
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of molten sodium nitrate on the decomposition pathways of hydrated magnesium hydroxycarbonate to magnesium oxide probed by in situ total scattering†
Margarita
Rekhtina
 a,
Alessandro
Dal Pozzo
ab,
Dragos
Stoian
c,
Andac
Armutlulu
a,
Felix
Donat
a,
Maria V.
Blanco
c,
Zhu-Jun
Wang
d,
Marc-Georg
Willinger
d,
Alexey
Fedorov
a,
Paula M.
Abdala
*a and
Christoph R.
Müller
*a
a,
Alessandro
Dal Pozzo
ab,
Dragos
Stoian
c,
Andac
Armutlulu
a,
Felix
Donat
a,
Maria V.
Blanco
c,
Zhu-Jun
Wang
d,
Marc-Georg
Willinger
d,
Alexey
Fedorov
a,
Paula M.
Abdala
*a and
Christoph R.
Müller
*a
aLaboratory of Energy Science and Engineering, Department of Mechanical and Process Engineering, ETH Zürich, Leonhardstrasse 21, 8092 Zürich, Switzerland. E-mail: abdalap@ethz.ch; muelchri@ethz.ch
bLaboratory of Industrial Safety and Environmental Sustainability, Department of Civil, Chemical, Environmental and Materials Engineering, Alma Mater Studiorum–Università di Bologna, Via Terracini 28, 40131 Bologna, Italy
cEuropean Synchrotron Radiation Facility, 71 Avenue des Martyrs, Grenoble, France
dScientific Center for Optical and Electron Microscopy, ETH Zürich, Auguste-Piccard-Hof 1, 8093 Zurich, Switzerland
First published on 27th May 2020
Abstract
The effect of NaNO3 and its physical state on the thermal decomposition pathways of hydrated magnesium hydroxycarbonate (hydromagnesite, HM) towards MgO was examined by in situ total scattering. Pair distribution function (PDF) analysis of these data allowed us to probe the structural evolution of pristine and NaNO3-promoted HM. A multivariate curve resolution alternating least squares (MCR-ALS) analysis identified the intermediate phases and their evolution upon the decomposition of both precursors to MgO. The total scattering results are discussed in relation with thermogravimetric measurements coupled with off-gas analysis. MgO is obtained from pristine HM (N2, 10 °C min−1) through an amorphous magnesium carbonate intermediate (AMC), formed after the partial removal of water of crystallization from HM. The decomposition continues via a gradual release of water (due to dehydration and dehydroxylation) and, in the last step, via decarbonation, leading to crystalline MgO. The presence of molten NaNO3 alters the decomposition pathways of HM, proceeding now through AMC and crystalline MgCO3. These results demonstrate that molten NaNO3 facilitates the release of water (from both water of crystallization and through dehydroxylation) and decarbonation, and promotes the crystallization of MgCO3 and MgO in comparison to pristine HM. MgO formed from the pristine HM precursor shows a smaller average crystallite size than NaNO3-promoted HM and preserves the initial nano-plate-like morphology of HM. NaNO3-promoted HM was decomposed to MgO that is characterized by a larger average crystallite size and irregular morphology. Additionally, in situ SEM allowed visualization of the morphological evolution of HM promoted with NaNO3 at a micrometre scale.
Introduction
The development of efficient solid sorbents for CO2 capture plays a key role in carbon capture storage and utilization (CCSU) technologies, which are considered strategic in reducing greenhouse emissions and mitigating global warming.1–4 MgO-based materials are attractive CO2 sorbents for operation at moderate temperatures (200–450 °C)5 due to their high theoretical capture capacity of 1.09 g CO2 per g MgO6 and for being economically promising,7 environmentally benign and naturally abundant.8 Despite these advantages, the application of MgO as a CO2 sorbent has been hampered by its slow carbonation kinetics. In recent years, a breakthrough in the development of effective MgO-based sorbents has been achieved through their promotion by molten salts.9 The presence of molten alkali metal nitrates (such as LiNO3, NaNO3, KNO3 and mixtures thereof) drastically accelerates the rate of MgO carbonation;10–17 however, an in-depth understanding of the role of molten salts in the carbonation of MgO has yet to be obtained. It has been proposed that molten salts act as a phase transfer catalyst in the reaction between MgO and CO2.16 The dissolution of MgO in the molten salt, leading to the formation of solvated Mg–O pairs, may help to overcome the large lattice enthalpy of crystalline MgO and thereby facilitate carbonation. The solvated pairs can react with CO2 adsorbed on the MgO surface at the triple phase boundaries between solid, gaseous and the molten phases. Other studies have postulated that the molten alkali metal nitrates act primarily as a diffusion medium for CO2, preventing the formation of a CO2-impermeable unidentate carbonate and promote the rapid generation of carbonate ions.17 Elucidation of the mechanism behind the promoting effect of molten salts on the CO2 capture of MgO is key for the rational design of more effective MgO-based CO2 sorbents.In addition to the accelerated carbonation of MgO, the presence of molten salts promotes also the decomposition of magnesium carbonate (regeneration of the CO2 sorbent) and hydrated magnesium hydroxycarbonates.16–19 For example, based on thermogravimetric analysis (TGA) the decomposition temperature of MgCO3 mixed with NaNO3 was determined to be lowered by 50 °C with respect to un-promoted MgCO3.16 Similarly, it was observed that the presence of alkali nitrates also affects the decomposition of hydromagnesite, Mg5(CO3)4(OH)2·4H2O (HM).11
HM is a naturally occurring form of magnesium carbonate20 and is widely employed as a precursor for the synthesis of MgO-based sorbents.11,16,19,21 Upon thermal treatment, HM decomposes via the loss of water of crystallization, dehydroxylation and decarbonation, leading ultimately to the formation of crystalline MgO.22–24 The overall decomposition process involves amorphous intermediate phases, yet, their structure, i.e., the local atomic arrangement of the intermediates, is still unknown.25–27 Previous TGA-based experiments11,19 have shown a decrease in the decomposition temperature of HM in the presence of NaNO3. The crystallite size of the resulting MgO, obtained in the presence of molten nitrates, was also larger than that of the decomposition of unpromoted HM.11 It was suggested that NaNO3 in its molten state facilitates the release of CO2.19 However, there is limited understanding of the effect of NaNO3 and its physical state during the formation of MgO from HM.
Key tools to understand in more detail the effect of alkali salt promoters on the decomposition of MgCO3 and HM and the carbonation of MgO, are in situ studies. In this regard, the pair distribution function (PDF) analysis of total scattering data is of great interest as it provides information about both crystalline and amorphous intermediates.28,29 PDF analysis is based on high-energy X-rays, which allows for high penetration depth in versatile sample environments including reaction cells, rendering it ideal for in situ studies. Interrogation of materials consisting of solely light elements by PDF is particularly advantageous30,31 when compared to other techniques for probing the local-structure such as X-ray absorption spectroscopy (XAS) that requires high vacuum and a tailored cell owing to the tender X-ray energy of the absorption edges of Mg and Na.32,33 Advancements in time-resolved in situ and operando PDF analysis are, therefore, of great importance in the understanding and development of more active and stable MgO-based CO2 sorbents.
This work focuses on determining the role of NaNO3 on the decomposition pathways of HM leading to the formation of MgO and the physical state of NaNO3 (and changes thereof) during this process. To this end, in situ X-ray total scattering studies during the thermal treatment of pristine HM and NaNO3-promoted HM were carried out. Bulk NaNO3 was also investigated to determine the evolution of its structure when transitioning from the crystalline to the molten state. The analysis of the in situ total scattering data by the PDF technique, assisted by the multivariate curve resolution alternating least squares (MCR-ALS) method, provided information on the structural evolution in the local and medium-range order during the decomposition of HM. NaNO3 influences this transformation not only by accelerating dehydration, dehydroxylation and decarbonation of HM, but also by promoting crystallization of MgCO3 and MgO.
Experimental
Materials
Hydromagnesite (Mg5(CO3)4(OH)2·4H2O, Acros Organics, extra pure) and sodium nitrate (NaNO3, Sigma-Aldrich, ≥99.5%) were used as received. HM-20NaNO3 (with a Na![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mg molar ratio of 20:100) was obtained by a wet mixing method, as reported elsewhere.11 Mg5(CO3)4(OH)2·4H2O and NaNO3 were mixed in deionized water and stirred magnetically for 1 h at room temperature. The aqueous slurry was dried at 120 °C for 12 h and ground to a fine powder in an agate mortar.
:Mg molar ratio of 20:100) was obtained by a wet mixing method, as reported elsewhere.11 Mg5(CO3)4(OH)2·4H2O and NaNO3 were mixed in deionized water and stirred magnetically for 1 h at room temperature. The aqueous slurry was dried at 120 °C for 12 h and ground to a fine powder in an agate mortar.
TGA and heat flow measurements coupled with gas analysis
TGA and heat flow (HF) measurements (Mettler Toledo TGA/DSC3) were performed using ca. 70 mg of the specimen loaded into an alumina crucible (900 μl) and heated from 25 to 600 °C under a flow of nitrogen (80 mL min−1, measured and controlled at normal temperature and pressure, NTP) at a heating rate of 10 °C min−1. The rate of mass loss was calculated as the first derivative of the normalized weight loss with respect to time. An ABB EL3020 FTIR gas analyser and a Sensirion SHT31 humidity sensor were connected to the outlet of the TGA to measure the mole fractions of CO2 and H2O, respectively, in the off-gas stream. Integrating the mole fractions of CO2 and H2O over time and multiplying them by the molar flow rate of the gas yielded the total amounts of CO2 and H2O that evolved from the sample.In situ X-ray total scattering
In situ total scattering measurements were performed at the ID31 beamline at the European Synchrotron Radiation Facility (ESRF) in Grenoble, France. The incident X-rays were set to an energy of 70.0 keV (λ = 0.1771 Å) and were focused to a size of 0.6 mm × 0.6 mm. The data were collected using a 16-inch PerkinElmer 1621 2D detector. The sample-to-detector distance was set to ca. 26 cm to collect total scattering data I(Q)26; to acquire more resolved scattering patterns I(2θ)83 sample-to-detector distance was changed to ca. 83 cm (Fig. S2†). The materials were loaded in a quartz capillary reactor34 (external diameter = 1 mm, wall thickness = 0.01 mm), heated via an air blower. A temperature calibration was carried out first with a thermocouple placed inside the sample bed to determine the actual sample temperature as a function of the air blower temperature (the procedure was repeated twice to verify the reproducibility). The total scattering data were collected every 120 s using an exposure time of 10 s per acquisition while ramping up in the N2 flow (10 °C min−1, 5 mL min−1), more detailed scheme of data acquisition can be found in Fig. S2 and S3.† A CeO2 diffraction standard (National Institute of Standards and Technology, NIST) was used to determine the azimuthal parameters and the sample-to-detector distances. Azimuthal integration and normalization on the incident beam intensity were done using the pyFAI software package.35 The PDFGetX3 software package was used to subtract the background signal from an empty capillary (rpoly = 1.15 Å) and to obtain the coherent scattering function I(Q), reduced total scattering structure function F(Q) and the reduced pair distribution function G(r).36 The total scattering data were analysed up to Qmax = 22 Å−1 (Q = scattering vector). The PDF data of the CeO2 diffraction standard was used to determine the damping and broadening parameters of the PDF peaks due to the experimental resolution (Qdamp = 0.045 Å−1; Qbroad = 0.026 Å−1, Fig. S20†). These parameters were then fixed during the PDF refinements that were carried out with PDFgui37 using previously published structures from the Inorganic Crystal Structure Database (ICSD) database. Rietveld refinements of the XRD data (I(2θ)83) were carried out using FullProf software.38 Line broadening analysis was implemented in the Rietveld analysis; the instrumental resolution function was determined from the refinement of CeO2 reference (NIST). Additional information can be found in ESI.†MCR-ALS analysis was employed as a data reduction tool for the analysis of in situ, time-resolved PDF data obtained during the thermal treatment of HM and HM-20NaNO3 under N2. The MCR-ALS scripts, written in Matlab®, were used in the present work.39 MCR-ALS analysis was carried out in the r-range (r = interatomic distances) from 0 to 7 Å; the number of components used in the analysis was estimated based on a principal component analysis and prior knowledge of the systems. A more detailed description of the MCR-ALS analysis and deconvolution procedure can be found in the ESI.†
In situ scanning electron microscopy (SEM)
In situ SEM experiments were performed inside the chamber of a modified commercial ESEM (Thermo Scientific™ Quattro S). The vacuum system of the ESEM was modified and upgraded with oil-free prevacuum pumps. The instrument is equipped with a homemade laser heating stage, a gas supply unit (mass flow controllers from Bronkhorst), and a mass spectrometer (Pfeiffer OmniStar) for the analysis of the chamber atmosphere. After each sample loading, the chamber was pumped out to around 10−3 Pa, purged with pure nitrogen, and pumped again to 10−3 Pa successively for several times. The temperature was measured via a K-type thermocouple that was spot-welded onto the sample supporting substrate. In situ observation was performed at temperatures ranging from room temperature to 450 °C, with 46 Pa N2 (99.9995% purity). During the experiments, the microscope was operated at an acceleration voltage of 7.5 kV. Images were recorded by a large field detector. No influence of the electron beam on the sample could be observed during annealing process. The imaged regions and their respective surroundings showed similar behaviour, as evidenced by changing the magnification or by moving the sample under the beam. Furthermore, no electron beam induced contamination was observed.Transmission electron microscopy (TEM)
The morphology of the materials was studied by TEM (FEI Talos F200X) and HAADF-STEM (high-angle annular dark field – canning transmission electron microscope) equipped with a high-brightness field-emission gun, a HAADF detector, and a large collection-angle energy-dispersive X-ray spectroscopy (EDX) detector. The operation voltage of the instrument was set to 200 kV. The powders were dispersed onto Cu grids coated with lacey carbon.Results and discussion
First, we investigated the decomposition of the HM and HM-20NaNO3 materials using thermal gravimetric analysis with simultaneous heat flow (HF) measurements, coupled with the analysis of the off-gases (H2O, CO2). Additionally, in situ PDF analysis of the structural evolution of HM and HM-20NaNO3, as well as the reference NaNO3 during thermal treatment, allowed us to probe the decomposition pathways. Finally, the structural and morphological characteristics of the MgO produced from both precursors were determined by in situ XRD, PDF, SEM and ex situ HAADF-STEM.TGA/HF coupled gas analysis
Thermolysis of HM and HM-20NaNO3 in N2 was studied by TGA allowing the determination of changes in the sample mass (Fig. 1a and b) and the HF (Fig. 1c). The concentrations of H2O and CO2 in the off-gas were measured simultaneously by a humidity sensor and a FTIR gas analyser (Fig. 1d and e).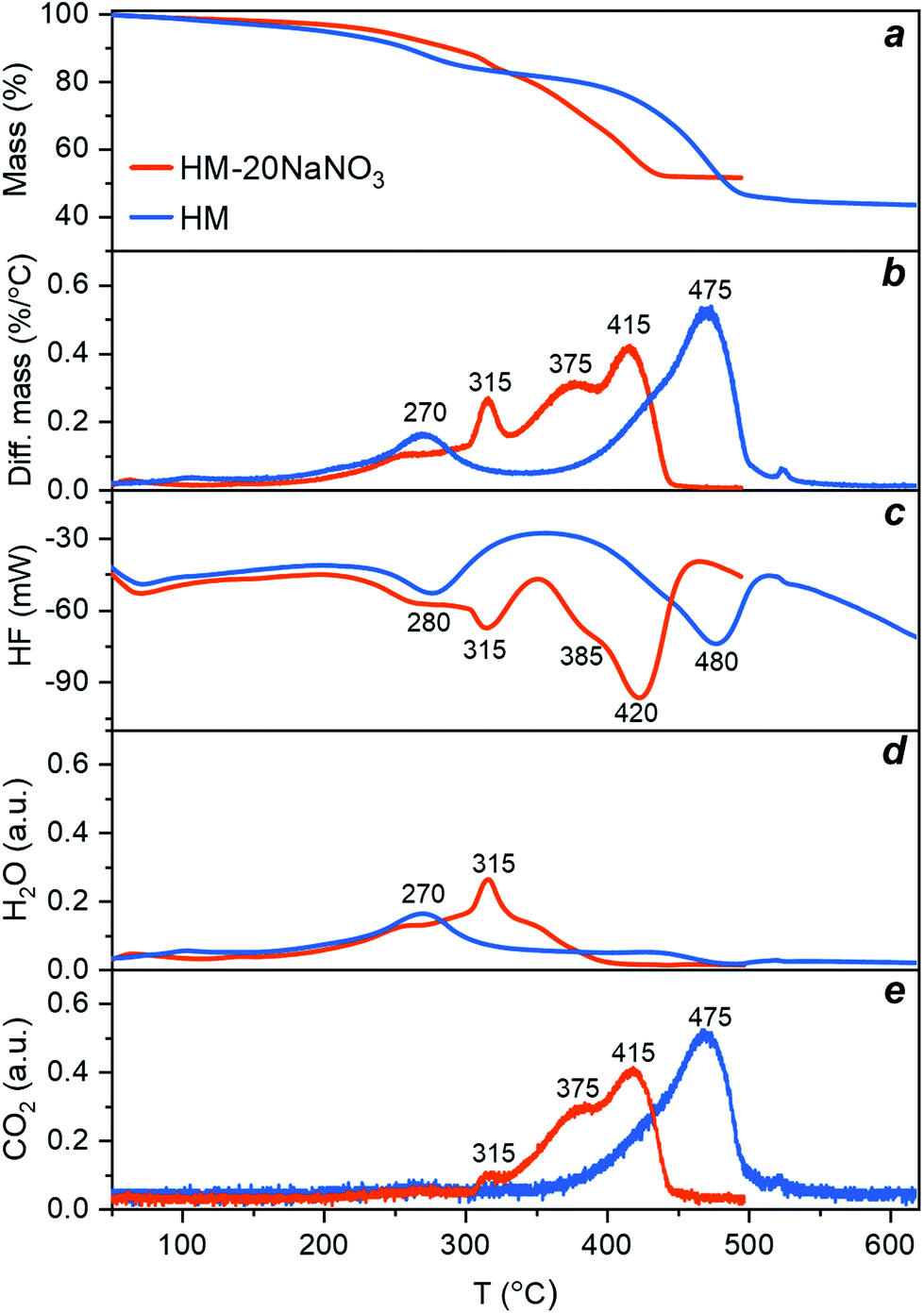 | ||
| Fig. 1 Thermal decomposition of HM (blue) and HM-20NaNO3 (red) during thermal treatment in N2 (10 °C min−1): (a) mass loss and (b) rate of mass loss determined by TGA; (c) heat flow and simultaneous measurement of (d) H2O and (e) CO2 released during the reaction. | ||
The differential change of the mass of HM exhibits two main peaks at 270 °C and 475 °C (Fig. 1b, blue trace), corresponding to two exothermic minima in the HF data (Fig. 1c, blue trace). The first peak corresponds to the release of water of crystallization (eqn (1)), according to the simultaneous analysis of the H2O released in the off-gas stream (Fig. 1d, blue trace). The long tailing of the H2O peak at higher temperatures, showing a continuous and gradual release of H2O between 270 and 500 °C, can be ascribed to the dehydroxylation of HM (eqn (2)). The second peak centred at 475 °C in the differential mass change curve is due to the release of CO2 through the decomposition of the carbonates (eqn (3)). This decomposition proceeds between 350–550 °C (Fig. 1e, blue trace). Table S1† summarizes the observed mass losses. The values obtained are in line with the following decomposition pathway of HM:23,26
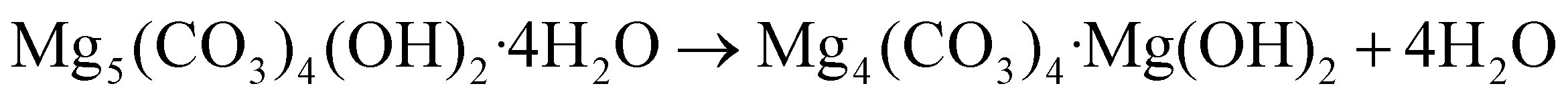 | (1) |
| Mg4(CO3)4·Mg(OH)2 → 4MgCO3 + MgO + H2O | (2) |
| MgCO3 → MgO + CO2 | (3) |
Turning now to HM promoted by NaNO3, we observe that the presence of NaNO3 significantly alters the decomposition profile of HM, as observed in both the differential mass change and HF profiles (Fig. 1b and c, red trace). The decomposition of HM-20NaNO3 was completed at 450 °C, i.e. 100 °C lower than for HM. In addition, the number of peaks increased. For HM-20NaNO3, the initial mass loss at ca. 260 °C, i.e., the release of water of crystallization, was not affected significantly by the presence of NaNO3. NaNO3 is still crystalline at this temperature (see section below and Fig. S1 and S4†). A more pronounced, and rather sudden release of water between 300 and 335 °C (with a maximum at 315 °C) correlates with the melting of NaNO3, as indicated by the HF signal of this sample and the reference experiment using pure NaNO3 (Fig. S1 and S4†). The dehydroxylation of HM is completed at 400 °C, i.e., 100 °C lower than for HM. Similarly, previous TGA and XRD studies have shown that the dehydroxylation of the brucite phase, Mg(OH)2, was facilitated by the presence of NaNO3.40 Therefore, these results suggest that the molten salt acts as a phase-transfer agent that facilitates the migration of H2O from HM into the gas phase, possibly due to the dissolution (solvation) of H2O in the melt. The subsequent release of CO2 splits into two main peaks centred at 375 °C and 415 °C (Fig. 1e) which are, respectively, 100 °C and 60 °C lower than the single peak observed for HM. We speculate that this splitting is related to different decomposition intermediates, which can be evidenced by in situ PDF studies (vide infra). Overall, these results indicate that (molten) NaNO3 facilitates the release of water of crystallization, dehydroxylation and the decomposition of the carbonates.
In situ PDF and XRD analyses
The structural changes occurring in HM and HM-20NaNO3 during the thermal heat treatment in N2 were also followed by PDF and XRD. However, the overlapping signals of the different phases in the PDF of HM-20NaNO3, make the identification of the dynamic phase changes in this material challenging. Therefore, an additional in situ experiment using pure NaNO3 and heating it from 40 °C to its melting point was performed as a reference.![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c space group, illustrated in Fig. 2b).41 The PDF of pristine NaNO3 at the lowest acquired temperature (ca. 40 °C) showed intense correlations in both the short (1 < r < 5 Å) and the medium r-ranges (5 < r < 25 Å), as expected for a crystalline material. According to the simulated PDF for NaNO3, the peaks in the short r-range, i.e., those centred at 1.25 Å and 2.20 Å, correspond to the, respectively, N–O and O–O intramolecular correlations of the NO3− group (Fig. S5†). The peaks at 2.46 Å and 3.32 Å are due to intermolecular pairs: the former corresponds to Na–O and the latter to the overlapping correlations of Na–N, Na–O and O–O. Upon heating to 290 °C, NaNO3 undergoes a second-order (order-disorder) phase transition, from an Rc to an Rm space group (marked by a gradual disappearance of the superstructure (113) reflection in the diffraction patterns, Fig. S4†),42–44 associated with an increase of the orientational freedom of nitrate groups. This transition appears as a gradual decrease in intensity of the intermolecular O–O and Na–O correlations in the short r-range of the PDF (i.e., an increasing disorder between the NO3− groups). In addition, the PDF data in Fig. 2a provide a direct evidence for the evolution of the Na–O distance, i.e., a continuous increase from 2.46 Å to 2.47 Å between 40 °C and 240 °C, followed by a sharper increase from 2.47 Å to 2.49 Å between 255 °C and 290 °C (Fig. S6†). The N–O distance remained largely unchanged during this order-disorder transformation (variations < 0.006 Å), evidencing the rigid structure of the NO3− group. The extent of the variations in the N–O distance during the second order phase transition has been a matter of debate in the literature.41,42 However, previous studies have determined the interatomic distances from XRD analysis and, therefore, they depend on the structural model used.41,42 Here, the PDF analysis provides direct information in real space, confirming that the N–O distance remains largely unchanged while the disorder between the nitrate groups increases gradually. When reaching 308 °C, the long-range ordering decreased, associated with vanishing correlations for r > 5 Å, which is in line with the melting (solid–liquid) transition of NaNO3. The melting of NaNO3 at 308 °C is supported by XRD analysis, showing the disappearance of all Bragg peaks due to NaNO3 (Fig. S4†). The PDF of pure, molten NaNO3 (i.e., the final measurement at 308 °C in Fig. 2a) shows peaks due to intramolecular N–O and O–O (1.25 Å and 2.20 Å) pairs, and weaker signals due to intermolecular correlations at 2.49 Å(Na–O) and between 2.5–5 Å (Na–Na, Na–N, Na–O, N–N, N–O, O–O).45 No correlations above 5 Å were present due to the lack of long-range order in molten salts.46,47
c space group, illustrated in Fig. 2b).41 The PDF of pristine NaNO3 at the lowest acquired temperature (ca. 40 °C) showed intense correlations in both the short (1 < r < 5 Å) and the medium r-ranges (5 < r < 25 Å), as expected for a crystalline material. According to the simulated PDF for NaNO3, the peaks in the short r-range, i.e., those centred at 1.25 Å and 2.20 Å, correspond to the, respectively, N–O and O–O intramolecular correlations of the NO3− group (Fig. S5†). The peaks at 2.46 Å and 3.32 Å are due to intermolecular pairs: the former corresponds to Na–O and the latter to the overlapping correlations of Na–N, Na–O and O–O. Upon heating to 290 °C, NaNO3 undergoes a second-order (order-disorder) phase transition, from an Rc to an Rm space group (marked by a gradual disappearance of the superstructure (113) reflection in the diffraction patterns, Fig. S4†),42–44 associated with an increase of the orientational freedom of nitrate groups. This transition appears as a gradual decrease in intensity of the intermolecular O–O and Na–O correlations in the short r-range of the PDF (i.e., an increasing disorder between the NO3− groups). In addition, the PDF data in Fig. 2a provide a direct evidence for the evolution of the Na–O distance, i.e., a continuous increase from 2.46 Å to 2.47 Å between 40 °C and 240 °C, followed by a sharper increase from 2.47 Å to 2.49 Å between 255 °C and 290 °C (Fig. S6†). The N–O distance remained largely unchanged during this order-disorder transformation (variations < 0.006 Å), evidencing the rigid structure of the NO3− group. The extent of the variations in the N–O distance during the second order phase transition has been a matter of debate in the literature.41,42 However, previous studies have determined the interatomic distances from XRD analysis and, therefore, they depend on the structural model used.41,42 Here, the PDF analysis provides direct information in real space, confirming that the N–O distance remains largely unchanged while the disorder between the nitrate groups increases gradually. When reaching 308 °C, the long-range ordering decreased, associated with vanishing correlations for r > 5 Å, which is in line with the melting (solid–liquid) transition of NaNO3. The melting of NaNO3 at 308 °C is supported by XRD analysis, showing the disappearance of all Bragg peaks due to NaNO3 (Fig. S4†). The PDF of pure, molten NaNO3 (i.e., the final measurement at 308 °C in Fig. 2a) shows peaks due to intramolecular N–O and O–O (1.25 Å and 2.20 Å) pairs, and weaker signals due to intermolecular correlations at 2.49 Å(Na–O) and between 2.5–5 Å (Na–Na, Na–N, Na–O, N–N, N–O, O–O).45 No correlations above 5 Å were present due to the lack of long-range order in molten salts.46,47
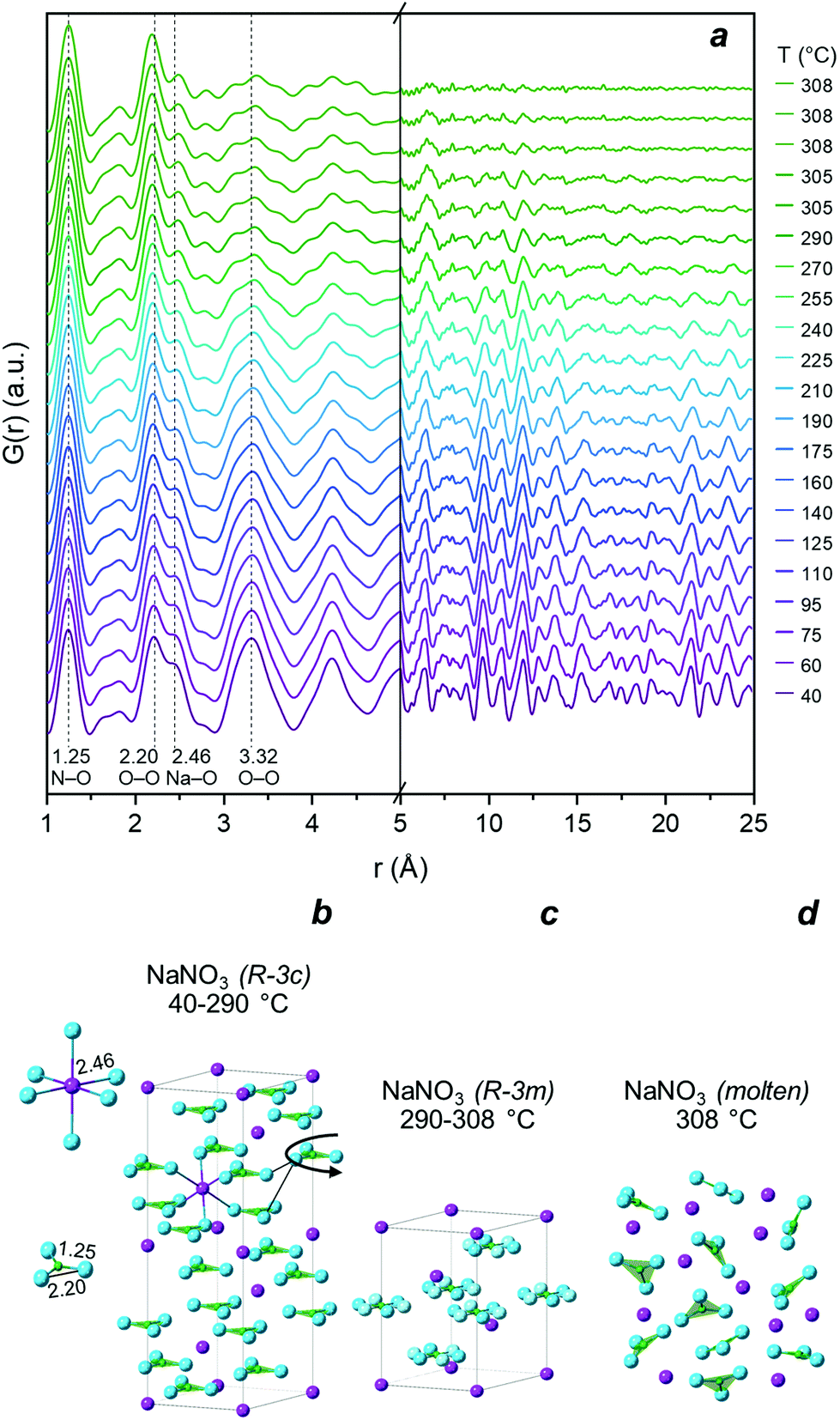 | ||
| Fig. 2 (a) PDF of NaNO3 heated from 40 °C to 308 °C in N2 and hold at 308 °C for 10 min; (b) illustration of the NaNO3Rc (ICSD: 180912) crystalline structure and short-range correlations below 2.5 Å; (c) Rm (ICSD: 180920) crystalline structure; (d) schematic representation of the molten NaNO3 state. Color legend: blue – oxygen, purple – sodium, green – nitrogen. | ||
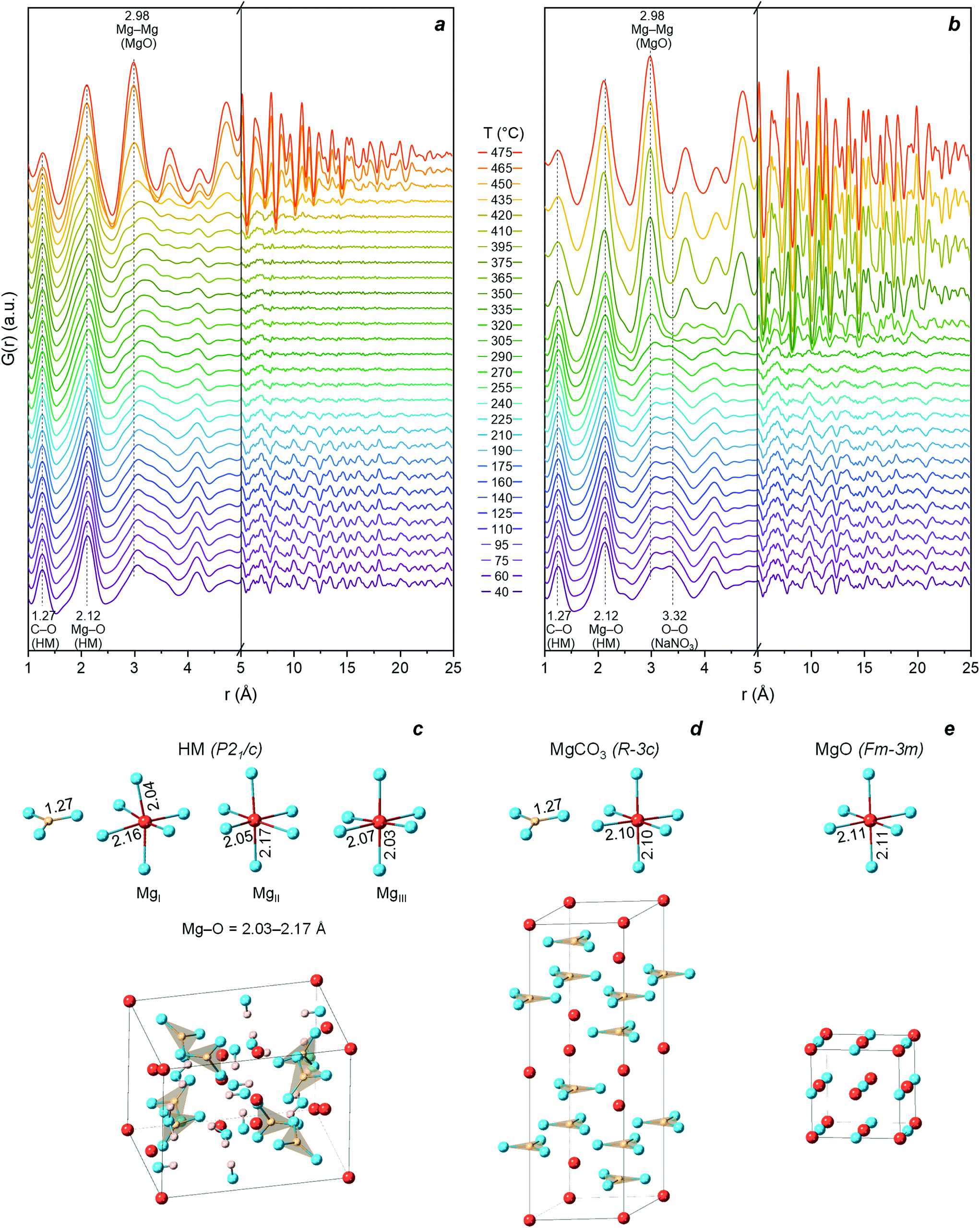 | ||
| Fig. 3 PDF obtained while heating (a) HM and (b) HM-20NaNO3 from 40 °C to 475 °C in N2. Illustration of the crystal structure and short-range correlations below 2.5 Å of (c) HM (P21/c, ICSD: 920); (d) crystalline-MgCO3 (Rc, ICSD: 40117); (e) MgO (Fmm, ICSD: 9863). Color legend: blue – oxygen, red – magnesium, pink – hydrogen, yellow – carbon. | ||
Above 190 °C, the disappearing correlations for r > 5 Å in HM reveal the loss of long-range order (i.e., amorphization), which coincides with the release of water of crystallization (based on the TGA results, Fig. 1d).
The gradual removal of bound water from the structure disrupts the hydrogen bonding network between water molecules,48,50 leading to an amorphous (partially hydrated and hydroxylated) magnesium carbonate (AMC). The material remains amorphous during the continuous loss of water of crystallization and dehydroxylation. Indeed, the intensity of the peak at ca. 3.0 Å (mainly ascribed to O–O distance between H2O molecules and between H2O and CO32− in the HM structure) decreased gradually. The loss of water was accompanied by a gradual change in the local structure, with notably, the Mg–O correlation (initially at ca. 2.12 Å) evolving towards a more asymmetric peak (inset in Fig. S9a and c†) and shifting ultimately towards a higher r (ca. 2.15 Å) and forming a shoulder at r < 2.0 Å. Between 350–450 °C, i.e., in the dehydroxylation temperature range (Fig. 1d), the splitting of the Mg–O peak in AMC intensified. This shows that during the dehydration and dehydroxylation of AMC, the distribution of the Mg–O distances becomes broader compared to HM. A similar behaviour for the first Ca–O coordination sphere upon release of water has been observed previously in the case of amorphous calcium carbonate (ACC).30 This observation can probably be linked to a re-arrangement of carbonate ions, compensating for the loss of water and hydroxyl groups around Mg, as has been shown previously for ACC by PDF and empirical potential structural refinement (EPSR) studies.51,52
The onset of MgO crystallization occurred at ca. 450 °C, marked by the growth of a Mg–O and Mg–Mg correlation at 2.11 Å and 2.98 Å that are characteristic for cubic MgO. Furthermore, the appearance of correlations for r > 5 Å indicates a long-range order in the MgO formed. The formation of a peak centered at 2.11 Å (and the decrease of the shoulder at ca. 1.5–2 Å) agrees with the formation of Mg–O octahedra within cubic MgO. Some degree of structural disorder seems to remain in MgO (see further characterization in the following section). Further crystal growth of MgO occurred upon heating to 475 °C, as confirmed by the intensification of correlations for r > 5 Å (cut-off point ca. 25 Å at 475 °C). In parallel to occurrence of the Mg–O and Mg–Mg peaks arising from crystalline MgO, the intensity of the peak attributed to C–O of the CO32− group (according to the crystallographic model of HM, Fig. S8†) decreased due to the decomposition of the carbonate to the corresponding oxide (Fig. S9c and d†). However, the low r-range of the PDF data is affected by the truncation effect using a finite Q-range, resulting in termination ripples.53 Therefore, even after the complete decomposition of the carbonate there is still a small peak at r of approximately 1.27 Å (the intensity of the low-r peaks as a function of temperature are shown in Fig. S10a and b†). A confirmation of these qualitative observations is given further below when using MCR-ALS analysis to obtain a quantitative assessment of the phase evolution with temperature.
Turning now to HM-20NaNO3 (Fig. 3b), at the initial temperature (40 °C) it consists of a mixture of crystalline HM and NaNO3. The atomic correlations of NaNO3 overlapped with those of HM and the latter dominated the PDF signal; however, the prominent peak at ca. 3.32 Å can be assigned to crystalline NaNO3. As in the case of pristine HM, during the release of water of crystallization, amorphization and a splitting of the Mg–O distance was observed (Fig. S9b and d†). Reaching 305 °C, intense atomic correlations that are assigned to both crystalline-MgCO3 and MgO appeared in the PDF data (Fig. S11 and S13†). The formation of MgO and MgCO3 atomic correlations is confirmed by the appearance of diffraction peaks in the XRD data (Fig. S11†). Importantly, the formation of crystalline phases occurs simultaneously with the melting of NaNO3 (confirmed by XRD in Fig. S4†), demonstrating that molten NaNO3 changed the crystallization behaviour of MgO and MgCO3 from HM.
Subsequently, we performed MCR-ALS analysis of the in situ PDF data to compare quantitatively the structural evolution of HM and HM-20NaNO3 upon their thermal decomposition. MCR-ALS allows to resolve the compositional changes of complex mixtures without requiring references or structural information. Using MCR-ALS analysis, a complex system with multiple evolving components is reduced to the least number of components that can describe the initial dataset. This makes MCR-ALS especially suitable for the analysis of large datasets generated during in situ and/or operando experiments, e.g., IR,54 Raman,55 XAS,56 XRD,57 or PDF.58,59 MCR-ALS provides the characteristic profiles of the individual components and the evolution of these components with time (time-concentration profiles).59 Comprehensive overviews on MCR-ALS analysis can be found in the literature39,60 and ESI† section.
The evolution of HM upon thermal treatment in N2 is well described by three components, as determined by MCR-ALS (Fig. 4a). Importantly, the components obtained by MCR-ALS analysis have a physical meaning (Fig. S13†), enabling their identification by comparison with references. We could assign the three components predicted by MCR-ALS as C1 = HM, C2 = AMC and C3 = MgO. The evolution of these components is plotted in Fig. 4a. A significant fraction of AMC appeared at ca. 200 °C and coincided with an appreciable release of water of crystallization from the HM structure (Fig. 1d). The final transformation of AMC to MgO started at 450 °C and was completed at 475 °C. Hence, the PDF experiment revealed that the thermal decomposition of HM to MgO proceeded through an AMC intermediate that formed during the continuous loss of water of crystallization.
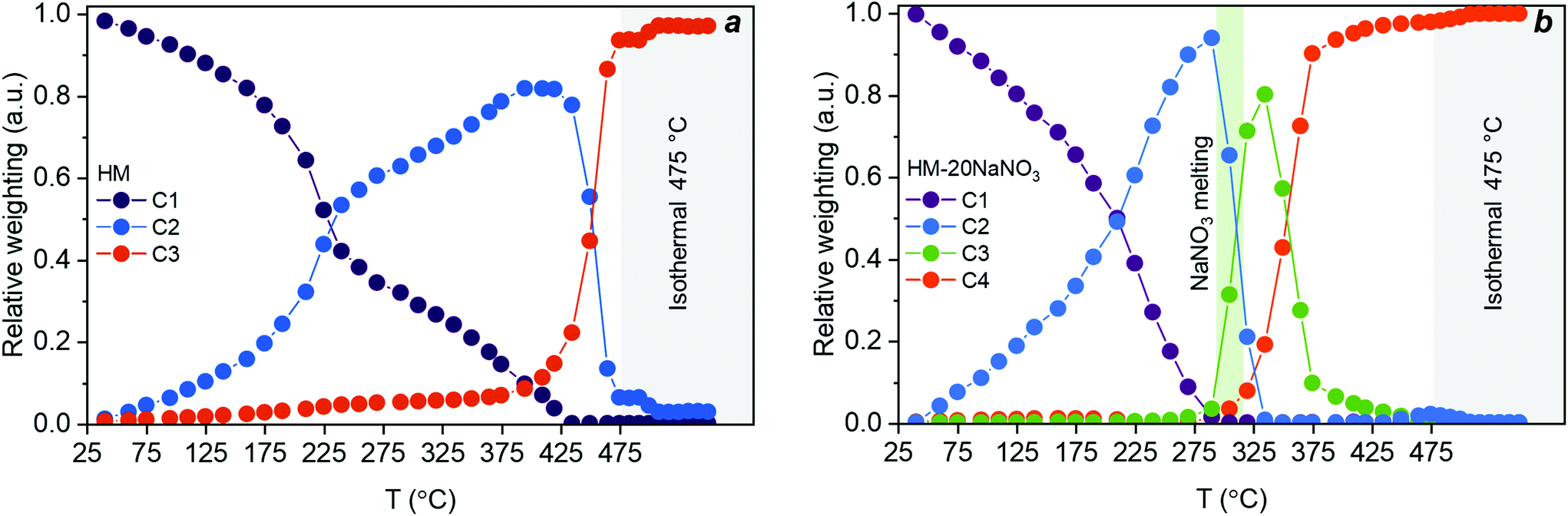 | ||
| Fig. 4 Concentration profiles extracted by MCR-ALS analysis for (a) the decomposition of HM with components assigned to HM (C1), AMC (C2), and MgO (C3); and (b) the decomposition of HM-20NaNO3 with components assigned to HM-20NaNO3 (C1), AMC-20NaNO3 (C2), crystalline-MgCO3-20NaNO3 (C3), and MgO-20NaNO3 (C4). The green vertical area in Fig. 4b corresponds to the meting point of NaNO3 while grey vertical areas in Fig. 4a and b correspond to isothermal conditions. The lack of data fit (LOF) and the standard deviation were 2.2 and 1.7% and 9.6 × 10−3 and 7.6 × 10−3 for HM and HM-20NaNO3, respectively. | ||
According to MCR-ALS analysis, the decomposition of HM-20NaNO3 can be described by four components, confirming an alteration of the decomposition pathway in the presence of (molten) NaNO3. These four components extracted from MCR-ALS analysis are ascribed to: C1 = HM-20NaNO3, C2 = AMC-20NaNO3, C3 = crystalline-MgCO3-20NaNO3 and C4 = MgO-20NaNO3. Hence, the thermal decomposition of HM-20NaNO3 proceeds via the intermediates AMC and crystalline-MgCO3, yielding ultimately MgO mixed with molten NaNO3. It should be noted that MCR-ALS analysis did not provide information on the NaNO3 phase transition, possibly due to the highly overlapping signal in the PDF and the relatively low content of NaNO3 with respect to HM. However, the XRD data showed that the melting of NaNO3 occurred between 305 °C and 308 °C (Fig. S4 and S11†). The structural changes of NaNO3 are visualized in Fig. 2. The temporal evolution of these four components is shown in Fig. 4b. In the first stage of the release of water of crystallization, the formation of AMC takes place. Subsequently, at ca. 305 °C, AMC converted rapidly to crystalline-MgCO3 upon melting of NaNO3. The formation of crystalline MgCO3 was facilitated by the rapid release of H2O from the structure upon melting of the salt, as observed in Fig. 1d, that acts as a phase-transfer agent. MgO started to form at 305 °C, in parallel to the formation of crystalline-MgCO3, which decomposed fully to MgO at ca. 410 °C.
In summary, the PDF/XRD results show that crystalline MgO formed from HM-20NaNO3 through the AMC and crystalline MgCO3 intermediates. This is in contrast to the thermal decomposition of HM, which proceeds through AMC only. The presence of molten NaNO3 accelerated the release of both H2O (from water of crystallization and through dehydroxylation) and CO2, affecting not only the decomposition pathways, but also promoting the crystallization of MgO at lower temperatures compared to HM.
| Material | Average crystallite size, nm | Cell parameter a, Å | U, Å2 |
|---|---|---|---|
| HM(MgO-308) | 3.8 | 4.229(4) | 0.020(1) |
| HM-20NaNO3(MgO-308) | 10.7 | 4.224(1) | 0.016(1) |
The slightly larger unit cell parameter observed for HM(MgO-308) with respect to HM-20NaNO3(MgO-308) was likely due to the smaller crystallite size of HM(MgO-308).64–66 A higher degree of structural disorder in HM(MgO-308) compared to HM-20NaNO3(MgO-308) is also evidenced by larger refined atomic displacement parameters (U in Tables 1 and 2). The results of PDF data fitting are in line with the XRD analysis, exhibiting the same trends in crystallite sizes, cell parameter and structural disorder (Table 2, Fig. S19 and S21†). However, slightly smaller values of the crystallite sizes were obtained by PDF analysis when compared to the values obtained by XRD, possibly due to the different influence of the instrumental resolution on the peak broadening. Nonetheless, it should be noted that the values determined by the two different analyses represent an estimation of the average crystallite size, and a distribution of sizes and shapes can also be expected (further discussion about particle shape will be inferred based on HAADF-STEM, vide infra). Comparison of the PDFs of the samples revealed that the peaks were sharper and more well-defined for HM-20NaNO3(MgO-308) than HM(MgO-308) (Fig. S19†). The width of the PDF peaks provides information about the static and dynamic disorder of the atoms involved in the atomic correlations.67 This is reflected by the larger ADPs of the PDF data (Table 2, Fig. S21†) for HM(MgO-308) compared to HM-20NaNO3(MgO-308). In addition, the Mg–O peak in HM(MgO-308) shows a higher asymmetry (Fig. S19b and S21c, d†) which suggests that a distorted Mg–O local environment remains in HM(MgO-308). This remaining distortion may arise from disordered regions at the surface of the nanoparticles. This is in line with Rietveld refinement and can be attributed to an increased structural disorder of HM(MgO-308) due to the smaller size of its MgO crystallites. Additionally, the dampening of the PDF oscillations at high r relates to the finite size effect of the MgO particles, i.e., the smaller the crystallites, the shorter the r at which the PDF peaks decays to zero.68 We observed a more pronounced dampening of correlations in HM(MgO-308) than in HM-20NaNO3(MgO-308), indicative of a smaller coherence length (i.e., a smaller crystallite size) of the MgO in HM(MgO-308).
| Material | Crystallite size diameter, nm | Cell parameter a, Å | U iso(O), Å2 | U iso(Mg), Å2 |
|---|---|---|---|---|
| HM(MgO-308) | 3.5(2) | 4.231(3) | 0.014(2) | 0.010(1) |
| HM-20NaNO3(MgO-308) | 10(2) | 4.220(2) | 0.012(1) | 0.009(1) |
Morphological characterization of the produced MgO by electron microscopy
To compare the morphology of the final MgO particles and initial precursor at a submicron scale, ex situ HAADF-STEM method was used. HAADF-STEM images of HM and HM-20NaNO3 prior to and after their thermal decomposition at 450 °C (N2, 10 °C min−1 performed in a TGA) are shown in Fig. 5 and the corresponding TEM images in Fig. S24.† MgO particles obtained from HM retained the plate-like morphology of HM (Fig. 5a and b) which is likely related to their transformation through an amorphous intermediate with a gradual release of water of crystallization and dehydroxylation. On the other hand, the morphology of MgO produced from HM-20NaNO3 was considerably different from that of HM in terms of size and shape of the MgO particles. MgO particles of irregular shape (10–80 nm) and aggregates were formed from HM-20NaNO3 (Fig. 5c and d and Fig. S24†).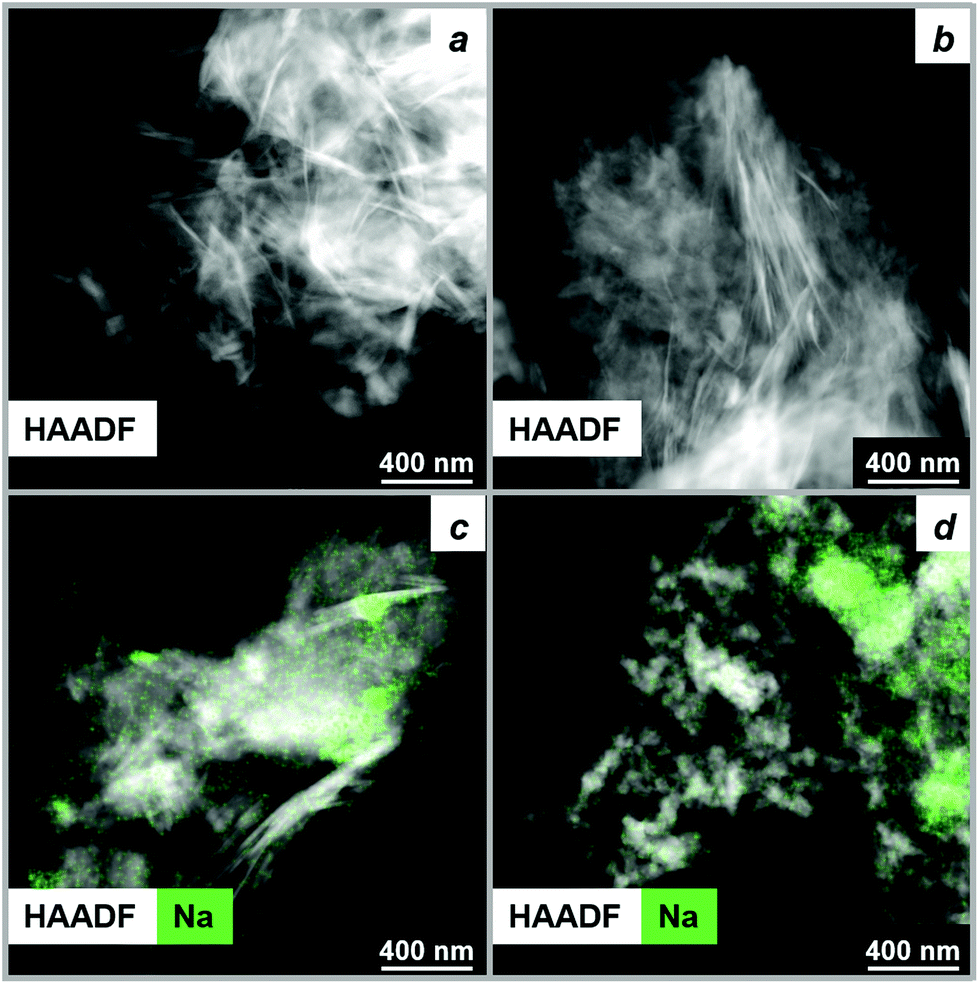 | ||
| Fig. 5 HAADF-STEM images along with Na EDX maps of (a) as-received HM, (b) HM after thermal treatment in N2 at 450 °C, (c) as-prepared HM-20NaNO3 and (d) HM-20NaNO3 after thermal treatment in N2 at 450 °C. | ||
The surface area of decomposed HM-20NaNO3 is significantly lower than that of decomposed HM (33 and 306 m2 g−1 respectively, according to N2 physisorption, Table S7†). We attribute the morphological differences of MgO produced from HM and HM-20NaNO3 to their transitioning through different intermediates, as determined by MCR-ALS analysis. Previous studies have also shown that the decomposition pathway of the precursors strongly determines the final MgO morphology.40,63,72,73
Visualization of MgO formation from HM by in situ SEM
In situ SEM allowed us to visualize the evolution of HM and HM-20NaNO3 precursors at the micron scale during their thermal decomposition in N2 between 150 and 450 °C (46 Pa, 10 °C min−1, Fig. 6 and Movie S1†). HM exhibits the typical plate-like morphology forming larger agglomerates with sizes of several microns. The morphology of HM does not change significantly with heating up to 450 °C, confirming the continuous nature of the HM decomposition (Fig. 6a–d). In HM-20NaNO3, NaNO3 particles with polyhedral shape can be identified (marked as a green area in Fig. 6, panels e-h and also visible in Movie S1†) among HM particles. The shrinkage of HM particles is observed at 200 °C and is likely related to the release of water of crystallization as indicated by TGA results discussed above. NaNO3 starts to melt at ca. 300 °C and this process is associated with a formation of cavities and channels at the surface of the NaNO3 particles. Close to 400 °C, NaNO3 particles are no longer observed by in situ SEM, explained by their melting. The melting of NaNO3 affects the morphology of the nearby HM particles, which we interpret as the formation of MgO in the proximity of molten NaNO3 (in correlation with TGA and PDF studies). The change is rather sudden, which is in line with the promoted formation of MgO in the presence of molten NaNO3 (Movie S1†). | ||
| Fig. 6 In situ SEM images obtained while heating (a–d) HM and (e–h) HM-20NaNO3 from 100 °C to 450 °C in 46 Pa of N2. The areas highlighted in yellow in Fig. 6a–h shows the HM particle agglomerates, while areas highlighted in green in Fig. 6e–h emphasizes the thermal evolution of typical NaNO3 particle. | ||
Conclusions
TGA and HF coupled with gas analysis showed that molten NaNO3 accelerated the decomposition of HM through the dehydration, dehydroxylation and decarbonation steps. In the presence of NaNO3, the temperature for the complete conversion of HM to MgO was lowered by ca. 100 °C. The PDF analysis of in situ total scattering data, complemented by MCR-ALS analysis, demonstrated that the presence of molten NaNO3 has a crucial influence on the decomposition pathways of HM and the structure of the resulting MgO. In the case of pristine HM, the decomposition occurred through an amorphous intermediate that formed upon the release of water of crystallization. Importantly, the amorphization process was accompanied by a rearrangement of the local structure resulting in the splitting of the Mg–O correlation. The Mg–O splitting intensified upon dehydroxylation at higher temperatures. Below the melting point of NaNO3, the decomposition of the promoted sample occurred in a similar fashion as for pristine HM and an amorphous intermediate was formed. However, melting of NaNO3 induces a rapid crystallization of the amorphous intermediate into crystalline MgCO3 and MgO, which was followed by the rapid decomposition of MgCO3 to MgO. MCR-ALS analysis further confirmed the lowering of the decomposition temperature of promoted HM by ca. 100 °C.Concerning the structure and morphology of the MgO formed, the presence of NaNO3 led to an increase of its average crystallite size reaching approximately 10.7 nm, whereas the pristine sample yielded MgO with an average crystallite size of 3.8 nm only, and with a higher degree of structural disorder. The larger average crystallite size is in line with the morphological differences of MgO observed via ex situ HAADF-STEM, showing the formation of highly crystalline nanoparticles in the presence of NaNO3. In situ SEM allowed to visualize the morphological changes occurring in HM promoted with NaNO3, at the micrometre scale.
We expect that the obtained atomic level insights on the effect of molten NaNO3 on the decomposition pathways of HM and the morphology and properties of the resultant MgO will aid the design of improved MgO-based sorbents and set the basis for further investigations of the mechanism of MgCO3 formation and growth. Improving the performance of MgO-based CO2 sorbents will rely critically on the rational selection of the most appropriate promoter. On the other hand, optimization of the surface area and crystallite size of MgO do not seem to be the prime parameters to optimize the CO2 capture performance of the sorbent. The presence of a promoter is a pre-requisite for MgO to act as a practical CO2 sorbent, therefore, determining the exact role of the promoter in carbonation and calcination reactions is key for the manufacture of more effective sorbents. Additionally, we have showcased the potential of PDF analysis for materials research on CO2 sorbents. Forthcoming studies shall shed light onto the growth of MgCO3 and the nature of the interaction between MgO and NaNO3 at the interface by surface sensitive techniques.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme grant agreement No. 819573 and from Swiss National Science Foundation (SNSF, 200020_156015). The SNSF Project 206021_183290 “Multi-Scale in situ observation of functional materials in their relevant state” is acknowledged. We thank the Scientific Centre for Optical and Electron Microscopy (ScopeM) for providing training on and access to electron microscopes. The ESRF is acknowledged for providing access to beam-time through proposal MA-3415. P. M. Abdala would like to acknowledge the ETH SEED grant scheme (SEED-02 17-2) and J. Drnec is acknowledged for his assistance during total scattering measurements.Notes and references
- J.-M. Barnola, D. Raynaud, Y. S. Korotkevich and C. Lorius, Nature, 1987, 329, 408–414 CrossRef CAS.
- Trends in Atmospheric Carbon Dioxide, https://www.esrl.noaa.gov/gmd/ccgg/trends, (accessed January 2020).
- B. D. Santer, K. Taylor, T. Wigley, T. Johns, P. Jones, D. Karoly, J. Mitchell, A. Oort, J. Penner and V. Ramaswamy, Nature, 1996, 382, 39–46 CrossRef CAS.
- International Energy Agency, CO2 emissions from fuel combustion – Highlights, IEA Publications, Paris, 2017 Search PubMed.
- A. Hassanzadeh and J. Abbasian, Fuel, 2010, 89, 1287–1297 CrossRef CAS.
- X. Zhao, G. Ji, W. Liu, X. He, E. J. Anthony and M. Zhao, Chem. Eng. J., 2018, 332, 216–226 CrossRef CAS.
- J. Chi, L. Zhao, B. Wang, Z. Li, Y. Xiao and Y. Duan, Int. J. Hydrogen Energy, 2014, 39, 6479–6491 CrossRef CAS.
- F. Goff and K. Lackner, Environ. Geosci., 1998, 5, 89–101 CrossRef.
- W. Gao, L. Sun and Q. Wang, in Pre-combustion Carbon Dioxide Capture Materials, ed. Q. Wang, The Royal Society of Chemistry, Cambridge, 2018, ch. 2, pp. 61–143 Search PubMed.
- Y. Qiao, J. Wang, Y. Zhang, W. Gao, T. Harada, L. Huang, T. A. Hatton and Q. Wang, Ind. Eng. Chem. Res., 2017, 56, 1509–1517 CrossRef CAS.
- A. Dal Pozzo, A. Armutlulu, M. Rekhtina, P. M. Abdala and C. R. Müller, ACS Appl. Energy Mater., 2019, 2, 1295–1307 CrossRef.
- Y. Hu, Y. Guo, J. Sun, H. Li and W. Liu, J. Mater. Chem. A, 2019, 7, 20103–20120 RSC.
- X. Yang, L. Zhao, Y. Liu, Z. Sun and Y. Xiao, Ind. Eng. Chem. Res., 2016, 56, 342–350 CrossRef.
- M. L. T. Triviño, V. Hiremath and J. G. Seo, Environ. Sci. Technol., 2018, 52, 11952–11959 Search PubMed.
- A.-T. Vu, Y. Park, P. R. Jeon and C.-H. Lee, Chem. Eng. J., 2014, 258, 254–264 CrossRef CAS.
- K. Zhang, X. S. Li, W. Z. Li, A. Rohatgi, Y. Duan, P. Singh, L. Li and D. L. King, Adv. Mater. Interfaces, 2014, 1, 1400030 CrossRef.
- T. Harada, F. Simeon, E. Z. Hamad and T. A. Hatton, Chem. Mater., 2015, 27, 1943–1949 CrossRef CAS.
- H. Lee, M. L. T. Triviño, S. Hwang, S. H. Kwon, S. G. Lee, J. H. Moon, J. Yoo and J. G. Seo, ACS Appl. Mater. Interfaces, 2018, 10, 2414–2422 CrossRef CAS PubMed.
- S.-I. Jo, Y.-I. An, K.-Y. Kim, S.-Y. Choi, J.-S. Kwak, K.-R. Oh and Y.-U. Kwon, Phys. Chem. Chem. Phys., 2017, 19, 6224–6232 RSC.
- Hydromagnesite, http://www.handbookofmineralogy.org/pdfs/hydromagnesite.pdf, (accessed January 2020).
- S. Jin, K. Ho, A.-T. Vu and C.-H. Lee, Energy Fuels, 2017, 31, 9725–9735 CrossRef CAS.
- Y. Sawada, K. Uematsu, N. Mizutani and M. Kato, Thermochim. Acta, 1978, 27, 45–59 CrossRef CAS.
- D. Bhattacharjya, T. Selvamani and I. Mukhopadhyay, J. Therm. Anal. Calorim., 2012, 107, 439–445 CrossRef CAS.
- C. Janet, B. Viswanathan, R. Viswanath and T. Varadarajan, J. Phys. Chem. C, 2007, 111, 10267–10272 CrossRef CAS.
- N. Koga and Y. Yamane, J. Therm. Anal. Calorim., 2008, 93, 963–971 CrossRef CAS.
- L. Hollingbery and T. Hull, Thermochim. Acta, 2010, 509, 1–11 CrossRef CAS.
- P. Ballirano, C. De Vito, S. Mignardi and V. Ferrini, Chem. Geol., 2013, 340, 59–67 CrossRef CAS.
- S. J. Billinge and M. G. Kanatzidis, Chem. Commun., 2004, 7, 749–760 RSC.
- C. E. White, N. J. Henson, L. L. Daemen, M. Hartl and K. Page, Chem. Mater., 2014, 26, 2693–2702 CrossRef CAS.
- M. Albéric, L. Bertinetti, Z. Zou, P. Fratzl, W. Habraken and Y. Politi, Adv. Sci., 2018, 5, 1701000 CrossRef PubMed.
- A. E. Morandeau and C. E. White, J. Mater. Chem. A, 2015, 3, 8597–8605 RSC.
- A. Van der Eerden, J. A. van Bokhoven, A. Smith and D. Koningsberger, Rev. Sci. Instrum., 2000, 71, 3260–3266 CrossRef CAS.
- J. Xto, R. Wetter, C. N. Borca, C. Frieh, J. A. van Bokhoven and T. Huthwelker, RSC Adv., 2019, 9, 34004–34010 RSC.
- W. van Beek, O. V. Safonova, G. Wiker and H. Emerich, Phase Transitions, 2011, 84, 726–732 CrossRef CAS.
- J. Kieffer and D. Karkoulis, J. Phys.: Conf. Ser., 2013, 425, 202012 CrossRef.
- P. Juhás, T. Davis, C. L. Farrow and S. J. Billinge, J. Appl. Crystallogr., 2013, 46, 560–566 CrossRef.
- C. Farrow, P. Juhas, J. Liu, D. Bryndin, E. Božin, J. Bloch, T. Proffen and S. Billinge, J. Phys.: Condens. Matter, 2007, 19, 335219 CrossRef CAS PubMed.
- J. Rodriguez-Carvajal and T. Roisnel, IUCrJ, 1998, 20, 35–36 Search PubMed.
- J. Jaumot, A. de Juan and R. Tauler, Chemom. Intell. Lab. Syst., 2015, 140, 1–12 CrossRef CAS.
- A. Shkatulov, T. Krieger, V. Zaikovskii, Y. Chesalov and Y. Aristov, ACS Appl. Mater. Interfaces, 2014, 6, 19966–19977 CrossRef CAS PubMed.
- P. Ballirano, Phys. Chem. Miner., 2011, 38, 531–541 CrossRef CAS.
- S. Antao, I. Hassan, W. Mulder and P. Lee, Phys. Chem. Miner., 2008, 35, 545–557 CrossRef CAS.
- W. Gonschorek, W. W. Schmahl, H. Weitzel, G. Miehe and H. Fuess, Z. Kristallogr., 1995, 210, 843–849 CAS.
- W. W. Schmahl and E. Salje, Phys. Chem. Miner., 1989, 16, 790–798 CrossRef CAS.
- S. Tahara, H. Toyama, H. Shimakura and T. Fukami, EPJ Web Conf., 2017, 151, 01004 CrossRef.
- H. Ohno and K. Furukawa, J. Chem. Soc., Faraday Trans. 1, 1978, 74, 1861–1870 RSC.
- M. C. Wilding, M. Wilson, M. C. Ribeiro, C. J. Benmore, J. Weber, O. Alderman, A. Tamalonis and J. Parise, Phys. Chem. Chem. Phys., 2017, 19, 21625–21638 RSC.
- M. Akao and S. Iwai, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 1273–1275 CrossRef.
- M. Akao, F. Marumo and S. Iwai, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 2670–2672 CrossRef.
- S. Bach, E. Visnow, M. Panthöfer, T. Gorelik, A. G. Buzanich, A. Gurlo, U. Kolb, F. Emmerling, C. Lind and W. Tremel, Eur. J. Inorg. Chem., 2016, 2016, 2072–2081 CrossRef CAS.
- A. C. Jensen, S. Imberti, S. F. Parker, E. Schneck, Y. Politi, P. Fratzl, L. Bertinetti and W. J. Habraken, J. Phys. Chem. C, 2018, 122, 3591–3598 CrossRef CAS.
- H. Du and E. Amstad, Angew. Chem., Int. Ed., 2020, 59, 1798–1816 CrossRef CAS PubMed.
- X. Qiu, E. S. Božin, P. Juhas, T. Proffen and S. J. Billinge, J. Appl. Crystallogr., 2004, 37, 110–116 CrossRef CAS.
- M. Khanmohammadi, S. Amani and A. B. Garmarudi, Anal. Methods, 2016, 8, 2799–2804 RSC.
- J. P. Smith, E. C. Holahan, F. C. Smith, V. Marrero and K. S. Booksh, Analyst, 2019, 144, 5425–5438 RSC.
- A. Tsoukalou, P. M. Abdala, D. Stoian, X. Huang, M.-G. Willinger, A. Fedorov and C. R. Müller, J. Am. Chem. Soc., 2019, 141, 13497–13505 CrossRef CAS PubMed.
- A. Taris, M. Grosso, M. Brundu, V. Guida and A. Viani, J. Appl. Crystallogr., 2017, 50, 451–461 CrossRef CAS.
- M. Roelsgaard, A.-C. Dippel, K. A. Borup, I. G. Nielsen, N. L. N. Broge, J. T. Röh, O. Gutowski and B. B. Iversen, IUCrJ, 2019, 6, 299–304 CrossRef CAS PubMed.
- F. M. Michel, V. Barrón, J. Torrent, M. P. Morales, C. J. Serna, J.-F. Boily, Q. Liu, A. Ambrosini, A. C. Cismasu and G. E. Brown, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2787–2792 CrossRef CAS PubMed.
- J. Jaumot, R. Gargallo, A. de Juan and R. Tauler, Chemom. Intell. Lab. Syst., 2005, 76, 101–110 CrossRef CAS.
- S. Y. Yao, W. Q. Xu, A. C. Johnston-Peck, F. Z. Zhao, Z. Y. Liu, S. Luo, S. D. Senanayake, A. Martínez-Arias, W. J. Liu and J. A. Rodriguez, Phys. Chem. Chem. Phys., 2014, 16, 17183–17195 RSC.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- J. Green, J. Mater. Sci., 1983, 18, 637–651 CrossRef CAS.
- A. Cimino, P. Porta and M. Valigi, J. Am. Ceram. Soc., 1966, 49, 152–156 CrossRef CAS.
- P. M. Diehm, P. Ágoston and K. Albe, ChemPhysChem, 2012, 13, 2443–2454 CrossRef CAS PubMed.
- B. M. Maoz, E. Tirosh, M. B. Sadan, I. Popov, Y. Rosenberg and G. Markovich, J. Mater. Chem., 2011, 21, 9532–9537 RSC.
- A. S. Masadeh, J. Exp. Nanosci., 2016, 11, 951–974 CrossRef CAS.
- V. Petkov, in Characterization of Materials, ed. E. N. Kaufmann, Wiley, New York, 2012, pp. 1361–1372 Search PubMed.
- H. Cui, Q.-M. Zhang, Y. Hu, C. Peng, X. Fang, Z.-M. Cheng, V. V. Galvita and Z. Zhou, ACS Appl. Mater. Interfaces, 2018, 10, 20611–20620 CrossRef CAS PubMed.
- K. Zhang, X. S. Li, H. Chen, P. Singh and D. L. King, J. Phys. Chem. C, 2015, 120, 1089–1096 CrossRef.
- T. Harada and T. A. Hatton, Chem. Mater., 2015, 27, 8153–8161 CrossRef CAS.
- M. D. Susman, H. N. Pham, A. K. Datye, S. Chinta and J. D. Rimer, Chem. Mater., 2018, 30, 2641–2650 CrossRef CAS.
- T. Xu, X. Zhou, Z. Jiang, Q. Kuang, Z. Xie and L. Zheng, Cryst. Growth Des., 2008, 9, 192–196 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr01760d |
| This journal is © The Royal Society of Chemistry 2020 |