
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoscale tailoring of supramolecular crystals via an oriented external electric field†
Xingming
Zeng‡
a,
Sadaf Bashir
Khan‡
 ab,
Ayyaz
Mahmood
ab and
Shern-Long
Lee
*a
ab,
Ayyaz
Mahmood
ab and
Shern-Long
Lee
*a
aInstitute for Advanced Study, Shenzhen University, Shenzhen, Guangdong, China 518060. E-mail: sllee@szu.edu.cn
bKey Laboratory of Optoelectronic Devices and Systems of Ministry of Education and Guangdong Provence, College of Optoelectronic Engineering, Shenzhen University, Shenzhen, Guangdong, China 518060
First published on 4th May 2020
Abstract
The oriented external electric field of a scanning tunneling microscope (STM) has recently been adapted for controlling the chemical reaction and supramolecular phase transition at surfaces with molecular precision. However, to date, advance controls using such electric-fields for crystal engineering have not been achieved yet. Here, we present how the directional electric-field of an STM can be utilized to harness supramolecular crystallization on a solid surface. We show that a glass-like random-tiling assembly composed of p-terphenyl-3,5,3′,5′-tetracarboxylic acid can transform into close-packed periodic assemblies under positive substrate bias conditions at the liquid/solid interface. Importantly, the nucleation and subsequent crystal growth for such field-induced products can be artificially tailored at the early stage in a real-time fashion. Through this method, we were able to produce a two-dimensional supramolecular single crystal. The as-prepared crystals with apparent brightness are ascribed to a spectroscopic feature linked to the electron density of states, which is thus strongly STM bias dependent.
Introduction
The ultimate goal of solid-state supramolecular chemistry is to harness molecular building blocks for surface patterning and utilize these resulting assemblies for a range of applications, such as surface modification, crystal engineering, and molecular electronics.1 In this context, surface supramolecular self-assembly, which deals with noncovalent-bonding interactions, develops a fundamental strategy for the generation of well-defined architectures with emergent functions. The assembling process and outcome not only depend on the intelligently designed molecules but also on the given experimental conditions, including sample concentration, substrate used, and external stimuli.2 These various motifs show molecular polymorphism in two-dimensional (2D) assemblies.Crystallization is at the heart of various scientific disciplines.3 Scanning tunneling microscopy (STM) is the most powerful tool to visualize 2D supramolecular polymorphic structures. This analytical tool helps in exploring detailed information about the dynamics of molecular self-assembly taking place at a liquid–solid interface.4 In recent years, relevant studies have unveiled essential knowledge, primarily promoting the progress of supramolecular crystal engineering. In this field, special attention has been given to the nucleation and 2D growth of a molecular crystal, elucidating the mechanism and formation of self-assembled molecular monolayers. In this context, the Curtin–Hammett principle suggests that a kinetically favored crystal would generate faster because the activation energy barrier to the state is lower in comparison with the thermodynamically favored one which takes a longer time span to form due to the higher activation barrier.5 Furthermore, the latter would be more stable since the final energy state is lower. Based on this principle, polymorphism may be less likely to occur if a crystalline form of a molecule is favored both kinetically and thermodynamically.5
The phenomenon of molecular polymorphs is ubiquitous; it is also true in surface supramolecular chemistry. To date, predicting crystal structures from just the molecular formula has remained a formidable challenge in contemporary science.6 While the 2D surface may provide a simplified platform for such explorations, the challenges associated with polymorphism are not alleviated.7 The resulting outcomes can be complicated even just for a structurally simple molecule. For example, De Feyter et al. reported that the symmetric building block of hexadecyl substituted bis(dehydrobenzo[12]annulene) can lead to four polymorphs depending on the sample concentration.8 Matzger et al. reported that 2D crystallization from an asymmetric building block of an amide amphiphile could give rise to six concentration-dependent networks.9 In general, when a higher concentration of a sample solution is used, the higher density assemblies on a surface were obtained. Generally, using a high-concentration sample solution, multiple small domains are obtained, ascribed to both the fast nucleation and growth of a molecular crystal.5
Polymorphism refers to the ability of a solid material including molecular building blocks to exist in more than one form or as a crystal structure. Currently, one of the important topics is the controlled formation of one type from at least two types of 2D supramolecular polymorphs.7 Besides concentration effect,10 external stimuli such as temperature, sonication, ion triggers, electrochemical potentials, and solvent flow have been explored for controlling supramolecular self-assembly, achieving polymorph selection.2,11–21 The external stimuli can also induce the generation of new polymorphic structures.22–24 This was extensively investigated in these works by STM with sub-molecular resolution.22–24 Recently, it has been reported that the direction-oriented electric-field of an STM presents its unique and powerful ability for controlling supramolecular phase transformations as well as chemical reactions at surfaces.25–29 Although an STM can be operated at the liquid–solid interface, the nucleation and controlled growth of supramolecular assemblies have been less explored. Using a molecular building block of p-terphenyl-3,5,3′,5′-tetracarboxylic acid (TPTC),30 we report that an external, oriented electric-field of an STM can be utilized to generate STM-bias-related new 2D self-assembled structures, which would be otherwise inaccessible by drop-casting or spin-coating methods.
Results and discussion
It is reported that the target molecule can self-assemble into glass-like random-tiling motifs presenting five types of pores including a different number of molecular networks namely A[triangle], B[parallelogram], C[arrow], D[semicircle], and E[star] at the octanoic acid (OA)/highly oriented pyrolytic graphite (HOPG) interface (Fig. S1, Table S1 in the ESI†). Such glass-like structures may result from the multiplicity of distinctive hydrogen-bonding junctions and the rearrangements taking place from the defects within the assemblies, as observed by Beton et al.30 Using this elegant system, here we demonstrate that it is possible to tailor the supramolecular crystallization and produce electric-stimulation-induced on-surface polymorphic crystals by tuning the orientation and magnitude of an STM electric field. Besides being an analytical tool, the stimuli and manipulations through STM are local, allowing us not only to visualize but also to manipulate the nucleation and growth of supramolecular assemblies in a real-time fashion.31,32 With this methodology, we were able to grow 2D supramolecular architectures in a bottom-up single-crystal manner.Molecules with carboxylic acid groups, due to their ability to undergo self-assembly through non-covalent hydrogen bonding, render themselves ideal building blocks for the synthesis of a variety of nanoscale architectures based on supramolecular interactions.33 Trimesic acid (TMA) represents a simple molecular building block, and its polymorphs on a solid surface have been extensively explored either by computational simulations or via scanning probe microscopy.26,34 The electric-field-induced phase transitions of TMA were observed previously yet found to occur rather sporadically.26 Recently, we have demonstrated both experimentally and theoretically that water can play a crucial role in assisting the electrically triggered phase transition of TMA.29 Herein, we chose TPTC as its self-assemblies were randomly constituted by glass-like motifs, which may have low stability and thus have been expected to readily undergo phase transformations upon being subjected to external stimuli. As anticipated, such reversible phase transformations electrically triggered by STM were readily observed. The presence or absence of water did not lead to any significant difference (Fig. S2 and S3†). Applying a positive STM bias to the sample surface can induce the occurrence of close packing. The random tiling can re-generate again when the STM bias was reversed to negative at the OA/HOPG interface (Fig. 1b and c). The sample solution was prepared by dilution to 10% concentration of the saturated solution. To our knowledge, the close-packed phases have not been previously reported yet. Our high-resolution STM images revealed that the packing axis of the enantiomeric polymorphs runs along with one of the normal directions of HOPG underneath, and the molecular skeleton is 60° from the packing axis (m line, Fig. 1c). The skeleton of TPTC lies approximately along another normal direction (n line, Fig. 1c). It includes a mirror image, R and S forms, in which molecular axes adsorb symmetrically, that is, along two normal directions of HOPG, namely n line which is 60° from the m line and 120° from the m line (Fig. 1c). Another packing, a head-to-head packing, is expected (Fig. 1a). For the three types of close packing, they have the same unit cell dimensions.
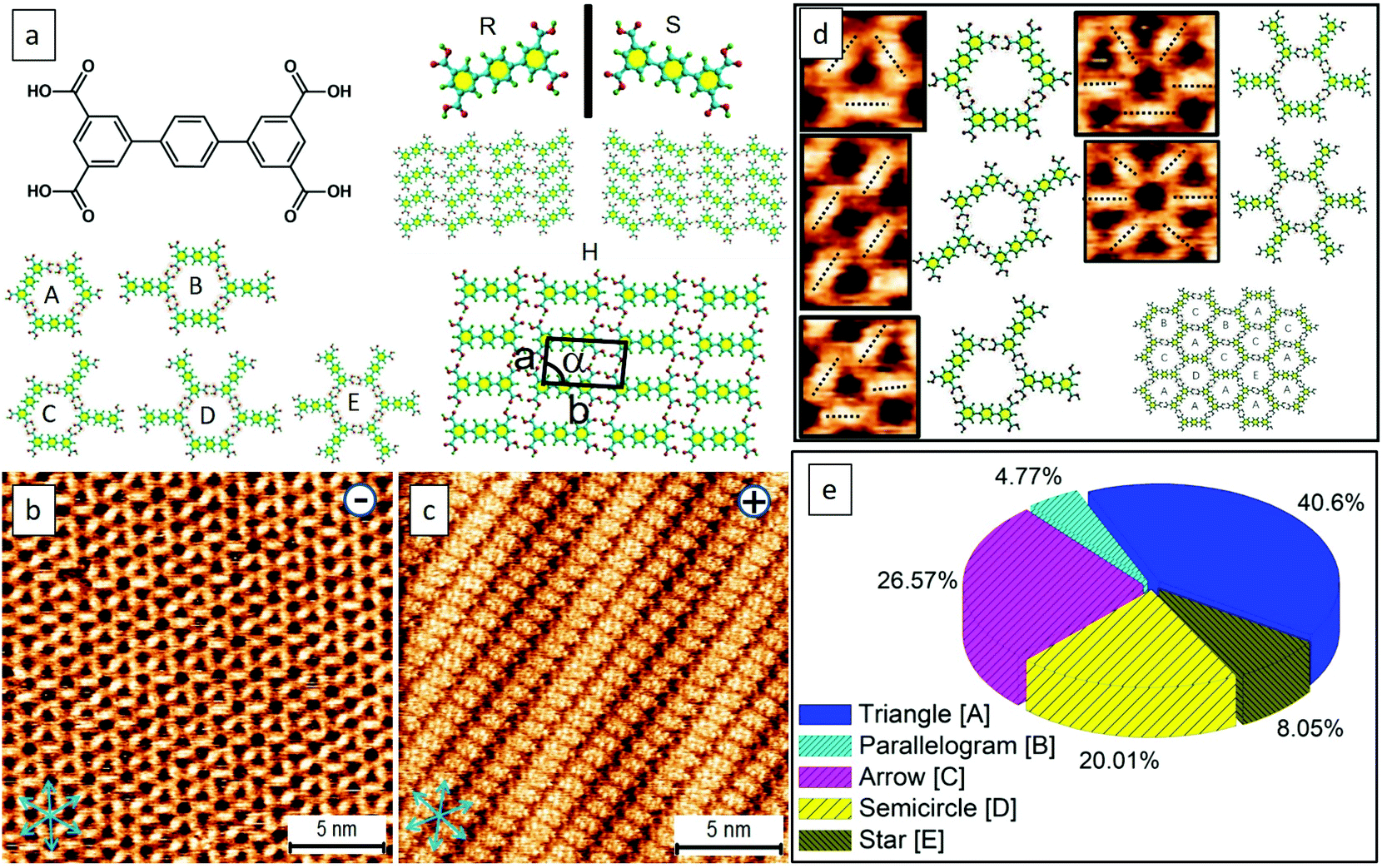 | ||
| Fig. 1 (a) Chemical structure of the target molecule of TPTC. All of the assembled types of TPTC at the OA/HOPG interface. (b,c) The close packed patterns are induced to generate at positive STM bias while the random tiling that includes cores from A to E appears at negative bias. The close-packed phase exists as a mirror image (R and S forms), and another one is expected to be an H (head-to-head) packing; all of them have the same unit cell dimensions. The packing axis of the close packing motif runs along one of the normal directions of HOPG underneath (m line) and a molecular skeleton is 60° from the packing axis so that TPTC lies approximately along another normal direction (n line). The blue arrows indicate the lattice structure direction of graphite. Imaging conditions (Ebias, itunneling) are 0.5 V and 70 pA. Panels (d) and (e) show the random tiling and the percentage of the five types of pores on the surface. Unit-cell parameters of a, b, and α: 0.9 (±0.3) nm, 1.7 (±0.3) nm, 87° (±2°). | ||
Scheme 1 illustrates the normal process of nucleation and the growth of a molecular crystal. In general, a molecular assembly results from nucleation seed(s) and, in turn, the subsequent crystal growth of the molecule, naturally producing multiple small domains oriented randomly at a flat atomic surface (e.g., HOPG). Considering the chirality of TPTC assemblies on the HOPG surface and taking into account the threefold symmetry of HOPG, three types of polymorphic forms and up to nine directions of packing in domains can exist on the surface. The significance of this work is to report a simple method for preparing a single-domain supramolecular assembly with nanometer precision based on the concept of the single-crystal-growth mode that is from a nucleation seed at its early stage and the tailored processes in crystal growth. This paper is organized as follows. Firstly, we present the normal formation of the close packing of TPTC at the liquid–solid interface under the control of the electric polarity of the substrate by selecting the sign of the STM bias. Secondly, we show how to manipulate the nucleation and subsequent growth of such field-induced self-assemblies to prepare supramolecular single crystals via real-time electric stimuli on a selected scan-area through STM. Meanwhile, we present how to use the local electric field for biasing the generation of one over other concomitant polymorphs. Finally, we discuss the mechanism underpinning the formation of the close packing of TPTC. We expect that this methodology for controlling surface molecular self-assembly toward supramolecular single crystals is universal for a wide range of molecular systems, especially for these molecules with carboxylic acid groups.
Fig. 2 shows the typical process of the molecular self-assembling of TPTC at a positive STM bias of 0.9 V at the OA/HOPG interface. The time-dependent STM images display the close-packed assemblies formed from fragmentary packing, namely, very small assemblies (Fig. 2a). Fig. 2b shows randomly oriented multiple small domains. The domain size of the packing is around 15 × 15 nm2 counted from 10 STM images with a scan size of 80 × 80, 100 × 100, and 200 × 200 nm2. More than 500 domains were analyzed carefully. Meanwhile, it is also noted that, within the margin of error, the percentage of the enantiomeric motifs on the surface was statistically equivalent (Fig. 2c). Fig. 2d exhibits another experimental session of an STM to show the consecutive process of yielding these typical assemblies. They are multiple small domains in general. Our experimental results demonstrate the presence of R and S form close packing; there are equal chances of their occurrence. Besides this, we also observe head-to-head close packing, which appears for a short period and seems to be a metastable state. We concluded this because, according to our experimental observations, head-to-head packing was absent eventually. Thus, we expected that it is relatively less stable, as described via models (Fig. 2e). It might have disappeared or transformed into a stable R or S form in the course of continuous STM scanning. The domain(s) of head-to-head close packing were not experimentally observed. The head-to-head orientation of TPTC only appears within R or S form assemblies (Fig. S2†).
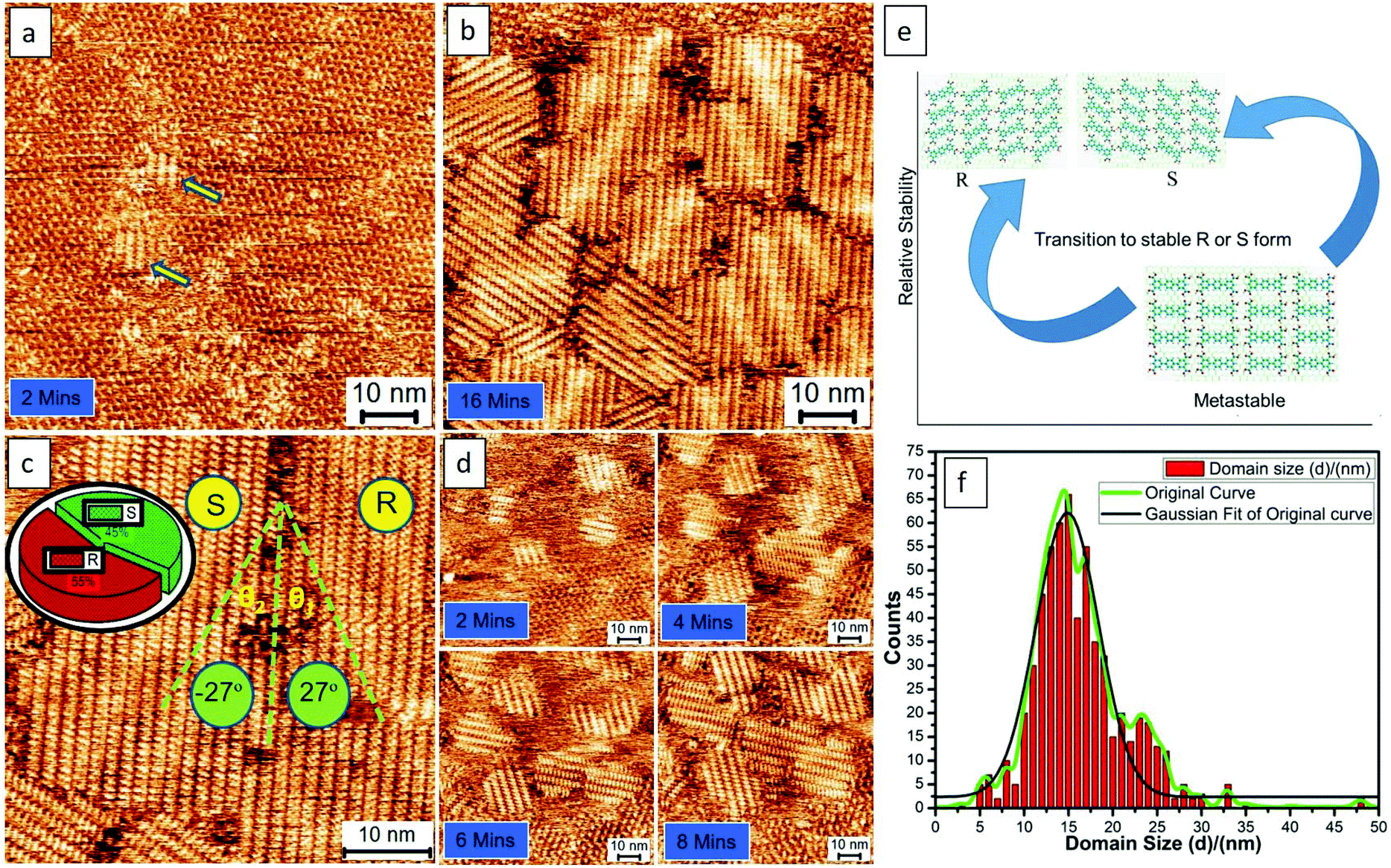 | ||
| Fig. 2 STM images and analyses showing the typical process of the molecular self-assembling of TPTC at positive STM bias at the OA/HOPG interface (0.9 V). The enantiomeric motifs which belong to R or S forms can be explicitly discriminated by the feature of the Moiré-pattern angle (e.g., R form: 27°; S form: −27° from the packing axis of the close packing). Imaging conditions (Ebias, itunneling) are ±0.9 V and 70 pA. Panels (a,b) and (d) exhibit the consecutive process of yielding typical assemblies with multiple small domains from 2 experimental sessions. Panels e and f show the relative stability of the R, S and head-to-head packing and the domain-size distribution of the motifs (bin size: 1.0 nm). | ||
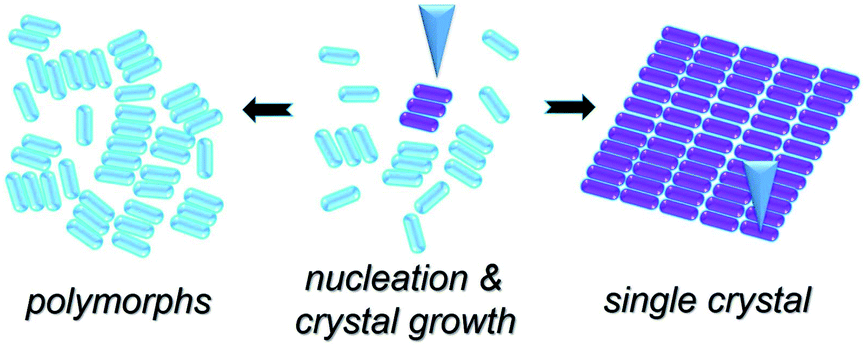 | ||
| Scheme 1 A scheme to show our method for preparing single crystals from selective nucleation and subsequent tailored growth of TPTC via STM. | ||
The feature of the Moiré-pattern angle can distinguish against the enantiomeric motifs which belong to R or S forms (e.g., R form: 27°; S form: −27° from the packing axis of the close packing, Fig. 2c).35 The contrast difference in an STM image depicts that the as-prepared crystals feature higher electric conductivity compared with that of the glass-like random-tiling assembly, which may result from the strong molecule–substrate interactions, namely, the Moiré-pattern effect. The as-prepared crystals with apparent brightness can also be ascribed to a spectroscopic feature linked to the electron density of states, which is thus strongly STM bias dependent. The histogram of the domain-size distribution is also presented (Fig. 2f), demonstrating a normal distribution, namely, the Gauss distribution. Experimentally, the crystal growth from these nucleation seeds was isokinetic, thereby exhibiting a specific domain size under the given experimental conditions.
After describing the packing types of the assemblies, we explore the possibility of tailoring them, aiming to enlarge their domain size and control their chirality. Based on the traditional principle and methods for preparing a single crystal, it is envisaged that using lower concentrations of a sample solution, increasing the environmental temperature or increasing high pressure may help to restrict nucleation or to develop the seed progression into crystal growth.18 However, we found that only the sample solutions having higher concentrations (e.g., sample-solution concentrations higher than 10% diluted from saturation) can generate the close-packed motifs under the positive STM bias conditions. Thus, the surface density of molecular assemblies is STM-bias dependent (Fig. S3 and S4†).
Using a hot sample solution pre-heated to 100 °C for the test, either the small-domain feature or the intrinsic chirality of the close packing in the assembly was observed. We reasoned that the assembling processes of the close packing were mainly controlled by the external oriented electric field of an STM. Thus, thermal annealing turns out to be unsuccessful for further tailoring. As such, we used a lower voltage bias of STM in our method. With this in mind and an intention to produce molecular assemblies with large domain size, we reversed the electric polarity of a substrate with STM from positive to negative to “clean out” the already-formed assemblies on a surface. By doing so and further lowering the positive voltage bias close packing can be re-generated. An example of the clean-out manipulation is presented in the ESI (Fig. S4†), demonstrating the reversibility of the patterning.
Fig. 3 shows the comparison results of the close packing formed using 0.9 V and 0.5 V. Remarkably, the assemblies formed via a lower voltage bias possess a larger domain size, by a factor around 2–3 times, than those obtained using a higher voltage bias. Although the extension of the molecular-assembly domain size was achieved using this strategy, the chiral separation or selection for the polymorphic phases was unsuccessful. In the case of low bias, the percentage of the enantiomeric motifs on the surface remains equivalent, and some misconnections between molecules exist in the domain boundaries, as indicated by a red circle in Fig. 3b.
 | ||
| Fig. 3 Using low STM bias to produce molecular assemblies with large domain size. Panels a and b show the assemblies produced by 0.9 V and 0.5 V, respectively. Clearly, lowering voltage bias produced assemblies with enlarged domain but the chiral separation or selection in polymorphic phases was unsuccessful. The misconnection between molecules takes place frequently in the domain boundaries as indicated by a red circle in panel (b). Imaging conditions (Ebias, itunneling) are 0.5 V, 70 pA. | ||
The analyses, as mentioned above, encouraged us to investigate further the use of a lower voltage bias for developing better controls. The close-packed motifs shown in Fig. 3b come from the process wherein 4–5 nucleation seeds were initially obtained, followed by subsequent crystal growth. Significantly, if a crystal formation results from single nucleation or a small amount of the TPTC aggregation with uniaxial and homo-chiral organization, it would be possible to construct a uniform single-domain assembly, which can be considered as a single crystal. Such assemblies can be considered virtually being produced via a single-crystal-growth mode and the products can be referred to as supramolecular single crystals. The subsequent crystal growth from a single nucleation seed is the key to success in synthesizing supramolecular single crystals. For TPTC, we have already found a suitable range of voltage bias that is from 0.16 V to 2.2 V to efficiently control nucleation and its crystal growth. Fig. 4 presents the occurrence of nucleation under such experimental conditions. Under 0.17 V, some aggregations occur as indicated by the arrows (Fig. 4a), and the electric stimuli are sufficient to proceed with their subsequent crystal growth. In our investigations, the minimum voltage was found to be 0.17 V to produce the close packing (Fig. 4b). Technically, the noteworthy mechanism is that when the small domains begin to appear, the electric stimuli should be focused on it locally (i.e., STM tip scans a small area, Fig. 4b) such that the growth of other undesired crystal domains can be cleverly circumvented.
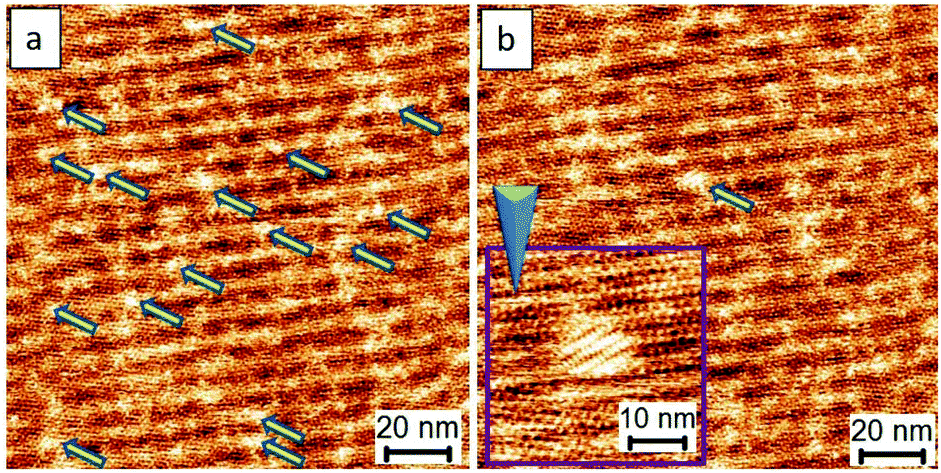 | ||
| Fig. 4 (a) and (b) demonstrates the control over initial nucleation and its early stage of the crystal growth for polymorph selection toward the formation of a supramolecular single crystal. Imaging conditions (Ebias, itunneling) are 0.17 V and 70 pA. | ||
Fig. 5 presents the subsequent growth of the close packing controlled with a selection from a nucleation seed presented in Fig. 4b. In principle, the strongest electric stimulation is near the center of an STM image, which is an area underneath the STM tip. An important step in our approach toward single crystal growth is that the STM scan size needs to initially start from a small zone. In our studies, we always start the scan size from a small area (e.g., changing the scan size from that in Fig. 5a to that in Fig. 5b). Later, we will extend the scan size to a large one gradually when the as-prepared assemblies grow and occupy the image spacing (e.g., changing the scan size from that in Fig. 5e to that in Fig. 5f and from that in Fig. 5g to that in Fig. 5h). An example of an uncontrolled scan, where mirror assemblies exist at the surface naturally, is already demonstrated in Fig. 3. During experimentation, we intentionally move our STM tip in the opposite direction (green arrows) to avoid the unwanted crystal growth (as indicated by blue arrows) and thus to avoid the formation of hetero-assemblies (Fig. 5c and f). Such simple manipulations were able to eliminate the occurrence of hetero-structure growth. This method can readily prepare supramolecular single-crystals with a domain size of at least 100 × 100 nm2. The as-prepared assemblies show a larger domain size, by a factor of ca. 10 times, compared with those obtained from the normal method (Fig. 2). The area which is out of the scan zone shows the absence of close packing, validating the nanoscale local manipulation of crystal growth (Fig. S5†).
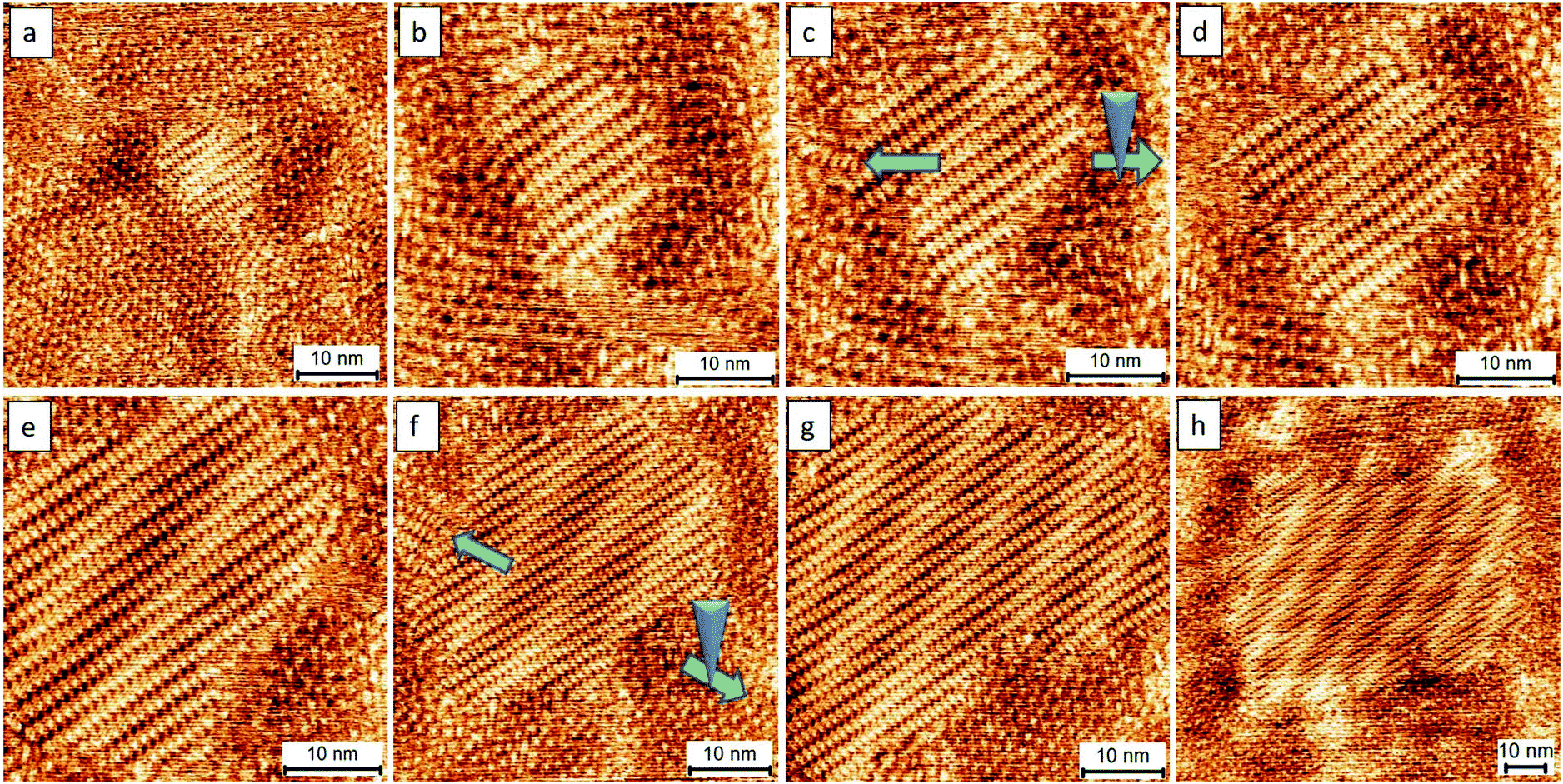 | ||
| Fig. 5 The consecutive images (a–h) showing the dynamics of the formation of a supramolecular single crystal. The green arrows indicate that the STM tip was deliberately moved in the opposite direction to avoid the growth of hetero-structures from the single-domain assembly. The preparation of the assembly took ca. 2 hours to create the pattern. Imaging conditions (Ebias, itunneling) are 0.17 V and 70 pA. | ||
Fig. 6a and b show the successful preparation of the R and S form single crystals, respectively (Fig. S6†). Based on our approach, they were readily prepared, demonstrating its potential and high efficiency for the preparation of a homo-chiral single crystal. The unbalanced enantiomeric domains result from the growth of one single domain of a given chiral orientation. Even though the polarity of STM can influence the packing manner at the liquid–solid interface, the close-packed motifs can stably exist on the surface for at least 24 hours and become less sensitive to the changes in the polarity of STM bias upon complete evaporation of the solvent (e.g., 6 hours later after drop-casting 5 μl sample solution using OA as the solvent). This is because of the limits of the degrees of the freedom of molecular flapping in a dried state, which makes these single-crystal patterns useful in further applications, for instance, in molecular electronics.
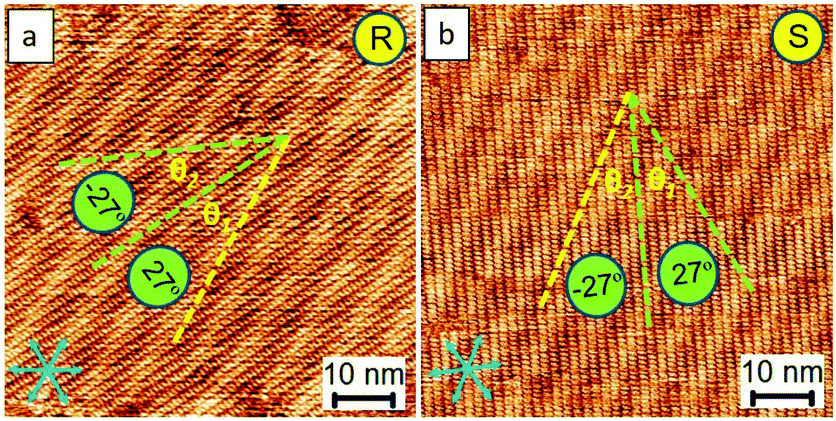 | ||
| Fig. 6 STM images show both R and S enantiomers in the single crystal fashion prepared via our approach. The enantiomeric motifs which belong to R or S forms can be explicitly discriminated by the feature of the Moiré-pattern angle (e.g., R form: 27°; S form: −27° from the packing axis of the close packing). The yellow lines in panels (a) and (b) indicate the directions parallel to the Moiré-pattern feature. The blue arrows indicate the lattice-structure direction of graphite. Imaging conditions (Ebias, itunneling) are 0.17 V and 70 pA. | ||
We now compare with the similarity for different target molecule systems. Trimesic acid (TMA), 1,3,5-tris(4-carboxyphenyl)-benzene (BTB), and the target molecule TPTC can be grouped because they are structurally decorated with carboxylic acid groups. Besides TPTC, we have thoroughly investigated and recently demonstrated that depending on the given experimental conditions, applying positive STM bias to the sample surface can induce either TMA or BTB molecules to undergo phase transition yielding the close-packed polymorphic assemblies. The electric-field triggered phase transition can be ascribed to the same mechanism, namely, the (partial) deprotonation of the functional groups of these molecules.34 The tendency of TPTC, TMA, and BTB under positive substrate bias is to maximize the utilization of the spacing of an HOPG surface leading to close packing that optimizes intermolecular interactions. However, STM revealed that the molecules adsorbed on the surface in a face-on orientation, ascribed to the strong π-attraction between the aromatics of these molecules and the surface of HOPG.
Based on thermodynamic viewpoints, adsorbates tend to maximize their interactions with the substrate, thereby leading to close packing.36 Thus, the tendency of the phase transition is to naturally occur from random packing to close packing(s) that includes head to head, R, and S form patterns. The possibility of kinetic control to be the main driving force for the resulting outcomes can be ruled out, as these assemblies are well-ordered. The ordered phases of the close packing are ascribed to the enthalpy control, whereas random tilling is mainly associated with the entropy control.37 When talking about the effect of the electrical polarity of the substrate, the dipole moment of the functional groups of the carboxylic acids of TPTC can be sensitive to the direction of the electric field of an STM. Therefore, under a positive STM bias, more molecules arise from solution to the gap between the STM tip and substrate and thus the tendency of close packing takes place.32 This is also consistent with the mechanism for the phase transitions, namely, the occurrence of the (partial) deprotonation of the functional groups under a positive STM bias. Similar results were concluded from the TMA systems in our recent work.34
Furthermore, the concentration or density of surface molecules can be controlled by the applied voltage bias; thus, only a low bias can produce supramolecular single crystals in our methodology (e.g., 0.17 V). However, it should be noted that close packing only occurs when high-concentration sample solutions were used. Besides this, in general low set points of tunneling current helps in obtaining better quality of STM images (Fig. S7†). The differences may result from the distance of an STM tip from the sample surface and thus the degree of perturbation caused by the tip. One more difference when compared with other methods is that an electrochemical controller can determine the potentials of a substrate, and thus can influence the global change of molecular patterning. Our present approach demonstrates the local effect, that is, an STM electric field can bias the crystal growth of one single domain from a given chiral orientation. Thus, this method itself is a unique tool for preparing homo-chiral assemblies on a surface. Note that our approach is in stark contrast to the traditional solution method for single crystal growth, where it is well-established that “the lower, the better” for the concentration parameter so that one can avoid the occurrence of hetero-crystal nucleation and epitaxial growth in crystal-growth processes. In our experiments, we frequently found that some fuzzy surfaces named “fast dynamics” were observed when the tendency of close packing occurred. This suggests that some randomly adsorbed molecules were at the state of the fast dynamic adsorption and desorption. Such fuzzy features were abundantly observed both in the nucleation and crystal growth process (e.g., the dots in Fig. 4 and the edge of close packing in Fig. 5).
The occurrence of nucleation of TPTC packing under 0.16 V is provided in the ESI† (Fig. S8†), where the early nucleation of 2–3 molecules was unveiled at the molecular resolution level using STM. The nucleation and early crystal growth were revealed after the appearance of the fuzzy surfaces, namely, the fast dynamics. The fuzzy feature usually represents the concentration of molecules at the surface at least higher than a critical concentration for generating close packing. Importantly, in the present work, the crystalline process toward the formation of supramolecular single crystals occurs based on the precise controls via the electric field of an STM rather than molecular aggregation due to the slow evaporation of solvents. Hence, in view of this aspect, our methodology is superior to traditional strategies.
Last but not least, we demonstrate that there is no significant difference in the results using other solvents such as heptanoic acid (HA) or 1-phenyloctane [261 °C to 263 °C] for this study. While HA [223 °C] with a lower boiling point in comparison to OA may evaporate faster, causing some small defects or enantiomeric phases (3–5%), taking advantage of the self-repairing ability based on the supramolecular re-assembly, the tailored architectures can virtually remain a homo-chiral single domain. Fig. 7a–c shows the sequential images enlightening the re-assembly dynamics of the self-repairing in the inner structure of the close packing. Fig. 7d shows the R-form single domain prepared from a solvent with a lower boing point, namely, HA compared with that of OA. The results are virtually the same (Fig. 6a, 7d) indicative of less solvent effect. Fig. 7e is a histogram showing that the enantiomeric phases (3–5%, e.g., in Fig. 7a) disappear after running STM for a long time, suggesting the self-healing ability of the thin films. In a nutshell, the organization can spontaneously repair itself, the so-called Ostwald ripening, at the liquid–solid interface. Together with the advantages of supramolecular self-assembly, our methodology presents an effortless means toward the preparation of supramolecular single crystals.
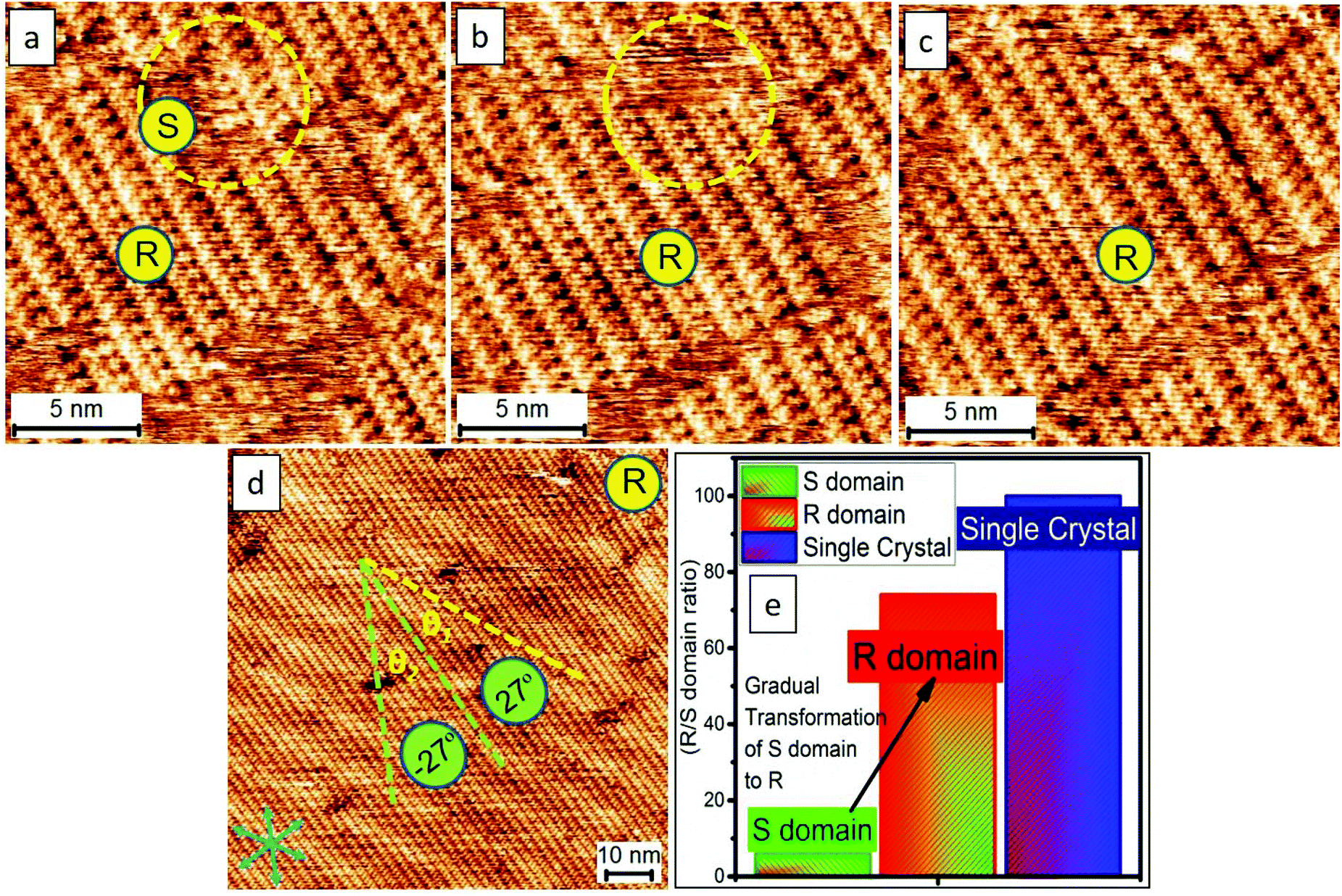 | ||
| Fig. 7 The consecutive images (a–c) of STM revealing the re-assembly dynamics of the self-repairing in the inner structure of the close-packing. We demonstrate that the homo-chiral close packing can be prepared easily which is, in part, benefited from the advantages of supramolecular self-assembly. The blue arrows in panel (d) indicate the lattice-structure direction of graphite. Imaging conditions (Ebias, itunneling) are 0.17 V and 70 pA. (e) Bar graph demonstrating the appearance of dominating phases generating a single crystal. | ||
Conclusions
In conclusion, we have reported an unprecedented, unconventional yet promising method to effortlessly prepare 2D supramolecular single crystals. While, in general, the molecular assemblies from some nucleation seeds and their subsequent crystal growth can produce multiple small domains randomly oriented on a surface, our methodology offers a powerful and reliable way to produce single-domain-size and homo-chiral assemblies. Using TPTC as an example, our method involves the manipulation of the nucleation and the following crystal growth via STM with its electric-polarity and scan-area determined at nanoscale precision. Although multiple nucleation seeds and, in turn, crystal growth can occur simultaneously under electric stimuli, it is possible to bias the crystal growth behavior of these small domains via local electric stimuli and manipulation using STM. We reasoned that these small assemblies were in their very early stages. Thus, one can accelerate a selected domain among others to grow specifically through scanning a zoomed-in area, including a targeted seed or small assembly. For other small domains that may result in polycrystalline structures, they can disappear due to their instability (i.e., in principle, the smaller-size assemblies have lower stability) or because of the reduction in electrical stimulation (i.e., area uncovered by the electric stimulation with STM resulting in the spontaneous collapse of small-domain assemblies or tentative aggregations). Moreover, we also showed that enantiomeric phases were present in the electric-stimulation-induced results. Our method is highly efficient for tailoring them into a homo-chiral single crystal via manipulating it in a preliminary stage (e.g., initial nucleation and bias the crystal growth via the electric field of an STM).11 We also discussed the mechanism as well as the details that can underpin our approach for efficient control over the tailored processes. Overall, based on the principle of supramolecular chemistry and the stimuli-responsive concept, we provide a novel strategy that can be useful for preparing 2D supramolecular single crystals, achieving molecular alignment and homo-chiral polymorph formation via a simple one-step process, namely, applying an external electric field through STM. The synergic effect combined with the electric manipulation and the advantages of molecular self-assembly including self-(re)organization and self-repairing makes our present methodology significant in the field of materials chemistry particularly supramolecular crystal engineering, beneficial, for example, for pharmaceutical industries, molecular electronics, and functional surfaces.Experimental
STM experiments were performed at an organic solvent/highly oriented pyrolytic graphite (HOPG) interface using a Keysight 5500 operating in the sample-biased and constant-current mode at room temperature (ca. 22 °C). The experiments were repeated several times. STM tips were mechanically cut Pt/Ir wires (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20%, diameter 0.25 mm). HOPG was purchased from Advanced Ceramics (ZYB grade). Imaging conditions of Ebias and iset ranged from −1.50 to 1.50 V (sample bias) and from 50 to 200 pA, respectively. The reported STM images were calibrated by the unit cell vectors of the underlying HOPG using either WSxM or SPIP software (scanning probe image processor, Image Metrology ApS). The chemicals were commercially available and were used as received, including p-terphenyl-3,5,3′,5′-tetracarboxylic acid (TPTC, ≥95%), octanoic acid (OA, ≥99%), heptanoic acid (HA, ≥99%), and 1-phenyloctane (PO, ≥98%). TPTC molecules were weighed and dissolved in organic solvents, and for all of the experiments, the sample concentration is 10% diluted from the saturated solution. A 5 μL sample solution in a liquid cell was used. We note that the sample volume lost is ca. 10–20% for one cycle of experiment (1 h). The modeled molecular packing structures were obtained using the HyperChem Professional 7.5 program based on the lattice-structure parameters. First, a molecular model for a single molecule was built, and then this model was duplicated. We constructed the model of the entire monolayer via placing the molecules following the intermolecular distances and angles obtained from calibrated STM images. The experimental errors were derived from five STM images. The imaging parameters are indicated in the figure caption: sample bias (Ebias) and tunneling current (itunneling). Fig. S9† explains how we measure the diameter for a domain size.
:20%, diameter 0.25 mm). HOPG was purchased from Advanced Ceramics (ZYB grade). Imaging conditions of Ebias and iset ranged from −1.50 to 1.50 V (sample bias) and from 50 to 200 pA, respectively. The reported STM images were calibrated by the unit cell vectors of the underlying HOPG using either WSxM or SPIP software (scanning probe image processor, Image Metrology ApS). The chemicals were commercially available and were used as received, including p-terphenyl-3,5,3′,5′-tetracarboxylic acid (TPTC, ≥95%), octanoic acid (OA, ≥99%), heptanoic acid (HA, ≥99%), and 1-phenyloctane (PO, ≥98%). TPTC molecules were weighed and dissolved in organic solvents, and for all of the experiments, the sample concentration is 10% diluted from the saturated solution. A 5 μL sample solution in a liquid cell was used. We note that the sample volume lost is ca. 10–20% for one cycle of experiment (1 h). The modeled molecular packing structures were obtained using the HyperChem Professional 7.5 program based on the lattice-structure parameters. First, a molecular model for a single molecule was built, and then this model was duplicated. We constructed the model of the entire monolayer via placing the molecules following the intermolecular distances and angles obtained from calibrated STM images. The experimental errors were derived from five STM images. The imaging parameters are indicated in the figure caption: sample bias (Ebias) and tunneling current (itunneling). Fig. S9† explains how we measure the diameter for a domain size.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the NSFC (21972095), Shenzhen University, Guangdong Government (2018A030313467), Shenzhen city (the overseas talent set-up funding; project: exploring multicomponent supramolecular systems, JCYJ2018-0305124732178); Shenzhen University and National Taipei University of Technology joint funding (2019008); and the China Postdoctoral Science Foundation (65th Batch-2019M653033) for their support.References
- S. Chen, P. Slattum, C. Wang and L. Zang, Chem. Rev., 2015, 115, 11967–11998 CrossRef CAS PubMed.
- J. A. A. W. Elemans, Adv. Funct. Mater., 2016, 26, 8932–8951 CrossRef CAS.
- M. Gemmi, E. Mugnaioli, T. E. Gorelik, U. Kolb, L. Palatinus, P. Boullay, S. Hovmöller and J. P. Abrahams, ACS Cent. Sci., 2019, 5, 1315–1329 CrossRef CAS PubMed.
- A. Ciesielski and P. Samori, Nanoscale, 2011, 3, 1397–1410 RSC.
- G. R. Desiraju, Nat. Mater., 2002, 1, 77–79 CrossRef CAS PubMed.
- C. A. Palma, M. Cecchini and P. Samori, Chem. Soc. Rev., 2012, 41, 3713–3730 RSC.
- S. L. Lee, J. Adisoejoso, Y. Fang, K. Tahara, Y. Tobe, K. S. Mali and S. De Feyter, Nanoscale, 2015, 7, 5344–5349 RSC.
- K. Tahara, S. Okuhata, J. Adisoejoso, S. Lei, T. Fujita, S. D. Feyter and Y. Tobe, J. Am. Chem. Soc., 2009, 131, 17583–17590 CrossRef CAS PubMed.
- S. Ahn and A. J. Matzger, J. Am. Chem. Soc., 2010, 132, 11364–11371 CrossRef CAS PubMed.
- S. Lei, K. Tahara, F. C. De Schryver, M. Van der Auweraer, Y. Tobe and S. De Feyter, Angew. Chem., Int. Ed., 2008, 47, 2964–2968 CrossRef CAS PubMed.
- I. Destoop, E. Ghijsens, K. Katayama, K. Tahara, K. S. Mali, Y. Tobe and S. De Feyter, J. Am. Chem. Soc., 2012, 134, 19568–19571 CrossRef CAS PubMed.
- S. L. Lee, Z. Yuan, L. Chen, K. S. Mali, K. Mullen and S. De Feyter, J. Am. Chem. Soc., 2014, 136, 7595–7598 CrossRef CAS PubMed.
- S. L. Lee, Z. Yuan, L. Chen, K. S. Mali, K. Mullen and S. De Feyter, J. Am. Chem. Soc., 2014, 136, 4117–4120 CrossRef CAS PubMed.
- T. Balandina, K. Tahara, N. Sandig, M. O. Blunt, J. Adisoejoso, S. Lei, F. Zerbetto, Y. Tobe and S. De Feyter, ACS Nano, 2012, 6, 8381–8389 CrossRef CAS PubMed.
- L. Piot, C. Marie, X. L. Feng, K. Mullen and D. Fichou, Adv. Mater., 2008, 20, 3854–3858 CrossRef CAS.
- B. E. Hirsch, K. P. McDonald, B. Qiao, A. H. Flood and S. L. Tait, ACS Nano, 2014, 8, 10858–10869 CrossRef CAS PubMed.
- B. E. Hirsch, S. Lee, B. Qiao, C.-H. Chen, K. P. McDonald, S. L. Tait and A. H. Flood, Chem. Commun., 2014, 50, 9827–9830 RSC.
- R. Gutzler, T. Sirtl, J. r. F. Dienstmaier, K. Mahata, W. M. Heckl, M. Schmittel and M. Lackinger, J. Am. Chem. Soc., 2010, 132, 5084–5090 CrossRef CAS PubMed.
- A. Ciesielski, S. Lena, S. Masiero, G. P. Spada and P. Samori, Angew. Chem., Int. Ed., 2010, 49, 1963–1966 CrossRef CAS PubMed.
- K. Cui, K. S. Mali, O. Ivasenko, D. Wu, X. Feng, M. Walter, K. Mullen, S. De Feyter and S. F. Mertens, Angew. Chem., Int. Ed., 2014, 53, 12951–12954 CrossRef CAS PubMed.
- S. Yoshimoto, T. Sawaguchi, W. Su, J. Jiang and N. Kobayashi, Angew. Chem., Int. Ed., 2007, 46, 1071–1074 CrossRef CAS PubMed.
- S. L. Lee, C. Y. J. Chi, M. J. Huang, C. h. Chen, C. W. Li, K. Pati and R. S. Liu, J. Am. Chem. Soc., 2008, 130, 10454–10455 CrossRef CAS PubMed.
- D. Rohde, C. J. Yan, H. J. Yan and L. J. Wan, Angew. Chem., Int. Ed., 2006, 45, 3996–4000 CrossRef CAS PubMed.
- K. S. Mali, D. Wu, X. Feng, K. Mullen, M. Van der Auweraer and S. De Feyter, J. Am. Chem. Soc., 2011, 133, 5686–5688 CrossRef CAS PubMed.
- Z. F. Cai, G. Zhan, L. Daukiya, S. Eyley, W. Thielemans, K. Severin and S. De Feyter, J. Am. Chem. Soc., 2019, 141, 11404–11408 CrossRef CAS PubMed.
- G. Velpula, J. Teyssandier, S. De Feyter and K. S. Mali, ACS Nano, 2017, 11, 10903–10913 CrossRef CAS PubMed.
- S. L. Lee, Y. Fang, G. Velpula, F. P. Cometto, M. Lingenfelder, K. Mullen, K. S. Mali and S. De Feyter, ACS Nano, 2015, 9, 11608–11617 CrossRef CAS PubMed.
- K. Y. Cheng, C. H. Lin, M. C. Tzeng, A. Mahmood, M. Saeed, C. h. Chen, C. W. Ong and S.-L. Lee, Chem. Commun., 2018, 54, 8048–8051 RSC.
- M. Saeed, A. Mahmood, A. S. Saleemi, X. Zeng and S. L. Lee, J. Phys. Chem. C, 2020, 124, 829–835 CrossRef CAS.
- M. O. Blunt, J. C. Russell, M. D. C. Giménez-López, J. P. Garrahan, X. Lin, M. Schroder, N. R. Champness and P. H. Beton, Science, 2008, 322, 1077–1081 CrossRef CAS PubMed.
- F. P. Cometto, K. Kern and M. Lingenfelder, ACS Nano, 2015, 9, 5544–5550 CrossRef CAS PubMed.
- J. Ubink, M. Enache and M. Stohr, J. Chem. Phys., 2018, 148, 174703 CrossRef CAS PubMed.
- M. Lackinger and W. M. Heckl, Langmuir, 2009, 25, 11307–11321 CrossRef CAS PubMed.
- A. Mahmood, M. Saeed, Y. Chan, A. S. Saleemi, J. Guo and S. L. Lee, Langmuir, 2019, 35, 8031–8037 CrossRef CAS PubMed.
- F. Silly, Nanotechnology, 2012, 23, 225603 CrossRef PubMed.
- U. Mazur and K. W. Hipps, Chem. Commun., 2015, 51, 4737–4749 RSC.
- S. Uemura, R. Tanoue, N. Yilmaz, A. Ohira and M. Kunitake, Materials, 2010, 3, 4252–4276 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr01946a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |