyrene base and the aromatic functional group are both parallel to the surface of graphene, whereas for PRD2 and PRD3, the functional group is energetically frustrated, leading to a decreased interaction with the graphene layer. This is further illustrated by the calculation of the tilt angle between the vector perpendicular to the pyrene base and the vector perpendicular to the graphene surface (Table S1†): the calculated tilt angles of PRD2 and PRD3 pyrene are much lower (166° and 154°, respectively) than that of PRD1 and PRD4 pyrenes (∼180°). Similar energetic profiles are expected for IMI family, as the IMI functional group is similar to that of PRD in terms of energy delocalisation and hence, the same mechanism is expected to govern the energetic interactions between graphene and IMI derivatives.
Our results show that higher exfoliation efficiency is obtained with pyrene derivatives with long chain length (n > 3). In case of short chain length, the exfoliation efficiency will strongly depend on the type of functional groups used, with IMI or PRD outperforming TMA due to the increased affinity to graphene.
3.3. Characterisation of the exfoliated graphene nanosheets
Fig. 2B (bottom) shows that all the graphene dispersions prepared in this study have a relatively high zeta potential of ∼40 mV. The zeta potential is typically used as an indicator of the stability of colloidal suspension: a zeta potential modulus higher than 30 mV is associated to a stable suspension in aqueous media.14 To further confirm the stability of the dispersions, the material was monitored for at least 10 months: no notable sedimentation was observed and no change in the zeta potential measured after 10 months’ storage was reported (Fig. S4†). This result shows that the dispersions are stable, regardless of which cationic pyrene is used for exfoliation. This is in contrast with the concentration, which strongly depends on the type of pyrene derivative used.Raman spectroscopy is one of the most popular techniques for characterisation of graphene.42,43 Fig. S8† shows representative spectra taken on ∼40 individual flakes drop-cast on silicon substrate for each dispersions. The Raman spectrum shows the characteristic G and D peak at ∼1580 cm−1 and ∼1350 cm−1, respectively.43 The D peak is characteristic of graphene produced by LPE2 and it is activated by the edges of the nanosheets,44 having smaller size than that of the laser spot. The 2D peak at 2680 cm−1 is also observed in the high energy region of the spectrum. Note that this peak is typically used to identify single-layer graphene, having a full width at half maximum (FWHM) of around 30 cm−1.43 However, this identification method does not work for graphene produced by LPE as further broadening of the 2D peak is observed as result of the nanosheets edges, adsorption of solvent and stabiliser molecules, and random re-stacking of the flakes.22,45 We performed qualitative thickness analysis by using a method previously tested in our group (Methods).22,31,33–35 This is based on the fit of the 2D peak with a Lorentzian lineshape. Fig. 2C and Table S2† show that all twelve dispersions contain more than 20% single layer graphene, which matches rather well with the typical value observed in graphene dispersions obtained with PS1 (∼30%).22 Notably, the highest single layer graphene percentage (>40%) is observed by using IMI4 and TMA4, suggesting that longer chains may help in achieving not only higher yield, but also better exfoliation, when used in combination with specific functional groups. In contrast, the concentration of single layer graphene obtained by the PRD family seems to be less sensitive to the chain length.
We further investigated the graphene flakes by atomic force microscopy (AFM) and transmission electron microscopy (TEM). Fig. 2C shows the mean flake size measured by AFM. The thickness and size distribution histogram for all the samples can be found in ESI (Fig. S5 and S6†). Note that the theoretical thickness of single layer graphene is estimated to be ∼0.34 nm, but because of surface adsorption of stabilisers and/or solvent molecules, AFM often shows higher thickness: typically, a thickness of 1–2 nm for single layer graphene produced by LPE has been reported.22,46,47 Thus, AFM cannot be used to derive quantitative information on the thickness. The AFM analysis shows that the lateral size distribution of the graphene nanosheets is between 50 nm and 400 nm, with average lateral size of ∼200 nm, for all the graphene dispersions prepared in this paper (Fig. 2C).
Three dispersions, obtained with TMA4, IMI1 and PRD1, were selected for TEM measurement, as they are characterised by the highest graphene concentration values for each cationic group-type. Distribution histograms of lateral size and number of layers of those dispersions are shown in Fig. 2D. The lateral size distribution is similar to that one measured by AFM (Fig. S5†). Compared to the other two dispersions, the dispersion prepared with TMA4 showed a higher percentage of smaller flakes (<300 nm), but still within the expected range for LPE graphene dispersions. The number of layers of graphene nanosheets was counted from individual flakes in the High Resolution TEM image. These measurements indicate that the dispersions are mostly composed by few layers graphene nanosheets (<7 layers), where the sample prepared with TMA4 shows the higher percentage of thinner layers, in accordance with the Raman analysis (Fig. 2C). The disagreement in the single layer graphene percentage between Raman and TEM measurements is attributed to the difference in resolution between the two techniques and in the different sample preparations. In any case, both techniques indicate that the dispersions mostly contain thin layers (<7 layers). These results show that the thickness and size distribution of the nanosheets is similar amongst the three graphene dispersions obtained with the most efficient cationic pyrene stabilisers.
4. Biological studies
It is well known that dimensionality, lateral size, charge, surface functionalisation and chemical composition of the nanomaterial can lead to radically different interactions with living systems.48–50 In particular, cationic nanomaterials are typically preferred for biomedical applications because, due to their positive charge, they show better interactions with the biological environment, such as better cellular internalisation due to stronger affinity to the negatively charged biomaterials such as small interfering ribonucleic acid (siRNA).51 However, the charge is also a significant contributor to the nanomaterial's toxicity.52 Thus, the development of non-toxic cationic nanomaterials is of fundamental importance in nanomedicine.In the framework of 2-dimensional materials, the most used method to produce cationic graphene for biomedical applications is based on the use of functionalisation of graphene oxide (GO) and reduced GO (rGO) with cationic polymers such as polyethylenimine (PEI) and polyethylene glycol (PEG). However, production of GO involves use of acid and washing steps,2 and the intrinsic negative surface charge of GO requires complex multiple steps for such functionalisation. Furthermore, its oxygen-containing functional groups may lead to oxidative stress in biological environment,53–56 for example it has been reported that PEI-functionalised GO shows cytotoxicity and reduced colloidal stability.57–59 Particularly, reduced colloidal stability of GO or functionalised GO in biological medium, especially in presence of protein serum, can lead to different cytotoxicity and cellular uptake due to the agglomeration by biocoronation.57–60 Hence, alternative oxygen-free graphene-based materials are extremely attractive for biomedical studies.
For the biomedical study, we selected graphene dispersions with the highest concentration of graphene flakes, namely those produced with TMA3, IMI1 and PRD1. Note that instead of TMA4, TMA3 was used since it gives similarly high concentration of graphene flakes with easier synthesis of the stabiliser. Graphene dispersions obtained using PS125 were also added for comparison. Note that the thickness and size distribution are similar in all dispersions, i.e. average size of ∼200 nm and thickness of less than 7 layers for most of the flakes. Note that other graphene-based materials with similar thickness and size have been shown to be able to penetrate the membrane, but it is unclear if and how the surface chemistry and charge may impact on the intracellular uptake. The samples are labelled as follows: Gr-TMA3, Gr-IMI1, Gr-PRD1 and Gr-PS1. The first three samples contain graphene flakes with positive surface charge (Gr+), while the last one contains graphene with negative surface charge (Gr−).
4.1. Stability of graphene in cell culture medium
Let us first focus on stability of the graphene dispersions in biological solutions, such as cell culture medium, as this can govern both cellular interactions and subsequent responses.61–63 Furthermore, the aggregation of nanomaterials in biological media is often associated with data misinterpretation and artifacts,64,65 making characterisation of nanomaterials in biologically relevant media as a prerequisite for accurate interpretation of the results. Excellent colloidal stability of nanomaterials is crucial for successful biomedical applications and clinical translation.The stability of the graphene dispersions was therefore assessed after incubation in complete RPMI1640 cell culture medium using DLS and surface charge measurements (Fig. S11 and Table S3†). Although DLS characterisation is valid only for spherical nanoparticles, it can be used to detect changes in size and polydispersibility of graphene dispersions,53,66,67 in combination with other characterisation techniques. Table S3† shows that all graphene dispersions have monodisperse size distribution upon incubation in complete cell culture medium for 24 h. This is confirmed by the lack of sedimentation/precipitation of material upon incubation in the cell culture medium (Fig. S11,† insets). Interestingly, the overall surface charge of all four types of graphene after incubation was found to be negative. However, it is important to note that the zeta potential measurement is done upon incubation in complete cell culture medium (in acellular conditions), followed by centrifugation, washing and finally re-dispersing in water. As the zeta potential cannot be measured in situ, the sign of the zeta potential measured may not be indicative of the zeta potential of the material when in contact with the cells. Furthermore, the change in surface charge is not specific to graphene based materials: the same result has been reported for other positively charged nanomaterials and attributed to their coating with proteins from the serum.68,69
4.2. In vitro uptake and biocompatibility
The assessment of cytotoxicity and cellular uptake is fundamental to establish the safety profile, but also to explore potential therapeutic applications (such as delivery of small therapeutic molecules or gene therapies) of nanomaterials.70 The cytotoxicity profile has been established using optical microscopy and further quantified using flow cytometry on two cell lines: Beas-2B and HeLa (Fig. S12,†Fig. 4B and Fig. S13B†). BEAS-2B cell line was selected as a representative of healthy lung epithelium, commonly used in nanotoxicology to assess the safety of nanomaterials.71,72 HeLa cell line was chosen as a representative cancer cell line, commonly used in the studies focusing on applications of nanomaterials (in particular graphene-based) for drug delivery.73 The two cell lines were exposed to increasing concentrations of graphene in complete cell culture medium, i.e. RPMI1640 cell culture medium containing 10% fetal bovine serum (FBS). The healthiness of the untreated or monolayers cells exposed to 50 μg mL−1 of the four types of graphene has been compared (Fig. S12A†). No evident morphological changes on the cellular level (e.g. rounding up of the cells, detachment from the support, loss of intercellular connections, swelling of the lysosomes) were observed, demonstrating that the graphene flakes were not cytotoxic even at the highest concentration used (50 μg mL−1). Cell viability after the treatment with increasing concentrations of graphene materials was further quantified using Annexin V/Propidium Iodide assay. The cells appeared unstained for indicators of apoptotic and/or necrotic cell death, confirming that graphene material is not cytotoxic up to the concentration of 100 μg mL−1 (Fig. 4B and Fig. S13B†). Thus, no matter the charge or the functional groups, graphene produced by pyrene derivatives (at least those investigated in this work) shows exceptional biocompatibility. To the best of our knowledge there are not studies on positively charged graphene flakes in vitro.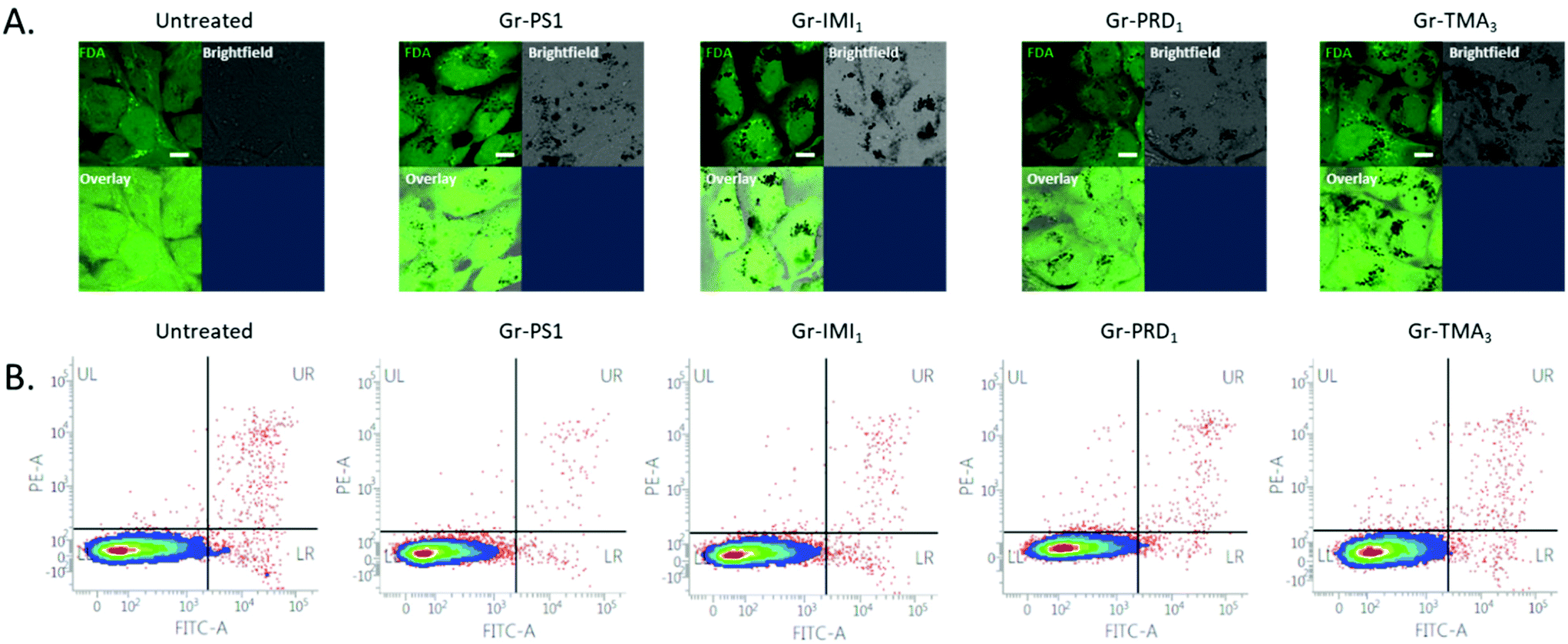 | ||
| Fig. 4 Uptake and cytotoxicity of Gr in BEAS-2B cells. Uptake and cytotoxicity of Gr in BEAS-2B cells. (A) BEAS-2B cells were exposed to 50 μg mL−1 of Gr-PS1, Gr-IMI1, Gr-PRD1 or Gr-TMA3 for 24 h. After the treatment, cells were stained using fluorescein diacetate (FDA) dye and the uptake of the flakes was estimated using confocal microscopy, by exploiting fluorescence quenching of the FDA dye due to the presence of the flakes inside the cells. Staining of the cells: FDA – green. Scale bar is 10 μm. Assessment of the cytotoxicity of Gr by flow cytometry using Propidium Iodide/Annexin V staining for BEAS-2B cells were exposed to 100 μg mL−1 of Gr for 24 h. In the bivariate plots, live cells are represented in the lower left (LL) quadrant, early apoptotic in the lower right (LR), late apoptotic and/or necrotic cells are shown in the upper right (UR) and necrotic cells are in the upper left (UL) quadrant. | ||
Moving to the intracellular uptake, due to the high light absorption capacity of graphene, the material can be easily detected using bright-field imaging. The optical inspection already shows that Gr+ interact more with the two cell lines, compared to Gr− (Fig. S12A,† bright-field images). For more explicit determination of intracellular uptake of the material, fluorescein diacetate dye (FDA) was used to stain the inside of the cells with green fluorescent signal, which will be quenched where graphene is localised intracellularly (Fig. 4A, Fig. S13A and S14,† black spots in images showing FDA stained cells). This approach clearly shows that the uptake occurs for all four types of graphene. However, Gr− is the least internalised by the cells, whereas all Gr+ are taken up more efficiently, even though to a different extent, after 24 h of treatment at the used dose of 50 μg mL−1 (Fig. 4A, Fig. S13A and S14† overlay images), possibly related to the different surface chemistry between the three Gr+. This result confirms that positively charged nanomaterials are better taken up by the cells due to the higher affinity of interaction with negatively charged proteoglycans on the plasma membrane of the cells.74–76
Our results clearly demonstrate that defect-free and cationic graphene flakes produced by pyrene derivatives have excellent stability in the medium, and exceptional biocompatibility, after being efficiently internalised by the cells: this is a very remarkable finding as alternative materials with positive surface charge, such as cationic nanoparticles and polymers, have been shown to induce cell death via lysosomal or rupture of the plasma membrane.47,74,75
5. Conclusions
In this study, various cationic pyrene derivatives were designed and used to produce stable, concentrated, positively charged and defect-free graphene dispersions in water via a simple one-pot LPE process. By performing a systematic study using pyrene derivatives with different functional groups and chain lengths, we concluded that aromatic functional groups (e.g. 3-methylimidazolium and pyridinium) enhance the adsorption of pyrene on graphene surface but at the expense of lower dispersibility in water. On the other hand, the use of the trimethylammonium functional group allowed better exfoliation efficiency with increased distance due to improved separation of adsorption of the pyrene base to the graphene surface and solubilisation of localised charge in water.The graphene dispersions show excellent colloidal stability in cell culture medium and exceptional biocompatibility (up to 100 μg mL−1) in both non-cancer and cancer cell lines. In particular, cationic graphene shows very high internalisation by both cell types, opening potential use of this material for a variety of biomedical applications, in particular for intracellular delivery of small therapeutic molecules and drugs.
Author contributions
I. L., K. K., and C. C. devised the project and supervised the work. X. J. B. performed pyrene derivative synthesis and characterisation, Y. S. performed graphene exfoliation and characterisation, and S. V. performed biocompatibility and uptake study. S. M. G. and Y. S. performed and analysed the data from the simulation study, under the supervision of D. B., A. G. performed TEM measurement under the supervision of E. P.The paper was written by Y. S., X. J. B., S. V. and C. C. with contribution from all authors.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work has been supported by the EPSRC in the framework of the project 2D Health (EP/P00119X/1) and the M-ERA.NET project MODIGLIANI (project2110). Computer time for this study was provided by the Consortium des Equipements de Calcul Intensif (CECI, http://www.ceci-hpc.be), and particularly those of the Technological Platform on High-Performance Computing, for which we gratefully acknowledge the financial support of the FNRS-FRFC (Conventions No. 2.4.617.07.F and 2.5020.11). DB is FNRS Research Director. CC strongly acknowledges very useful discussions with Lucia Delogu.References
- Y. Hernandez, et al., High-yield production of graphene by liquid-phase exfoliation of graphite, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- K. S. Novoselov, et al., A roadmap for graphene, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Liquid Exfoliation of Layered Materials, Science, 2013, 340, 1226419–1226419 CrossRef.
- K. Kostarelos, Translating graphene and 2D materials into medicine, Nat. Rev. Mater., 2016, 1, 16084 CrossRef CAS.
- G. Hu, et al., Functional inks and printing of two-dimensional materials, Chem. Soc. Rev., 2018, 47, 3265–3300 RSC.
- Y. Hernandez, M. Lotya, D. Rickard, S. D. Bergin and J. N. Coleman, Measurement of multicomponent solubility parameters for graphene facilitates solvent discovery, Langmuir, 2010, 26, 3208–3213 CrossRef CAS PubMed.
- U. Khan, A. O'Neill, M. Lotya, S. De and J. N. Coleman, High-concentration solvent exfoliation of graphene, Small, 2010, 6, 864–871 CrossRef CAS PubMed.
- G. Eda and M. Chhowalla, Chemically derived graphene oxide: Towards large-area thin-film electronics and optoelectronics, Adv. Mater., 2010, 22, 2392–2415 CrossRef CAS PubMed.
- S. Stankovich, et al., Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- M. Lotya, et al., Liquid phase production of graphene by exfoliation of graphite in surfactant/water solutions, J. Am. Chem. Soc., 2009, 131, 3611–3620 CrossRef CAS PubMed.
- A. Ciesielski and P. Samorì, Graphene via sonication assisted liquid-phase exfoliation, Chem. Soc. Rev., 2014, 43, 381–398 RSC.
- M. Lotya, P. J. King, U. Khan, S. De and J. N. Coleman, High-concentration, surfactant-stabilized graphene dispersions, ACS Nano, 2010, 4, 3155–3162 CrossRef CAS PubMed.
- R. J. Smith, M. Lotya and J. N. Coleman, The importance of repulsive potential barriers for the dispersion of graphene using surfactants, New J. Phys., 2010, 12(12), 125008 CrossRef.
- A. A. Green and M. C. Hersam, Solution phase production of graphene with controlled thickness via density differentiation, Nano Lett., 2009, 9, 4031–4036 CrossRef CAS PubMed.
- L. Guardia, et al., High-throughput production of pristine graphene in an aqueous dispersion assisted by non-ionic surfactants, Carbon, 2011, 49, 1653–1662 CrossRef CAS.
- A. B. Bourlinos, et al., Aqueous-phase exfoliation of graphite in the presence of polyvinylpyrrolidone for the production of water-soluble graphenes, Solid State Commun., 2009, 149, 2172–2176 CrossRef CAS.
- T. Skaltsas, et al., Graphene exfoliation in organic solvents and switching solubility in aqueous media with the aid of amphiphilic block copolymers, J. Mater. Chem., 2012, 22, 21507–21512 RSC.
- A. S. Wajid, et al., Polymer-stabilized graphene dispersions at high concentrations in organic solvents for composite production, Carbon, 2012, 50, 526–534 CrossRef CAS.
- A. Ghosh, K. V. Rao, S. J. George and C. N. R. Rao, Noncovalent Functionalization, Exfoliation, and Solubilization of Graphene in Water by Employing a Fluorescent Coronene Carboxylate, Chem. – Eur. J., 2010, 16, 2700–2704 CrossRef CAS PubMed.
- J. M. Englert, et al., Soluble Graphene: Generation of Aqueous Graphene Solutions Aided by a Perylenebisimide-Based Bolaamphiphile, Adv. Mater., 2009, 21, 4265–4269 CrossRef CAS.
- D. Parviz, et al., Dispersions of non-covalently functionalized graphene with minimal stabilizer, ACS Nano, 2012, 6, 8857–8867 CrossRef CAS PubMed.
- H. Yang, et al., A simple method for graphene production based on exfoliation of graphite in water using 1-pyrenesulfonic acid sodium salt, Carbon, 2013, 53, 357–365 CrossRef CAS.
- A. Schlierf, et al., Nanoscale insight into the exfoliation mechanism of graphene with organic dyes: effect of charge, dipole and molecular structure, Nanoscale, 2013, 5, 4205 RSC.
- Q. Su, et al., Composites of Graphene with Large Aromatic Molecules, Adv. Mater., 2009, 21, 3191–3195 CrossRef CAS.
- D. McManus, et al., Water-based and biocompatible 2D crystal inks for all-inkjet-printed heterostructures, Nat. Nanotechnol., 2017, 12, 1–19 CrossRef PubMed.
- W. Li, D. McManus, H. Liu, C. Casiraghi and S. J. Webb, Aqueous dispersions of nanostructures formed through the self-assembly of iminolipids with exchangeable hydrophobic termini, Phys. Chem. Chem. Phys., 2017, 19, 17036–17043 RSC.
- M. Zhang, et al., Production of graphene sheets by direct dispersion with aromatic healing agents, Small, 2010, 6, 1100–1107 CrossRef CAS PubMed.
- Y. Shin, et al., Charge-tunable graphene dispersions in water made with amphoteric pyrene derivatives, Mol. Syst. Des. Eng., 2019, 4(3), 503–510 RSC.
- R. J. Hunter, Zeta potential in colloid science : principles and applications. Colloid science, Academic Press, 1981, vol. 2 Search PubMed.
- R. Pecora, Dynamic Light Scattering: Applications of Photon Correlation Spectroscopy. Physics and Astronomy, Plenum Press, 1985. DOI:10.1007/978-1-4020-4465-6_13.
- A. Ciesielski, et al., Harnessing the Liquid-Phase Exfoliation of Graphene Using Aliphatic Compounds: A Supramolecular Approach, Angew. Chem., Int. Ed., 2014, 53, 10355–10361 CrossRef CAS PubMed.
- Y. Shin, et al., Synthesis and characterization of composite membranes made of graphene and polymers of intrinsic microporosity, Carbon, 2016, 102, 357–366 CrossRef CAS.
- S. Haar, et al., A supramolecular strategy to leverage the liquid-phase exfoliation of graphene in the presence of surfactants: Unraveling the role of the length of fatty acids, Small, 2015, 11, 1691–1702 CrossRef CAS PubMed.
- S. Haar, et al., Enhancing the Liquid-Phase Exfoliation of Graphene in Organic Solvents upon Addition of n-Octylbenzene, Sci. Rep., 2015, 5, 16684 CrossRef CAS PubMed.
- S. Conti, et al., Perchlorination of Coronene Enhances its Propensity for Self-Assembly on Graphene, ChemPhysChem, 2016, 17, 352–357 CrossRef CAS PubMed.
- J. C. Phillips, et al., Scalable molecular dynamics with NAMD, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and testing of a general amber force field, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- A. Pizzirusso, et al., Order and Conformation of Biphenyl in Cyanobiphenyl Liquid Crystals: A Combined Atomistic Molecular Dynamics and 1H NMR Study, ChemPhysChem, 2014, 15, 1356–1367 CrossRef CAS PubMed.
- D. J. Price and C. L. Brooks, A modified TIP3P water potential for simulation with Ewald summation, J. Chem. Phys., 2004, 121, 10096–10103 CrossRef CAS PubMed.
- J. Comer, et al., The Adaptive Biasing Force Method: Everything You Always Wanted To Know but Were Afraid To Ask, J. Phys. Chem. B, 2015, 119, 1129–1151 CrossRef CAS PubMed.
- Physicochemical properties of surfactants used in the definition of hydrophile-lipophile balance, in Studies in Interface Science, Elsevier, 2000, ch. 1, vol. 9, pp. 4–99 Search PubMed.
- A. Jorio, R. Saito, G. Dresselhaus and M. S. Dresselhaus, Raman Spectroscopy in Graphene Related Systems. Raman Spectroscopy in Graphene Related Systems, 2011. DOI:10.1002/9783527632695.
- A. C. Ferrari, et al., Raman spectrum of graphene and graphene layers, Phys. Rev. Lett., 2006, 97, 1–4 Search PubMed.
- C. Casiraghi, et al., Raman spectroscopy of graphene edges, Nano Lett., 2009, 9, 1433–1441 CrossRef CAS PubMed.
- V. Carozo, et al., Resonance effects on the Raman spectra of graphene superlattices, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 1–10 CrossRef.
- K. R. Paton, et al., Scalable production of large quantities of defect-free few-layer graphene by shear exfoliation in liquids, Nat. Mater., 2014, 13, 624–630 CrossRef CAS PubMed.
- C. Backes, et al., Guidelines for Exfoliation, Characterization and Processing of Layered Materials Produced by Liquid Exfoliation, Chem. Mater., 2017, 29, 243–255 CrossRef CAS.
- S. Harper, C. Usenko, J. E. Hutchison, B. L. S. Maddux and R. L. Tanguay, In vivo biodistribution and toxicity depends on nanomaterial composition, size, surface functionalisation and route of exposure, J. Exp. Nanosci., 2008, 3, 195–206 CrossRef CAS.
- B. Fadeel, et al., Safety Assessment of Graphene-Based Materials: Focus on Human Health and the Environment, ACS Nano, 2018, 12, 10582–10620 CrossRef CAS PubMed.
- P. Wick, et al., Classification Framework for Graphene-Based Materials, Angew. Chem., Int. Ed., 2014, 53, 7714–7718 CrossRef CAS PubMed.
- A. Schroeder, C. G. Levins, C. Cortez, R. Langer and D. G. Anderson, Lipid-based nanotherapeutics for siRNA delivery, J. Intern. Med., 2010, 267, 9–21 CrossRef CAS PubMed.
- T. Xia, M. Kovochich, M. Liong, J. I. Zink and A. E. Nel, Cationic Polystyrene Nanosphere Toxicity Depends on Cell-Specific Endocytic and Mitochondrial Injury Pathways, ACS Nano, 2008, 2, 85–96 CrossRef CAS PubMed.
- S. Vranic, et al., Live Imaging of Label-Free Graphene Oxide Reveals Critical Factors Causing Oxidative-Stress-Mediated Cellular Responses, ACS Nano, 2018, 12, 1373–1389 CrossRef CAS PubMed.
- V. C. Sanchez, A. Jachak, R. H. Hurt and A. B. Kane, Biological interactions of graphene-family nanomaterials: An interdisciplinary review, Chem. Res. Toxicol., 2012, 25, 15–34 Search PubMed.
- Y. Zhang, et al., Cytotoxicity effects of graphene and single-wall carbon nanotubes in neural phaeochromocytoma-derived pc12 cells, ACS Nano, 2010, 4, 3181–3186 CrossRef CAS PubMed.
- S. Gurunathan, J. W. Han, A. Abdal Dayem, V. Eppakayala and J. H. Kim, Oxidative stress-mediated antibacterial activity of graphene oxide and reduced graphene oxide in Pseudomonas aeruginosa, Int. J. Nanomed., 2012, 7, 5901–5914 CrossRef CAS PubMed.
- C. Xu and J. Wang, Delivery systems for siRNA drug development in cancer therapy, Asian J. Pharm. Sci., 2015, 10, 1–12 CrossRef.
- T. Ren, et al., Engineered polyethylenimine/graphene oxide nanocomposite for nuclear localized gene delivery, Polym. Chem., 2012, 3, 2561 RSC.
- H. Kim, R. Namgung, K. Singha, I.-K. Oh and W. J. Kim, Graphene Oxide–Polyethylenimine Nanoconstruct as a Gene Delivery Vector and Bioimaging Tool, Bioconjugate Chem., 2011, 22, 2558–2567 CrossRef CAS PubMed.
- G. Reina, A. Ruiz, D. Murera, Y. Nishina and A. Bianco, ‘Ultramixing’: A Simple and Effective Method To Obtain Controlled and Stable Dispersions of Graphene Oxide in Cell Culture Media, ACS Appl. Mater. Interfaces, 2019, 11, 7695–7702 CrossRef CAS PubMed.
- T. L. Moore, et al., Nanoparticle colloidal stability in cell culture media and impact on cellular interactions, Chem. Soc. Rev., 2015, 44, 6287–6305 RSC.
- A. Albanese and W. C. W. Chan, Effect of gold nanoparticle aggregation on cell uptake and toxicity, ACS Nano, 2011, 5, 5478–5489 CrossRef CAS PubMed.
- V. Hirsch, J. Salaklang, B. Rothen-Rutishauser and A. Petri-Fink, Influence of serum supplemented cell culture medium on colloidal stability of polymer coated iron oxide and polystyrene nanoparticles with impact on cell interactions in vitro, IEEE Trans. Magn., 2013, 49, 402–407 Search PubMed.
- J. C. Y. Kah, Stability and aggregation assays of nanoparticles in biological media, Methods Mol. Biol., 2013, 1025, 119–126 CrossRef CAS PubMed.
- A. M. Alkilany, et al., Misinterpretation in Nanotoxicology: A Personal Perspective, Chem. Res. Toxicol., 2016, 29, 943–948 Search PubMed.
- Y. Chen, et al., Dynamic interactions and intracellular fate of label-free GO within mammalian cells: role of lateral sheet size, bioRxiv, 2019, 805200, DOI:10.1101/805200.
- R. Li, et al., Surface Oxidation of Graphene Oxide Determines Membrane Damage, Lipid Peroxidation, and Cytotoxicity in Macrophages in a Pulmonary Toxicity Model, ACS Nano, 2018, 12, 1390–1402 CrossRef CAS PubMed.
- M. S. Ehrenberg, A. E. Friedman, J. N. Finkelstein, G. Oberdörster and J. L. McGrath, The influence of protein adsorption on nanoparticle association with cultured endothelial cells, Biomaterials, 2009, 30, 603–610 CrossRef CAS PubMed.
- A. M. Alkilany, et al., Cellular uptake and cytotoxicity of gold nanorods: Molecular origin of cytotoxicity and surface effects, Small, 2009, 5, 701–708 CrossRef CAS PubMed.
- M. Orecchioni, R. Cabizza, A. Bianco and L. G. Delogu, Graphene as cancer theranostic tool: Progress and future challenges, Theranostics, 2015, 5, 710–723 CrossRef CAS PubMed.
- J. M. Veranth, E. G. Kaser, M. M. Veranth, M. Koch and G. S. Yost, Cytokine responses of human lung cells (BEAS-2B) treated with micron-sized and nanoparticles of metal oxides compared to soil dusts, Part. Fibre Toxicol., 2007, 4(1), 2 CrossRef PubMed.
- G. Vales, L. Rubio and R. Marcos, Long-term exposures to low doses of titanium dioxide nanoparticles induce cell transformation, but not genotoxic damage in BEAS-2B cells, Nanotoxicology, 2015, 9, 568–578 CrossRef CAS PubMed.
- M. U. Farooq, et al., Gold Nanoparticles-enabled Efficient Dual Delivery of Anticancer Therapeutics to HeLa Cells, Sci. Rep., 2018, 8(1), 1–12 CrossRef CAS PubMed.
- E. Fröhlich, The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles, Int. J. Nanomed., 2012, 7, 5577–5591 CrossRef PubMed.
- S. Behzadi, et al., Cellular uptake of nanoparticles: journey inside the cell, Chem. Soc. Rev., 2017, 46, 4218–4244 RSC.
- A. Baeza-Squiban, S. Vranic and S. Boland, Fate and Health Impact of Inorganic Manufactured Nanoparticles, in Nanomaterials: A Danger or a Promise?, Springer London, 2013, pp. 245–267. DOI:10.1007/978-1-4471-4213-3_9.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of pyrene derivatives; MD simulation results; dispersion stability; AFM analysis; Raman analysis; TEM images; biocompatibility test. See DOI: 10.1039/d0nr02689a |
| ‡ Shared first authorship. |
| This journal is © The Royal Society of Chemistry 2020 |