
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design-controlled synthesis of IrO2 sub-monolayers on Au nanoflowers: marrying plasmonic and electrocatalytic properties†
Isabel C.
de Freitas
a,
Luanna S.
Parreira
a,
Eduardo C. M.
Barbosa
 a,
Barbara A.
Novaes
a,
Tong
Mou
b,
Tiago. V.
Alves
c,
Jhon
Quiroz
d,
Yi-Chi
Wang
e,
Thomas J.
Slater
ef,
Andrew
Thomas
e,
Bin
Wang
b,
Sarah J.
Haigh
e and
Pedro H. C.
Camargo
*ad
a,
Barbara A.
Novaes
a,
Tong
Mou
b,
Tiago. V.
Alves
c,
Jhon
Quiroz
d,
Yi-Chi
Wang
e,
Thomas J.
Slater
ef,
Andrew
Thomas
e,
Bin
Wang
b,
Sarah J.
Haigh
e and
Pedro H. C.
Camargo
*ad
aDepartamento de Química Fundamental, Instituto de Química, Universidade de São Paulo, Avenida Prof. Lineu Prestes, 748, 05508-000 São Paulo, SP, Brazil
bCenter for Interfacial Reaction Engineering and School of Chemical, Biological, and Materials Engineering, Gallogly College of Engineering, The University of Oklahoma, Norman, Oklahoma 73019, USA
cDepartamento de Físico-Química, Instituto de Química, Universidade Federal da Bahia Rua Barão de Jeremoabo, 147, 40170-115, Salvador, BA, Brazil
dDepartment of Chemistry, University of Helsinki, A.I. Virtasen aukio 1, Helsinki, Finland. E-mail: pedro.camargo@helsinki.fi
eSchool of Materials, University of Manchester, Manchester M13 9PL, UK
fElectron Physical Sciences Imaging Centre, Diamond Light Source Ltd., Oxfordshire OX11 0DE, UK
First published on 14th April 2020
Abstract
We develop herein plasmonic–catalytic Au–IrO2 nanostructures with a morphology optimized for efficient light harvesting and catalytic surface area; the nanoparticles have a nanoflower morphology, with closely spaced Au branches all partially covered by an ultrathin (1 nm) IrO2 shell. This nanoparticle architecture optimizes optical features due to the interactions of closely spaced plasmonic branches forming electromagnetic hot spots, and the ultra-thin IrO2 layer maximizes efficient use of this expensive catalyst. This concept was evaluated towards the enhancement of the electrocatalytic performances towards the oxygen evolution reaction (OER) as a model transformation. The OER can play a central role in meeting future energy demands but the performance of conventional electrocatalysts in this reaction is limited by the sluggish OER kinetics. We demonstrate an improvement of the OER performance for one of the most active OER catalysts, IrO2, by harvesting plasmonic effects from visible light illumination in multimetallic nanoparticles. We find that the OER activity for the Au–IrO2 nanoflowers can be improved under LSPR excitation, matching best properties reported in the literature. Our simulations and electrocatalytic data demonstrate that the enhancement in OER activities can be attributed to an electronic interaction between Au and IrO2 and to the activation of Ir–O bonds by LSPR excited hot holes, leading to a change in the reaction mechanism (rate-determinant step) under visible light illumination.
Introduction
Plasmonic catalysis relies on harvesting the energy generated by localized surface plasmon resonant (LSPR) excitations in plasmonic nanoparticles to drive, accelerate, and/or control molecular transformations.1–6 Following LSPR excitation in plasmonic nanoparticles, non-radiative plasmon decay can lead to the formation of LSPR-excited charge carriers.7,8 These LSPR-excited hot electrons and holes can electronically or vibrationally excite molecular adsorbates at the metal–molecule interface via direct or indirect mechanisms.9,10 This can lead not only to improved reaction rates relative to the reaction in the absence of LSPR excitation, but also provide new reaction pathways for the control over reaction selectivity relative to traditional thermochemically-driven processes.11–15 Gold (Au) and silver (Ag) nanoparticles are amongst the strongest plasmonic structures, supporting LSPR excitation in the visible and near-infrared ranges with wavelengths that are tunable via the control of shape, size, composition and structure.16,17 Consequently plasmonic catalysis has emerged as an attractive approach for solar to chemical energy conversion,1,3,18–22 with Au, Ag, and aluminum (Al) nanoparticles having been applied as plasmonic catalysts towards a variety of molecular transformations under visible-light excitation.20,23–27Among several important chemical transformations, the water splitting reaction to produce hydrogen (H2) and oxygen (O2) has attracted massive attention for energy conversion and storage applications.28–31 Unfortunately, this reaction is limited by significant efficiency loss and high overpotentials (η) as a result of the sluggish kinetics for the oxygen evolution reaction (OER, which represents the oxidative half-reaction).32,33 It has been established that iridium and ruthenium oxides (IrO2 and RuO2, respectively) are among the best OER electrocatalysts, and IrO2 is often used in proton exchange membrane water electrolyzers as a result of its higher durability relative to RuO2.34,35 However, to meet our future energy demands there is an urgent need to develop materials displaying improved OER electrocatalytic activites. For example, iridium and ruthenium are expensive so bulk oxide materials do not make the most efficient use of the material. Furthermore, IrO2 and RuO2 both require relatively high overpotentials and a reduction in the overpotential, and therefore an improvement in the energy efficiency of the OER is highly desirable.36,37
In this context, we believe that the harvesting of plasmonic effects represents an attractive strategy for the improvement of OER performances.38–40 This approach has the potential to enable the use of solar light as an abundant and sustainable energy input to enhance OER rates. In fact, several plasmonic materials have been employed to enhance OER, hydrogen evolution reaction, and oxygen reduction reaction rates under light illumination.41–45 Unfortunately one of the best OER materials, RuO2 and IrO2, do not support LSPR excitation in the visible or near infrared ranges.46 However, the range of materials that support LSPR excitation in the visible range is limited to Ag, Au, Cu, Mg, and Al.8,47–49 In order to bridge the gap between materials with the desired catalytic and LSPR properties, the synthesis of multimetallic nanoparticle architectures that enable one to combine catalytic and plasmonic components (and thus catalytic and plasmonic properties) has emerged as an effective approach.11,50,51 In these plasmonic–catalytic nanoparticles, the goal is to use the plasmonic metal to harvest energy from light excitation, so that the generated LSPR charge-carriers can be transferred or dissipated to the surface of the catalytic material, where it can be further utilized to perform plasmon-driven chemistry.8,52
Inspired by this approach, we describe herein the development of a plasmonic–catalytic core–shell multimetallic nanostructure composed of Au and IrO2 as the plasmonic and catalytic components, respectively. More specifically, the synthesized Au–IrO2 plasmonic–catalytic nanoparticles displayed a tortuous flower-like morphology, in which several branches are closely spaced to each other and each branch is composed of Au partially covered by an ultrathin (1 nm) Ir-based shell. These features are very attractive to address the OER for a variety of reasons: (i) IrO2 represents one of the most active species towards the OER;53,54 (ii) the ultrathin and incomplete IrO2 shell at the surface of each branch maximizes the light harvesting by Au and the subsequent flow of charge carriers from Au to the Ir-based shell;55 (iii) the ultrathin and incomplete Ir-based shell maximises the IrO2 surface area and therefore minimizes the loading and utilization of this expensive metal; and (iv) the high curvature and plasmonic hybridization between the closely spaced plasmonic branches allows for the generation of a high density of electromagnetic hot spots, i.e., areas of high electric field enhancements at the junctions of the branches as a result of the LSPR excitation that can be felt by the catalytic Ir-based shells.11,50 These high electric field enhancements, for instance, can decay via absorption and lead to the formation of energetic charge carriers (hot electrons and holes) at the IrO2, as described in antenna-reactor complex nanoparticle designs.12,50,56,57 The success of this approach is demonstrated by the significant improvement in the OER activity for the multimetallic Au–IrO2 nanoflowers due to LSPR excitation with visible light. The effect of the light excitation in determining the OER activity supports the role of the LSPR excitation in this transformation. Our data demonstrates that electronic/charge transfer interaction between Au and IrO2 and the activation of Ir–O bonds by the LSPR excited hot holes contributes to the catalysts improved performance under visible light irradiation.
Results and discussion
Plasmonic–catalytic nanostructures have been synthesized in which the plasmonic and catalytic components are in direct contact and where the two components are separated by a small distance (∼up to 5 nm).12,50,51,58,59 Both these scenarios can allow for the acceleration of reaction rates and control over reaction selectivity over the surface of the catalytic metal8,11,60,61 but the two architectures have different benefits.51 In nanoparticle designs in which plasmonic and catalytic components are not in direct contact, it has been demonstrated that the catalytic component may be exposed to regions of local electric fields induced by the LSPR excitation of the plasmonic metal, enhancing catalytic activity.10,12,56,62 In contrast with nanostructures where the catalytic and plasmonic components are in direct contact, electronic effects and charge flow from the plasmonic to the catalytic component occurs upon LSPR excitation.11,50,57 Inspired by these recent findings, we have developed nanoparticle architectures in which the plasmonic and catalytic components are in direct contact by using Au as the plasmonic component, IrO2 as the catalytic component, and adopting a complex core–shell nanoflower morphology.The synthesis of the multimetallic Au–IrO2 nanoflowers was performed by the co-reduction of Au and Ir precursors (AuCl4− and IrCl3·xH2O, respectively) in the presence of sodium citrate as both reducing agent and stabilizer.55Fig. 1A–E shows scanning electron microscope (SEM) images (Fig. 1A), high-angle annular dark field (HAADF) scanning transmission electron microscope (STEM) images (Fig. 1B and D), and STEM energy dispersive X-ray spectroscopy (EDX) elemental maps (Fig. 1C and E) for the Au–IrO2 nanoflowers obtained by this approach. Here, the molar ratio between the AuCl4− and IrCl3·xH2O precursors employed during the synthesis corresponded to AuCl4−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :IrCl3·xH2O 1:1.5. It can be observed from the SEM images (Fig. 1A) that the nanoflowers displayed an overall spherical morphology with a highly tortuous branched surface structure. The nanoflower particles have an overall average diameter corresponding to 93.2 ± 9 nm and thus a relatively monodisperse size distribution. The branched morphology is even more apparent in the HAADF-STEM images shown in Fig. 1B and D. These images indicate that the branches are closely spaced and each branch has an approximately spherical cross section with a diameter of around 5 nm. HRTEM imaging confirms the nanoflowers are polycrystalline with all the observed lattice spacings being assigned to fcc Au (Fig. S1†). This is further confirmed by the electron diffraction pattern from an individual nanoflower as shown in Fig. S2.†
:IrCl3·xH2O 1:1.5. It can be observed from the SEM images (Fig. 1A) that the nanoflowers displayed an overall spherical morphology with a highly tortuous branched surface structure. The nanoflower particles have an overall average diameter corresponding to 93.2 ± 9 nm and thus a relatively monodisperse size distribution. The branched morphology is even more apparent in the HAADF-STEM images shown in Fig. 1B and D. These images indicate that the branches are closely spaced and each branch has an approximately spherical cross section with a diameter of around 5 nm. HRTEM imaging confirms the nanoflowers are polycrystalline with all the observed lattice spacings being assigned to fcc Au (Fig. S1†). This is further confirmed by the electron diffraction pattern from an individual nanoflower as shown in Fig. S2.†
 | ||
| Fig. 1 (A–E): SEM (A), HAADF-STEM (B and D) and STEM-EDX (C and E) images of Au–IrO2 nanoflowers obtained by co-reduction of AuCl4− and IrCl3·xH2O precursors in a 1:1.5 molar ratio. The Au and Ir at% in the samples corresponded to 85 and 15, respectively. (F–J): SEM (F), HAADF-STEM (G and I) and STEM-EDX (H and J) images for Au–IrO2 nanoflowers obtained by co-reduction of AuCl4− and IrCl3·xH2O precursors in a 1:0.25 molar ratio. The Au and Ir at% in the samples corresponded to 96 and 4, respectively. The elemental distributions for Au and Ir are shown in red and green, respectively, in the STEM-EDX maps. Where both green and red signals are overlapping the colour map appears yellow. | ||
The STEM-EDX elemental mapping shown in Fig. 1C and E indicates that the nanoflower is composed of a core–shell morphology with a Au core covered by an ultrathin (1 nm or thinner) Ir rich surface layer. Closer inspection of the high-resolution elemental map (Fig. 1E) demonstrates that the Ir-based ultrathin layer does not completely cover the surfaces of the Au branches (Fig. S3† shows an additional STEM-EDX high resolution elemental map where regions of exposed Au surface are highlighted). The overall nanoparticle composition from the STEM-EDX compositional maps in Fig. 1C corresponded to Au 87 at% and Ir 13 at% which is in good agreement with the value obtained from flame atomic absorption spectroscopy (FAAS) (Au 86 at% and Ir 14 at%).
In order to investigate the effect of the molar ratio of AuCl4− and IrCl3·xH2O precursors used during the synthesis on the morphological and compositional features of the nanoflowers, we varied the precursor molar ratios in the co-precipitation synthesis to AuCl4−:IrCl3·xH2O 1:0.25 (previously 1:1.5). Fig. 1F–J shows the SEM (Fig. 1F) and HAADF-STEM (Fig. 1G and I) images and STEM-EDX elemental maps (Fig. 1H and J) for the resulting nanoflowers. A similar morphology as described in Fig. 1A–E is produced; nanoflowers with an overall spherical shape, a relatively monodisperse size distribution, closely spaced branches, and with each branch comprised of Au partially covered by an ultrathin and incomplete Ir-rich shell. However, two important differences were detected. As expected the nanoflowers contained less Ir, with an elemental composition of Au 96 at% and Ir 4 at% (data extracted from the STEM-EDX in Fig. 1H, which agrees with the values obtained from FAAS for these samples, corresponding to Au 95 at% and Ir 5 at%). In addition, the individual branches were more elongated, having a rod-like morphology, relative to the nanoflowers shown in Fig. 1A–E. This more-elongated, rod-like morphology for the branches can be also visualized by additional HAADF-STEM images and STEM-EDX elemental maps shown in Fig. S4.†
The XRD diffractograms obtained for the nanoflowers prepared employing AuCl4−:IrCl3·xH2O 1:1.5 and 1:0.25 molar ratios are shown in Fig. 2A (red and black traces, respectively). The results agree with the compositional and morphological variations in the samples. With the decrease in the IrCl3·xH2O precursor content employed in the synthesis, a decrease in the Ir content led to an increase in the intensity of the reflections assigned to fcc Au. Moreover, the XRD peaks from the fcc Au became less broad, consistent with the increased length of the dendrite branches observed by STEM, which will increase the size of the Au crystallites. Fig. S5† shows the XRD for Au–IrO2 having varying, intermediate compositions, which confirm this assignment. No peaks assigned to Ir or IrO2 phases could be detected in the samples, which is not unexpected given the ultrathin thickness of the Ir-based surface layer. Moreover, no XRD peaks at all were detected for a control sample prepared under identical conditions but in the absence of AuCl4− precursor (blue trace).
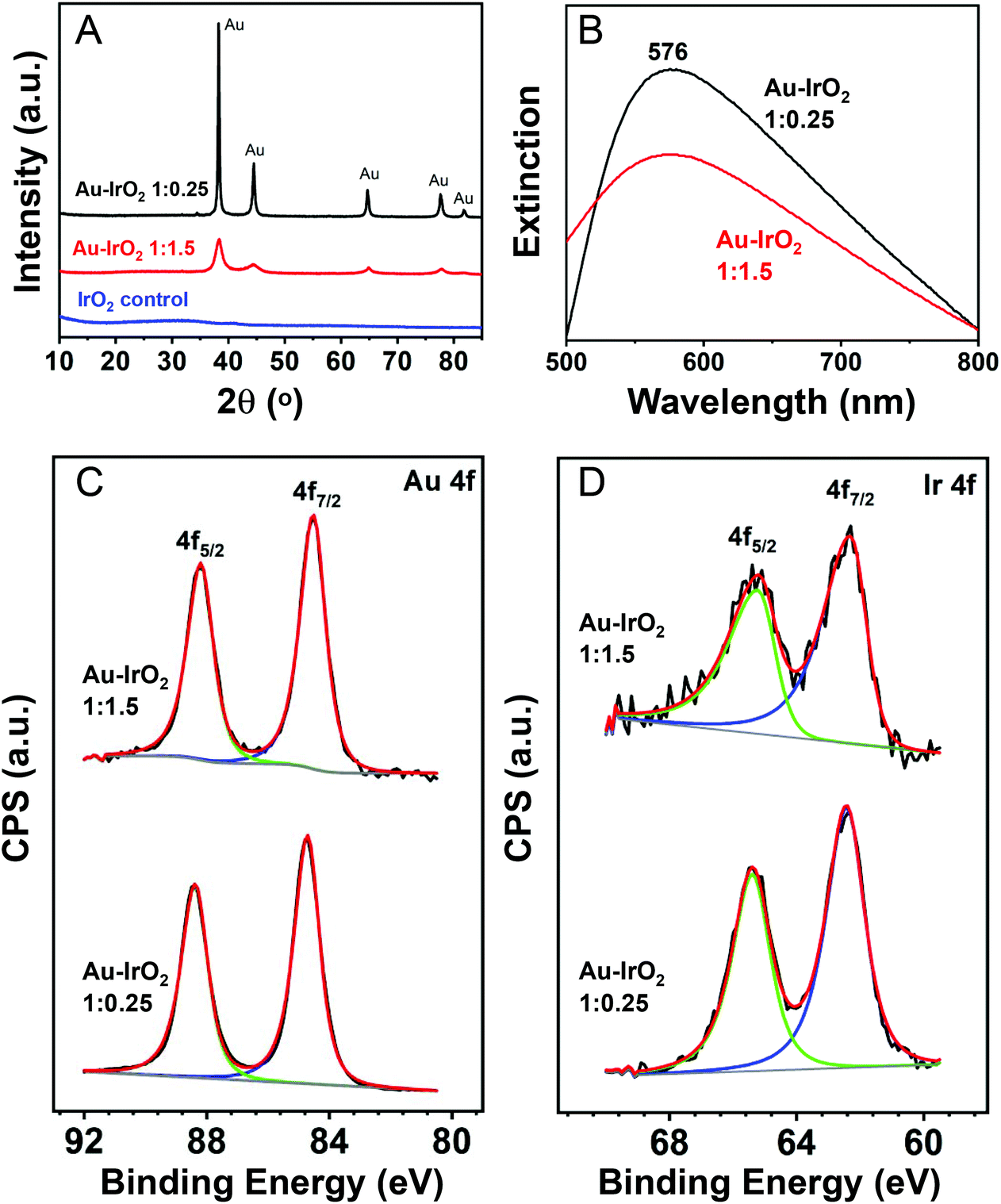 | ||
| Fig. 2 XRD diffractograms (A) and UV-VIS extinction spectra (B) recorded for Au–IrO2 nanoflowers obtained by co-reduction of AuCl4− and IrCl3·xH2O as precursors in a 1:1.5 (red trace) and 1:0.25 (black trace) molar ratios. (C) and (D) show the deconvoluted X-ray photoelectron spectra (XPS) of the Au 4f (C) and Ir 4f (D) core levels for the Au–IrO2 nanoflowers obtained by co-reduction of AuCl4− and IrCl3·xH2O as precursors in a 1:1.5 (top trace) and 1:0.25 (bottom trace) molar ratios. | ||
Fig. 2B depicts the UV-VIS extinction spectra recorded from aqueous suspensions containing the nanoflowers. It can be observed that both samples prepared employing AuCl4−:IrCl3·xH2O 1:1.5 and 1:0.25 molar ratios (red and black traces, respectively) displayed an extinction peak in the visible-range centered at 576 nm. A decrease in the extinction intensity for the sample with higher Ir content (AuCl4−:IrCl3·xH2O 1:1.5) was observed. This agrees with the fact that the larger Ir concentration in the samples prepared employing AuCl4−:IrCl3·xH2O 1:1.5 can lead to a stronger suppression of the LSPR excitation (absorption and scattering) from the plasmonic (Au) component relative to the sample prepared employing AuCl4−:IrCl3·xH2O 1:0.25 molar ratios (lower Ir content).63 Au NPs (obtained in the absence of IrCl3·xH2O precursor) displayed the characteristic LSPR dipolar band at 527 nm (Fig. S6A†),16 while IrO2 obtained in the absence of AuCl4− precursor displayed no bands in the visible region (Fig. S6B†).
To study the surface composition, we also characterized the samples by XPS. Fig. 2C and D depict the photoemission spectra in the Au 4f (Fig. 2C) and Ir 4f (Fig. 2D) core level regions for the nanoflowers prepared employing AuCl4−:IrCl3·xH2O 1:1.5 (top trace) and 1:0.25 (bottom trace) molar ratios. A summary of the binding energy (BE) values and calculated surface composition is shown in Table 1. The Au 4f region showed two intense photoelectron peaks with maxima at BE of 84 and 88 eV, ascribed to Au 4f7/2 and Au 4f5/2, respectively. These values are consistent with the presence of Au species in the metallic state.64 However, a slight negative shift from 88.4 and 84.7 eV to 88.2 and 84.5 eV in the samples prepared under AuCl4−:IrCl3·xH2O 1:0.25 and 1:1.5 molar ratios, respectively, was observed. These variations can be ascribed to intrinsic charge transfer between IrO2 and Au, which was more pronounced when the amount of IrO2 in the material was higher (1:1.5 sample). In the Ir 4f core level region, the XPS spectra displayed two photoelectron emission peaks at binding energies of around 62 and 65 eV (Fig. 2D and Table 1), corresponding to the Ir 4f7/2 and 4f5/2 states, respectively. The peak position and the asymmetry of the peaks evidences the presence of oxidized Ir species (Ir4+) consistent with the formation of IrO2 since the BE values reported for metallic Ir and IrO2 standards are 61.1/64.1 eV and 62.7/65.7 eV, respectively.65,66 The detected shifts in the Ir peak position in the two samples were very small, and both values are close to what is observed in IrO2 materials.67–70 The CV profiles (described later) also are in agreement with the presence of IrO2.67–70 This is important since STEM-EDS elemental mapping could not confirm whether the Ir was present as metallic Ir or IrO2 due to oxygen signal being overwhelmed by the presence of oxygen containing surface adsorbates. Finally, the surface composition estimated from the XPS spectra and Ir/Au ratio (Table 1) revealed an increase in the Ir content at the surface as the amount IrCl3·xH2O precursor relative to AuCl4− was increased during the synthesis. The Ir contents in Table 1 are both higher than the from STEM-EDX and FAAS, further demonstrating that the Ir exists as a surface layer.
| Au–IrO2/AuCl4−:IrCl3·xH2O molar ratios |
Au 4f (eV) | Ir 4f (eV) | Surface composition | ||||
|---|---|---|---|---|---|---|---|
| 4f5/2 | 4f7/2 | 4f5/2 | 4f7/2 | Au (%) | Ir (%) | Ir/Au | |
| 1:0.25 |
88.4 | 84.7 | 65.4 | 62.4 | 92.0 | 8.0 | 0.09 |
| 1:1.5 |
88.2 | 84.5 | 65.2 | 62.3 | 80.5 | 19.5 | 0.24 |
In the synthesis of the nanoflowers shown in Fig. 1, the Au and Ir precursors are firstly mixed at room temperature, which is accompanied by a change in the color of the solution from to green to blue. This change in color indicates the reduction of AuCl4− species to AuCl2− by IrCl63− (leading to the formation of Ir4+ species).55 In this case, we postulate that, as AuCl2− has a higher standard reduction potential relative to AuCl4−, this species is mainly responsible for the formation of the Au nuclei during the synthesis. Then, as further Au and Ir are produced from the reduction of precursors (co-reduction), growth takes place by precursor addition at the surface of the pre-formed nuclei/seeds as well as by their aggregation in the presence of citrate, which produces the so-called nanoflowers morphology observed herein. It is important to note that an oriented attachment mechanism has been previously reported during the formation of Au nanostructures in the presence of citrate, and has been recently been confirmed by liquid cell TEM studies.71,72
We performed a series of experiments in order to confirm this proposed mechanism. We started by monitoring the optical properties of the precursor solutions before and after they are mixed at room temperature. Fig. S7A† shows the absorption spectra for the AuCl4− and IrCl3·xH2O precursor solutions (red and blue traces, respectively). While AuCl4− has an absorption band at 310 nm, only an intense signal below 290 nm was detected for the IrCl3·xH2O solution. Therefore, we employed the band at 310 nm to monitor the AuCl4− reduction when the AuCl4− and IrCl3·xH2O precursors were mixed. Fig. S7B and C† show the UV-VIS spectra for the mixtures of the precursor solutions in the AuCl4−:IrCl3·xH2O 1:1.5 and 1:0.25 molar ratios, respectively (which corresponded to the synthesis conditions described in Fig. 1). In each case, the UV-VIS spectra were recorded at 5 s intervals following the mixture of the precursors. It can be observed that, in both cases, as the precursors were mixed, a gradual disappearance of the band assigned to AuCl4− was detected. This agrees with the AuCl4− being reduced to AuCl2−.73,74 No LSPR peaks assigned to Au nanoparticles were observed, indicating that no further reduction to Au0 takes place simply by mixing the precursors. Interestingly, under otherwise identical conditions, the AuCl4− reduction kinetics was strongly dependent on the amount of IrCl3·xH2O precursor employed during the synthesis. The AuCl4− reduction was much faster with a higher IrCl3·xH2O content and we postulate that this increased reduction kinetics favored in an increase in the number of Au nuclei, and hence a reduction in the overall size of the nanoflowers. In support of this hypothesis, we found it was possible to fine tune the size of the nanoflowers by controlling the AuCl4−:IrCl3·xH2O molar ratio during synthesis. Experiments employing AuCl4−:IrCl3·xH2O molar ratios of 1:1.25, 1:1, 1:0.75, and 1:0.5 led to nanoflowers having diameters of 98 ± 8, 101 ± 7, 116 ± 7, 133 ± 7, respectively (Fig. S8†).
We also studied the kinetics of the reaction by studying the nanoflowers formed at different time intervals following the addition of the solution containing the mixture of Ir and Au precursors to the boiling citrate solution. This was performed by removing aliquots from the reaction mixture, immersing in an ice bath, and isolating/washing the nanostructures by successive rounds of centrifugation and removal of the supernatant. SEM images for the Au–IrO2 nanoflowers extracted from the reaction mixture after 15 s, 1 min, 5 min, 10 min, and 20 min are shown in Fig. S9.† The flower-like morphology can already be observed in the products obtained after 15 s of synthesis. Moreover, the SEM images show that the nanoflowers gradually increase in size as a function of time, which agrees with a mechanism based on agregation.75 It is important to note that the nanoflower morphology was not observed when the synthesis was performed in the presence of only AuCl4− or IrCl3·xH2O precursors (Fig. S10†). In this case, spherical Au particles or irregular Ir-based materials were obtained. This result indicates that the presence of both precursors is required for formation of the nanoflower morphology, in agreement with the proposed mechanism based on the formation of AuCl2−, by IrCl3·xH2O, which then reduces to Au nuclei during synthesis.
It is also interesting to investigate how the choice of citrate as a stabilizer affects the morphology. Oriented attachment in Au nanocrystals stabilized by citrate has been reported to occur with attachment observed preferentially for {111} orientations71,72 Images of the products obtained under similar conditions but replacing citrate by PVP, hydroquinone, ascorbic acid, or borohydride led to the formation of irregular particles (Fig. S11†). This indicates that citrate, in addition to being a reducing and capping agent, is also an essential component for formation of the nanoflower morphology, playing a key role in their formation via aggregation in suspension.
Finally, the Au–IrO2 nanoflowers were employed as model systems to investigate how plasmonic effects in Au can be harnessed towards the enhancement of the electrocatalytic activity of IrO2 towards the OER. The plasmonic effects over electrocatalytic performance were studied in O2-saturated 0.1 M KOH electrolyte at room temperature via a typical three-electrode system at a scan rate of 10 mV s−1 at 532 nm irradiation (200 mW). Fig. 3A shows linear sweep voltammetry (LSV) tests for the nanoflowers obtained with 1:1.5 and 1:0.25 AuCl4−:IrCl3·xH2O molar ratios in the presence of light excitation (solid lines) and in the absence of light (dashed lines) (CVs are shown in Fig. S12†). The measured current for the bare glassy carbon electrode is also shown for comparison (black trace). It can be observed that the Au–IrO2 1:1.5 sample displayed higher current densities and an earlier onset potential relative to Au–IrO2 1:0.25, which can be ascribed to its superior OER activity associated with the higher Ir loading in this material. Importantly, when irradiated at 532 nm, the OER is accelerated in both samples. Here, a significant decrease in onset potential and increase in the detected current densities was detected under visible light illumination, assigned to the plasmonic enhancement of the OER.40 Specifically, the detected potentials required to achieve a current density of 10 mA cm−2, which represent an important metric in the solar synthesis of fuels, decreased from 0.72 to 0.69 for the Au–IrO2 1:1.5 sample under visible light illumination. For Au–IrO2 1:0.25, a decrease from 0.85 to 0.78 V took place. Considering the EOER value of 0.404 V in alkaline solution, the calculated overpotential at 10 mA cm−2 for Au–IrO2 1:1.5 and Au–IrO2 1:0.25 samples corresponded to 286 and 376 mV, respectively. Table 2 summarises the reported record values of overpotential for IrO2 and RuO2 electrocatalysts, which are typically in the 300–400 mV range. Therefore, an over potential of just 286 mV for the plasmonically assisted OER activity in the Au–IrO2 1:1.5 molar ratio represents a record value for IrO2 electrocatalysts which matches the most active reported electrocatalysts.32,40,55,76–81
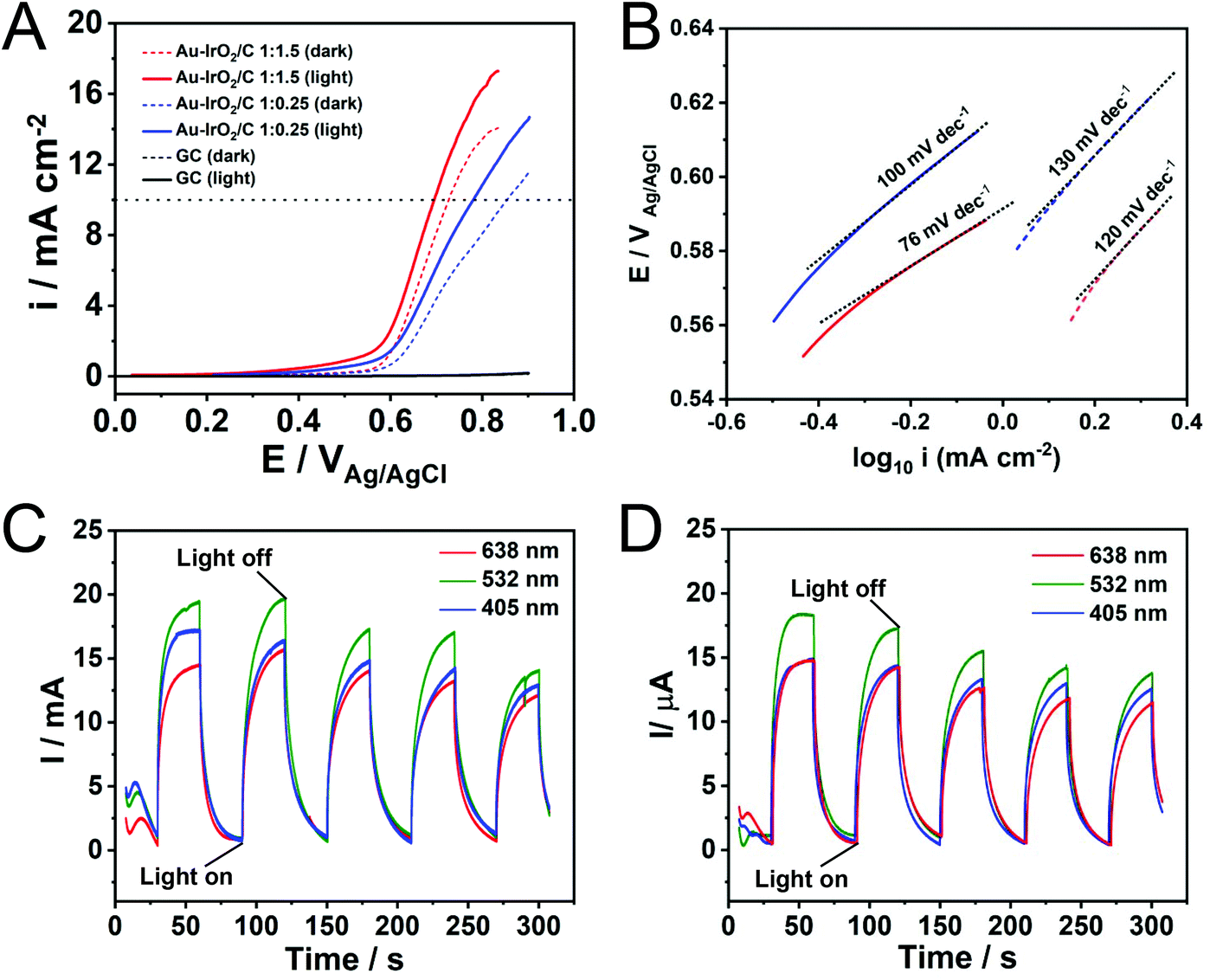 | ||
| Fig. 3 Electrochemical performances of the Au–IrO2 materials measured in a 0.1 M KOH electrolyte with and without LSPR excitation. OER linear scanning voltammetries recorded at a scan rate = 10 mV s−1 (A) and Tafel plots (B) for the Au–IrO2 nanoflowers obtained by co-reduction employing AuCl4− and IrCl3·xH2O as precursors in a 1:1.5 (red trace) 1:0.25 (blue trace) molar ratios with light excitation at 532 nm (solid line) and without light excitation (dashed line). The dotted horizontal line in (A) indicates a 10 mA cm−1 current. The measured current for the bare glassy carbon electrode is also shown for comparison (black trace). (C) and (D) show the light wavelength I–t curves recorded at 0.65 V (vs. Ag/AgCl) with light irradiation on/off for Au–IrO2 nanoflowers obtained by co-reduction employing AuCl4− and IrCl3·xH20 as precursors in a 1:1.5 (C) 1:0.25 (D) molar ratios under 405 (blue trace), 532 (green trace), and 638 nm (red trace) excitation. All the experiments were performed at room temperature. | ||
| Catalyst | η at j = 10 mA cm−2 (mV) | Ref. |
|---|---|---|
| Au–IrO2 1:1.5 (light) |
286 | This work |
| Au–IrO2 1:1.5 (dark) |
326 | This work |
| Au–IrO2 1:0.25 (light) |
376 | This work |
| Au–IrO2 1:0.25 (dark) |
446 | This work |
| IrOx/Au | 370 | 68 |
| IrOx[0.05]-Au nanoflowers | 481 | 55 |
| IrO2 | 330 | 76 |
| RuO2 | 305 | 77 |
| Au (light) | 455 | 40 |
| Au (dark) | 573 | 40 |
| CoFe2O4 | 370 | 78 |
| CaFeO3 | 390 | 79 |
| g-Ni0.87Fe0.13OOH | 390 | 32 |
| NiFe DH | 290 | 80 |
| Ni(OH)2-Au (light) | 270 | 40 |
| Ni(OH)2-Au (dark) | 330 | 40 |
| AuNP@Co/Ni-MOF | 330 | 81 |
In order to provide more insights into the effect of LSPR excitation on the kinetics of the OER, an analysis of the Tafel slope was performed. As seen from Fig. 3B, the Au–IrO2 1:1.5 sample exhibited a Tafel slope of 120 mV dec−1 in the dark (dashed red line). Upon plasmonic excitation, this value is sharply decreased to 76 mV dec−1 (solid red line), clearly showing that the kinetics of water oxidation are accelerated by visible light excitation.82 For the Au–IrO2 1:0.25 sample, a similar decrease in the Tafel slope value from 130 to 100 mV dec−1 was detected upon visible light illumination (dashed and solid blue lines, respectively).
In order to demonstrate the effect of the LSPR excitation over the electrocatalytic activity, we also collected the I–t curve at 0.65 V (vs. Ag/AgCl) for Au–IrO2 1:1.5 (Fig. 3C) and Au–IrO2 1:0.25 (Fig. 3D) samples under chopped light illumination for 3 different excitation wavelengths: 405 (blue trace), 523 (green trace), and 638 nm (red trace). The same illumination power was employed for all wavelengths. From Fig. 3C and D it can be seen that the Au–IrO2 samples displayed fast and reproducible current responses to on–off illumination cycles. The current densities were wavelength dependent, being greatest for 532 nm excitation, followed by 405 and 638 nm. However, these differences were not significative as both samples display a broad peak on the extinction spectra covering the 500–700 nm region (Fig. 2B). At 405 nm, IrO2 displays the onset of an absorption band associated with an O-p to Ir-d ligand-to-metal charge transfer process that can lead to the generation of charge carriers that also contribute to a photocatalytic enhancement.83Fig. 3C and D indicates that there was a decrease in the detected currents as a function of testing time under light irradiation. This behavior indicates loss of OER performance (Fig. S13†) as a result of changes in shape and surface composition under reaction conditions, indicating that stability of the Au–IrO2 nanostructures needs further optimizations.
It is recognized that the electrocatalyzed OER is a heterogeneous reaction comprising multiple elementary steps involving four electron transfer processes (multiple reaction channels) and generating several intermediates, such as OH*, O* and OOH*.32 Most of the proposed mechanisms include the formation of intermediates such as MOH and MO.82,84,85 Under basic conditions, the mechanism proceeds as described in eqn (1)–(5).32,82,84–86 In these processes, the bonding interactions (M–O) within the intermediates (MOH, MO and MOOH) are crucial for the overall electrocatalytic process.32,82,84–86
| M + OH− → MOH + e− | (1) |
| MOH + OH− → MO + H2O(l) + e− | (2) |
| MO + OH− → MOOH + e− | (3) |
| MOOH + OH− → MOO + H2O(l) + e− | (4) |
| MOO → M + O2(g) | (5) |
We performed DFT calculations in order to understand the reason that LSPR excitation produces increased OER activities for our photocatalytic materials. We calculated the binding energies and electronic structure of the O* and OH* intermediates for two slab models: (i) two IrO2 (110) layers (Fig. S14†), the thickness of which is ∼1 nm, matching the experimental observation (Fig. 1) and (ii) two IrO2 (110) layers supported on three Au (111) layers to simulate the Au–IrO2 material. Fig. 4A shows the two IrO2 (110) layers supported on three Au (111) layers employed in our model and Fig. 4B shows the O species adsorbed at the IrO2 surface. The calculated O* and OH* binding energies at the IrO2 and IrO2/Au surfaces are shown in Fig. 4C. We find that adsorption of both the O* and OH* is enhanced on the IrO2/Au surfaces relative to clean IrO2 surface. Note it has been recently demonstrated that the ΔG(O*) − ΔG(OH*) can be employed as a descriptor of OER activity, in which a volcano plot relationship between ΔG(O*) − ΔG(OH*) values and OER activity has been established.87,88 Our DFT calculations showed that ΔG(O*) − ΔG(OH*) for IrO2/Au was higher than for IrO2; 1.30 and 1.19 eV for IrO2/Au and IrO2 respectively (Fig. 4C). The increase in the value of ΔG(O*) − ΔG(OH*) brings it closer to the maximum of the volcano plot, indicating that the OER activity for Au–IrO2 should be higher than it on IrO2.88
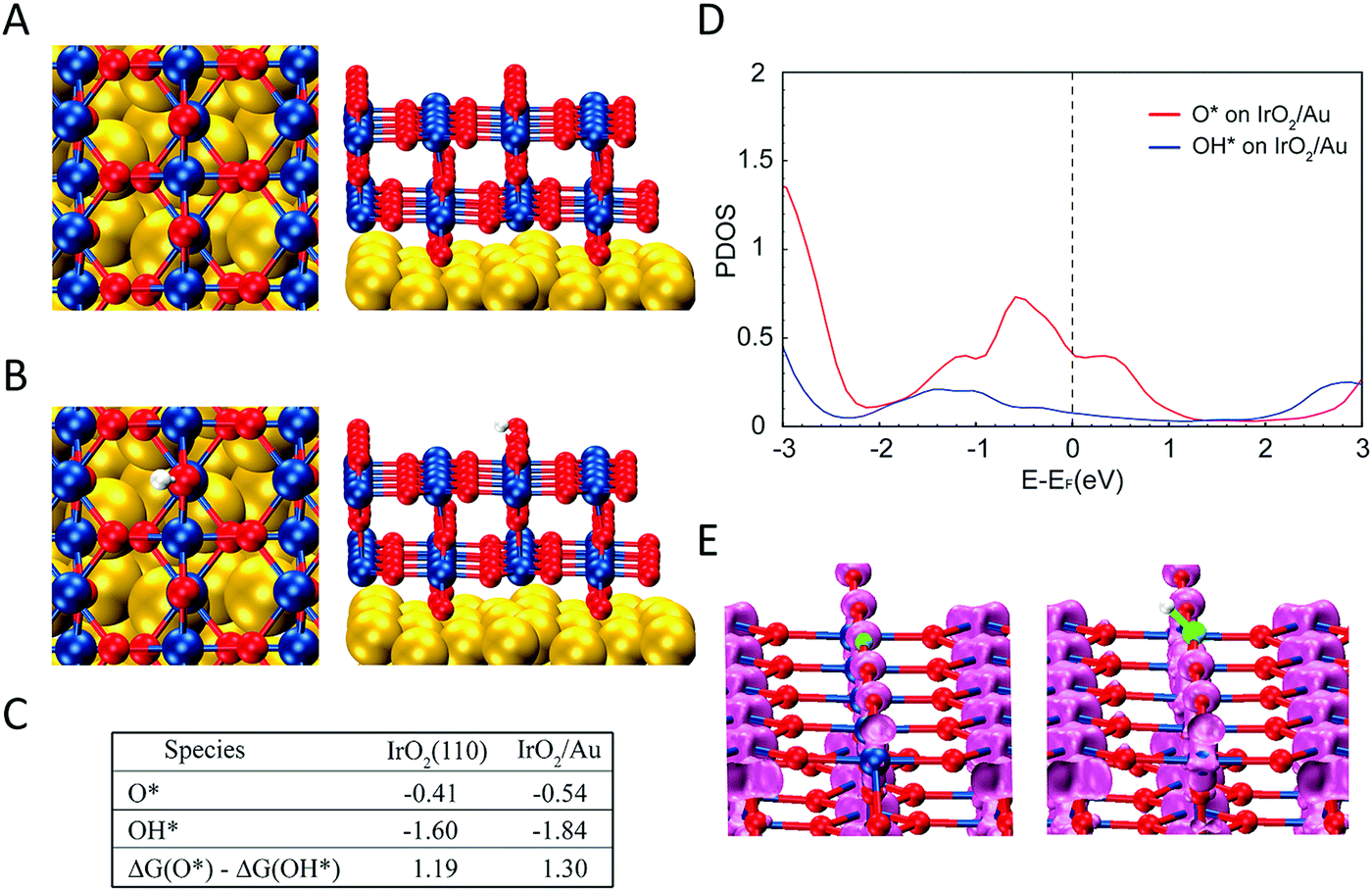 | ||
| Fig. 4 (A) The slab models employed in the DFT calculations consisting of two IrO2 (110) on three Au (111) layers. (B) Calculated atomic structure of the O species adsorbed on the IrO2 (110) surface in the Au–IrO2 hybrid structure. (C) Calculated binding energies of O* and OH* species and ΔG(O*) − ΔG(OH*) values on Au–IrO2 and the pure IrO2 slab models. When Au is present as the support for IrO2, the ΔG(O*) − ΔG(OH*) values (1.30 vs. 1.19 eV) is closer to the optimal point on the volcano plot for maximizing the OER activity. (D) Projected density of states of O and OH adsorbed on Au–IrO2. (E) Charge density analysis of energy range (−2.33, 0 eV) for O* and OH* on Au–IrO2, showing more occupied states on O*. | ||
Fig. 4D shows the calculated projected density of states (DOS) for O* and OH* adsorbed on Au–IrO2 (Fig. S15† shows the calculated DOS for O* and OH* adsorbed on IrO2 for comparison). It was found that the states between −2.33 eV (corresponding to the photon wavelength of 532 nm used in the experiments) and the Fermi level are more pronounced than the states above the Fermi level. This high population of states below the Fermi level may indicate a more pronounced mechanism based on the effect of the hot holes generated under LSPR excitation towards the enhanced OER activities.89,90 In Fig. 4E, the charge density analysis of an energy range from −2.33 to the Fermi level for O* and OH* on IrO2/Au shows the presence of more states on *O, which agrees with the DOS plot (Fig. 4D). Based on these results, we propose that when hot holes are LSPR-excited in the Au NPs and then transferred to O* (or the Ir–O antibonding state), the charge lowers the binding energy of O* increasing the value of ΔG(O*), and thus increases the OER activity. It is plausible that the hot holes may also lower the binding energy of OH*. However, because OH* has much less available states to populate the holes (Fig. 4D), this weakening of the binding energy of OH* is much less than the O* binding, which overall makes ΔG(O*) − ΔG(OH*) more positive under LSPR excitation, further shifting this value closer towards the maximum point in the volcano plot between ΔG(O*)-ΔG(OH*) and the OER activity.88
Our DFT calculations agree with the experimentally observed decrease in the Tafel slopes under visible light illumination, which indicated that the OER kinetics of the Au–IrO2 nanoflowers can be facilitated by LSPR excitation, in which hot holes can be generated at the Au NPs and flow to the IrO2, where they enhance the OER process. It is important to mention that this is agreement with several reports that shows the LSPR-driven hot carrier flow in the metal semiconductor systems, including its direct observation in Au/TiO2 materials.91–93 Fig. S16† shows the energy band diagrams for the Au–IrO2 nanoflowers to illustrate this proposed mechanism. In this case, hot holes generated from LSPR excitation in Au can be transferred to IrO2 conduction band which is located below the Au Fermi level.67 LSPR excited hot electrons, on the other hand, may be transferred to the glassy carbon substrate electrode across the ohmic interface between Au and GC with the assistance of external voltage.40,94
As the Tafel slopes depend on the strength of adsorption of the intermediate species, this would support our proposed mechanism in which hot holes can activate the M–O* bond at the surface during the OER, lowering its binding energy, which leads to a change in the rate limiting step.32,35,82,84,85 It has been reported that the OER reaction mechanism and rate-determining step cannot be unambiguously assigned simply according to the measured Tafel slope because surface intermediates, coverage, reaction pathways, and rate-determining steps may depend on the potential.32,35,82,84,85 Nevertheless, it has been proposed that if the first-electron transfer represents the rate-determining step, the corresponding Tafel slope should correspond to 120 mV dec−1. If the rate-determining step is the chemical reaction following a one-electron transfer process, the Tafel slope becomes 60 mV dec−1.32,35,82,84,85 One example is a process in which an OH surface species is rearranged via a surface reaction, as described in eqn (4). Therefore, our experimentally observed change in the Tafel slope from 120 to 76 mV dec−1 in the Au–IrO2 material as a result of visible light illumination may indicate that the OER mechanism and thus the rate determining step is changing from the first electron transfer reaction (eqn (1)) to the chemical reaction (eqn (4)).
It is important to note that, although we focused herein on the OER activities in alkaline conditions, we believe that the established the design principles can also be applied for developing plasmonic–catalytic electrocatalysts for the OER in neutral and acidic media. Though the adsorption energies of O and OH were found to shift to more positive energies with lower pH, it has been observed that the free energy shifts for OH* and O* preserves the universal scaling relation between free energy difference of O* and OH* with OER activity.95
Although the transfer of energy from the LSPR excitation to the reactant for the acceleration of the OER can occur via LSPR generated hot charges,8 temperature effects due to localized heating following LSPR decay may also play a role (together with LSPR hot carriers) over the OER activity enhancements. While the enhancement of electrocatalytic activity can originate from both photothermal effects and LSPR-generated charge carriers, previous results have shown that photothermally induced temperature rise does not fully account for the enhanced electrocatalytic activity, and that non-thermal effect play a significant role.94 However, from our current data, it is not possible to precisely quantify the relative contributions from LSPR-excited charge carriers and photothermal effects over the activities.
Conclusion
We have developed multimetallic Au–IrO2 plasmonic–catalytic nanoparticles and successfully demonstrated enhancement of the OER activity of IrO2via plasmonic catalysis under visible light illumination. The Au–IrO2 plasmonic–catalytic nanoparticles we synthesized were tunable for a range of sizes and compositions but all comprised of a core–shell Au–IrO2 flower-like nanoparticle morphology with closely spaced Au dendrite branches partially covered by an ultrathin (1 nm) IrO2 shell. These morphological and compositional features meet important design principles towards the optimization of OER activities and allow us to expand our understanding of enhancement mechanisms due to coupling with surface plasmon effects. The ultrathin and incomplete IrO2 shell at the surface of each branch enabled one to maximize the light harvesting by Au, and the plasmonic hybridization between the closely spaced plasmonic branches allow for the generation of electromagnetic hot spots to enhance light interactions. The OER activities we measured were light-excitation dependent, with the best values equaling the most active catalysts reported for OER. Furthermore, our calculations suggest that light generated hot holes play a dominant role in the mechanism of plasmonic enhancement. These LSPR excited holes could be transferred to Ir–O antibonding states, lowering the binding energy and accelerating the reaction. This was further confirmed by Tafel plot analysis, which suggested a change in the reaction mechanism (rate-determinant step) under LSPR excitation. We believe the results reported herein shed novel insights into the design principles required to develop plasmonic–catalytic nanoparticles capable of optimizing activities and also enable further mechanistic understanding over enhancement mechanisms that dictate plasmon-driven chemistry.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by FAPESP (Grant 2015/26308-7) and the Serrapilheira Institute (Grant Serra-1709-16900). This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brazil (CAPES) – Finance Code 001. L. S. P., J. Q. and E. C. M. B. thank FAPESP for the fellowships (Grants 2016/00819-8, 2016/17866-9, and 2015/11452-5, respectively). I. C. F. and B. A. N. thanks CNPq for the fellowship. T. M. and B. W. appreciate support from the Department of Energy (Grant No. DE-SC0020300) and computational resources at the OU Supercomputing Center for Education & Research (OSCER) at the University of Oklahoma. S. J. H. thanks the Engineering and Physical Sciences (UK) (Grants EP/M010619/1, EP/P009050/1) and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Grant ERC-2016-STG-EvoluTEM-715502).References
- Y. Zhang, S. He, W. Guo, Y. Hu, J. Huang, J. R. Mulcahy and W. D. Wei, Chem. Rev., 2018, 118, 2927–2954 CrossRef CAS PubMed.
- G. Baffou and R. Quidant, Chem. Soc. Rev., 2014, 43, 3898–3907 RSC.
- S. Linic, U. Aslam, C. Boerigter and M. Morabito, Nat. Mater., 2015, 14, 567–576 CrossRef CAS PubMed.
- R. Long, Y. Li, L. Song and Y. Xiong, Small, 2015, 11, 3873–3889 CrossRef CAS PubMed.
- J. G. Smith, J. A. Faucheaux and P. K. Jain, Nano Today, 2015, 10, 67–80 CrossRef CAS.
- S. Yu, A. J. Wilson, G. Kumari, X. Zhang and P. K. Jain, ACS Energy Lett., 2017, 2, 2058–2070 CrossRef CAS.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS PubMed.
- U. Aslam, V. G. Rao, S. Chavez and S. Linic, Nat. Catal., 2018, 1, 656–665 CrossRef.
- M. J. Kale, T. Avanesian and P. Christopher, ACS Catal., 2014, 4, 116–128 CrossRef CAS.
- K. Li, N. J. Hogan, M. J. Kale, N. J. Halas, P. Nordlander and P. Christopher, Nano Lett., 2017, 17, 3710–3717 CrossRef CAS PubMed.
- J. Quiroz, E. C. M. Barbosa, T. P. Araujo, J. L. Fiorio, Y.-C. Wang, Y.-C. Zou, T. Mou, T. V. Alves, D. C. De Oliveira, B. Wang, S. J. Haigh, L. M. Rossi and P. H. C. Camargo, Nano Lett., 2018, 18, 7289–7297 CrossRef CAS PubMed.
- D. F. Swearer, H. Zhao, L. Zhou, C. Zhang, H. Robatjazi, J. M. P. Martirez, C. M. Krauter, S. Yazdi, M. J. McClain, E. Ringe, E. A. Carter, P. Nordlander and N. J. Halas, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8916–8920 CrossRef CAS PubMed.
- A. Marimuthu, J. Zhang and S. Linic, Science, 2013, 340, 1590–1593 CrossRef PubMed.
- C. Hu, X. Chen, J. Jin, Y. Han, S. Chen, H. Ju, J. Cai, Y. Qiu, C. Gao, C. Wang, Z. Qi, R. Long, L. Song, Z. Liu and Y. Xiong, J. Am. Chem. Soc., 2019, 141, 7807–7814 CrossRef CAS PubMed.
- E. Peiris, S. Sarina, E. R. Waclawik, G. A. Ayoko, P. Han, J. Jia and H. Y. Zhu, Angew. Chem., Int. Ed., 2019, 58, 12032–12036 CrossRef CAS PubMed.
- J. L. Wang, R. A. Ando and P. H. C. Camargo, ACS Catal., 2014, 4, 3815–3819 CrossRef CAS.
- A. G. M. Da Silva, T. S. Rodrigues, J. Wang, L. K. Yamada, T. V. Alves, F. R. Ornellas, R. A. Ando and P. H. C. Camargo, Langmuir, 2015, 31, 10272–10278 CrossRef CAS PubMed.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- S. Yu and P. K. Jain, Nat. Commun., 2019, 10, 2022 CrossRef PubMed.
- Y. Kim, J. G. Smith and P. K. Jain, Nat. Chem., 2018, 10, 763–769 CrossRef CAS PubMed.
- M. Dhiman, A. Maity, A. Das, R. Belgamwar, B. Chalke, Y. Lee, K. Sim, J.-M. Nam and V. Polshettiwar, Chem. Sci., 2019, 10, 6594–6603 RSC.
- S. Atta, A. M. Pennington, F. E. Celik and L. Fabris, Chem, 2018, 4, 2140–2153 CAS.
- S. Mukherjee, F. Libisch, N. Large, O. Neumann, L. V. Brown, J. Cheng, J. B. Lassiter, E. A. Carter, P. Nordlander and N. J. Halas, Nano Lett., 2012, 13, 240–247 CrossRef PubMed.
- S. Mukherjee, L. Zhou, A. M. Goodman, N. Large, C. Ayala-Orozco, Y. Zhang, P. Nordlander and N. J. Halas, J. Am. Chem. Soc., 2014, 136, 64–67 CrossRef CAS PubMed.
- P. Christopher, H. Xin and S. Linic, Nat. Chem., 2011, 3, 467–472 CrossRef CAS PubMed.
- M. J. Landry, A. Gellé, B. Y. Meng, C. J. Barrett and A. Moores, ACS Catal., 2017, 7, 6128–6133 CrossRef CAS.
- P. Han, W. Martens, E. R. Waclawik, S. Sarina and H. Zhu, Part. Part. Syst. Charact., 2018, 35, 1700489 CrossRef.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- M. Tahir, L. Pan, F. Idrees, X. Zhang, L. Wang, J. J. Zou and Z. L. Wang, Nano Energy, 2017, 37, 136–157 CrossRef CAS.
- S. Bai, X. Li, Q. Kong, R. Long, C. Wang, J. Jiang and Y. Xiong, Adv. Mater., 2015, 27, 3444–3452 CrossRef CAS PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- H. Dotan, K. Sivula, M. Grätzel, A. Rothschild and S. C. Warren, Energy Environ. Sci., 2011, 4, 958–964 RSC.
- S. Trasatti, J. Electroanal. Chem., 1980, 111, 125–131 CrossRef CAS.
- E. Fabbri, A. Habereder, K. Waltar, R. Kötz and T. J. Schmidt, Catal. Sci. Technol., 2014, 4, 3800–3821 RSC.
- P. G. Hoertz, Y. Il Kim, W. J. Youngblood and T. E. Mallouk, J. Phys. Chem. B, 2007, 111, 6845–6856 CrossRef CAS PubMed.
- Y. Zhao, E. A. Hernandez-Pagan, N. M. Vargas-Barbosa, J. L. Dysart and T. E. Mallouk, J. Phys. Chem. Lett., 2011, 2, 402–406 CrossRef CAS.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef CAS PubMed.
- B. S. Yeo and A. T. Bell, J. Phys. Chem. C, 2012, 116, 8394–8400 CrossRef CAS.
- G. Liu, P. Li, G. Zhao, X. Wang, J. Kong, H. Liu, H. Zhang, K. Chang, X. Meng, T. Kako and J. Ye, J. Am. Chem. Soc., 2016, 138, 9128–9136 CrossRef CAS PubMed.
- L. Guo, K. Liang, K. Marcus, Z. Li, L. Zhou, P. D. Mani, H. Chen, C. Shen, Y. Dong, L. Zhai, K. R. Coffey, N. Orlovskaya, Y.-H. Sohn and Y. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 34970–34977 CrossRef CAS PubMed.
- F. Shi, J. He, B. Zhang, J. Peng, Y. Ma, W. Chen, F. Li, Y. Qin, Y. Liu, W. Shang, P. Tao, C. Song, T. Deng, X. Qian, J. Ye and J. Wu, Nano Lett., 2019, 19, 1371–1378 CrossRef PubMed.
- H.-X. Zhang, Y. Li, M.-Y. Li, H. Zhang and J. Zhang, Nanoscale, 2018, 10, 2236–2241 RSC.
- X. Guo, X. Li, S. Kou, X. Yang, X. Hu, D. Ling and J. Yang, J. Mater. Chem. A, 2018, 6, 7364–7369 RSC.
- J. E. Lee, F. Marques Mota, C. H. Choi, Y. R. Lu, R. Boppella, C. L. Dong, R. S. Liu and D. H. Kim, Adv. Mater. Interfaces, 2019, 6, 1801144 CrossRef.
- J. H. Weaver, Phys. Rev. B: Solid State, 1975, 11, 1416 CrossRef CAS.
- D. F. Swearer, H. Robatjazi, J. M. P. Martirez, M. Zhang, L. Zhou, E. A. Carter, P. Nordlander and N. J. Halas, ACS Nano, 2019, 13, 8076–8086 CrossRef CAS PubMed.
- H. Robatjazi, H. Zhao, D. F. Swearer, N. J. Hogan, L. Zhou, A. Alabastri, M. J. McClain, P. Nordlander and N. J. Halas, Nat. Commun., 2017, 8, 27 CrossRef PubMed.
- Z. Li, L. Shi, D. Franklin, S. Koul, A. Kushima and Y. Yang, Nano Energy, 2018, 51, 400–407 CrossRef CAS.
- U. Aslam, S. Chavez and S. Linic, Nat. Nanotechnol., 2017, 12, 1000–1005 CrossRef CAS PubMed.
- T. P. Araujo, J. Quiroz, E. C. M. Barbosa and P. H. C. Camargo, Understanding plasmonic catalysis with controlled nanomaterials based on catalytic and plasmonic metals, Elsevier, 2019, vol. 39 Search PubMed.
- E. Cortés, Adv. Opt. Mater., 2017, 5, 1700191 CrossRef.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- C. Zhao, Y. E. and L. Fan, Microchim. Acta, 2012, 178, 107–114 CrossRef CAS.
- C. Zhang, H. Zhao, L. Zhou, A. E. Schlather, L. Dong, M. J. McClain, D. F. Swearer, P. Nordlander and N. J. Halas, Nano Lett., 2016, 16, 6677–6682 CrossRef CAS PubMed.
- S. Chavez, U. Aslam and S. Linic, ACS Energy Lett., 2018, 3, 1590–1596 CrossRef CAS.
- L. Zhou, D. F. Swearer, C. Zhang, H. Robatjazi, H. Zhao, L. Henderson, L. Dong, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Science, 2018, 362, 69–72 CrossRef CAS PubMed.
- F. Wang, C. Li, H. Chen, R. Jiang, L.-D. Sun, Q. Li, J. Wang, J. C. Yu and C.-H. Yan, J. Am. Chem. Soc., 2013, 135, 5588–5601 CrossRef CAS PubMed.
- P. Christopher, H. Xin, A. Marimuthu and S. Linic, Nat. Mater., 2012, 11, 1044–1050 CrossRef CAS PubMed.
- S. Linic and M. A. Barteau, J. Am. Chem. Soc., 2003, 125, 4034–4035 CrossRef CAS PubMed.
- A. G. M. da Silva, T. S. Rodrigues, V. G. Correia, T. V. Alves, R. S. Alves, R. A. Ando, F. R. Ornellas, J. Wang, L. H. Andrade and P. H. C. Camargo, Angew. Chem., Int. Ed., 2016, 55, 7111–7115 CrossRef CAS PubMed.
- A. G. M. da Silva, T. S. Rodrigues, L. S. K. Taguchi, H. V. Fajardo, R. Balzer, L. F. D. Probst and P. H. C. Camargo, J. Mater. Sci., 2015, 51, 603–614 CrossRef.
- M. P. Seah, G. C. Smith and M. T. Anthony, Surf. Interface Anal., 1990, 15, 293–308 CrossRef CAS.
- R. G. Haverkamp, A. T. Marshall and B. C. C. Cowie, Surf. Interface Anal., 2011, 43, 847–855 CrossRef CAS.
- S. J. Freakley, J. Ruiz-Esquius and D. J. Morgan, Surf. Interface Anal., 2017, 49, 794–799 CrossRef CAS.
- S.-F. Hung, F.-X. Xiao, Y.-Y. Hsu, N.-T. Suen, H.-B. Yang, H. M. Chen and B. Liu, Adv. Energy Mater., 2016, 6, 1501339 CrossRef.
- S. Moon, Y. B. Cho, A. Yu, M. H. Kim, C. Lee and Y. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 1979–1987 CrossRef CAS.
- X. Yang, Y. Li, L. Deng, W. Li, Z. Ren, M. Yang, X. Yang and Y. Zhu, RSC Adv., 2017, 7, 20252–20258 RSC.
- Z. Ke, L. Li, Q. Jia, Y. Yang and H. Cui, Appl. Surf. Sci., 2019, 463, 58–65 CrossRef CAS.
- B. Jin, M. L. Sushko, Z. Liu, C. Jin and R. Tang, Nano Lett., 2018, 18, 6551–6556 CrossRef CAS PubMed.
- C. Zhu, S. Liang, E. Song, Y. Zhou, W. Wang, F. Shan, Y. Shi, C. Hao, K. Yin, T. Zhang, J. Liu, H. Zheng and L. Sun, Nat. Commun., 2018, 9, 1–7 CrossRef CAS PubMed.
- X. Zhao, Q. Wang, X. Zhang, Y. I. Lee and H. G. Liu, Phys. Chem. Chem. Phys., 2016, 18, 1945–1952 RSC.
- M. D. Urović, R. Puchta, Ž. D. Bugarčić and R. Van Eldik, Dalton Trans., 2014, 43, 8620–8632 RSC.
- H. You and J. Fang, Nano Today, 2016, 11, 145–167 CrossRef CAS.
- J. Huang, J. Chen, T. Yao, J. He, S. Jiang, Z. Sun, Q. Liu, W. Cheng, F. Hu, Y. Jiang, Z. Pan and S. Wei, Angew. Chem., Int. Ed., 2015, 54, 8722–8727 CrossRef CAS PubMed.
- P. Chen, K. Xu, T. Zhou, Y. Tong, J. Wu, H. Cheng, X. Lu, H. Ding, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2016, 55, 2488–2492 CrossRef CAS PubMed.
- M. Li, Y. Xiong, X. Liu, X. Bo, Y. Zhang, C. Han and L. Guo, Nanoscale, 2015, 7, 8920–8930 RSC.
- S. Yagi, I. Yamada, H. Tsukasaki, A. Seno, M. Murakami, H. Fujii, H. Chen, N. Umezawa, H. Abe, N. Nishiyama and S. Mori, Nat. Commun., 2015, 6, 8249 CrossRef PubMed.
- O. Diaz-Morales, I. Ledezma-Yanez, M. T. M. Koper and F. Calle-Vallejo, ACS Catal., 2015, 5, 5380–5387 CrossRef CAS.
- M. Wang, P. Wang, C. Li, H. Li and Y. Jin, ACS Appl. Mater. Interfaces, 2018, 10, 37095–37102 CrossRef CAS PubMed.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef PubMed.
- F. A. Frame, T. K. Townsend, R. L. Chamousis, E. M. Sabio, T. Dittrich, N. D. Browning and F. E. Osterloh, J. Am. Chem. Soc., 2011, 133, 7264–7267 CrossRef CAS PubMed.
- D. A. García-Osorio, R. Jaimes, J. Vazquez-Arenas, R. H. Lara and J. Alvarez-Ramirez, J. Electrochem. Soc., 2017, 164, E3321–E3328 CrossRef.
- Y.-H. Fang and Z.-P. Liu, ACS Catal., 2014, 4, 4364–4376 CrossRef CAS.
- E. Antolini, ACS Catal., 2014, 4, 1426–1440 CrossRef CAS.
- B. M. Tackett, W. Sheng, S. Kattel, S. Yao, B. Yan, K. A. Kuttiyiel, Q. Wu and J. G. Chen, ACS Catal., 2018, 8, 2615–2621 CrossRef CAS.
- I. C. Man, H.-Y. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- A. E. Schlather, A. Manjavacas, A. Lauchner, V. S. Marangoni, C. J. DeSantis, P. Nordlander and N. J. Halas, J. Phys. Chem. Lett., 2017, 8, 2060–2067 CrossRef CAS PubMed.
- P. Christopher and M. Moskovits, Annu. Rev. Phys. Chem., 2017, 68, 379–398 CrossRef CAS PubMed.
- X. C. Ma, Y. Dai, L. Yu and B. B. Huang, Light: Sci. Appl., 2016, 5, e16017 CrossRef CAS PubMed.
- Y. K. Lee, C. H. Jung, J. Park, H. Seo, G. A. Somorjai and J. Y. Park, Nano Lett., 2011, 11, 4251–4255 CrossRef CAS PubMed.
- H. Lee, H. Lee and J. Y. Park, Nano Lett., 2019, 19, 891–896 CrossRef PubMed.
- L. Huang, J. Zou, J.-Y. Y. Ye, Z.-Y. Y. Zhou, Z. Lin, X. Kang, P. K. Jain and S. Chen, Angew. Chem., Int. Ed., 2019, 58, 8794–8798 CrossRef CAS PubMed.
- D.-Y. Kuo, J. K. Kawasaki, J. N. Nelson, J. Kloppenburg, G. Hautier, K. M. Shen, D. G. Schlom and J. Suntivich, J. Am. Chem. Soc., 2017, 139, 3473–3479 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr01875a |
| This journal is © The Royal Society of Chemistry 2020 |