
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ speciation and spatial mapping of Zn products during pulsed laser ablation in liquids (PLAL) by combined synchrotron methods†
Stefan
Reich‡
 a,
Jörg
Göttlicher
a,
Anna
Ziefuss
b,
René
Streubel
b,
Alexander
Letzel
b,
Andreas
Menzel
c,
Olivier
Mathon
d,
Sakura
Pascarelli
d,
Tilo
Baumbach
ae,
Marcus
Zuber
a,
Bilal
Gökce
b,
Stephan
Barcikowski
*b and
Anton
Plech
*a
a,
Jörg
Göttlicher
a,
Anna
Ziefuss
b,
René
Streubel
b,
Alexander
Letzel
b,
Andreas
Menzel
c,
Olivier
Mathon
d,
Sakura
Pascarelli
d,
Tilo
Baumbach
ae,
Marcus
Zuber
a,
Bilal
Gökce
b,
Stephan
Barcikowski
*b and
Anton
Plech
*a
aInstitute for Photon Science and Synchrotron Radiation, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany. E-mail: anton.plech@kit.edu
bDepartment of Technical Chemistry I and Center for Nanointegration Duisburg-Essen, University of Duisburg-Essen, Universitätsstrasse 7, D-45141 Essen, Germany. E-mail: stephan.barcikowski@uni-due.de
cPaul-Scherrer-Institut, CH Villigen-PSI, Switzerland
dEuropean Synchrotron Radiation Facility, F-38043 Grenoble, France
eLaboratory for applications of synchrotron radiation, Karlsruhe Institute of Technology, Engesserstr. 15, D-76131 Karlsruhe, Germany
First published on 16th June 2020
Abstract
Pulsed laser ablation in liquids is a hierarchical multi-step process to produce pure inorganic nanoparticle colloids. Controlling this process is hampered by the partial understanding of individual steps and structure formation. In situ X-ray methods are employed to resolve macroscopic dynamics of nanosecond PLAL as well to analyse the distribution and speciation of ablated species with a microsecond time resolution. High time resolution can be achieved by synchrotron-based methods that are capable of ‘single-shot’ acquisition. X-ray multicontrast imaging by a Shack–Hartmann setup (XHI) and small angle X-ray scattering (SAXS) resolve evolving nanoparticles inside the transient cavitation bubble, while X-ray absorption spectroscopy in dispersive mode opens access to the total material yield and the chemical state of the ejecta. It is confirmed that during ablation nanoparticles are produced directly as well as reactive material is detected, which is identified in the early stage as Zn atoms. Nanoparticles within the cavitation bubble show a metal signature, which prevails for milliseconds, before gradual oxidation sets in. Ablation is described by a phase explosion of the target coexisting with full evaporation. Oxidation occurs only as a later step to already formed nanoparticles.
1 Introduction
Pulsed laser ablation in liquids1–5 is a route to produce pure nanoparticles, such as zinc oxide, without impurities that may interfere with biological, catalytic6,7 or photonic function.8–11 The primary process in PLAL happens on a sub-microsecond time scale, including material evaporation and spallation. While it has been observed that ablation of zinc produces partially metallic particles,12,13 which oxidize on a minute time scale,6,14 not much is known about the speciation in situ during the multistep ablation process. In general, it is established that during PLAL the target is heated far above the melting point such that explosive boiling and evaporation can eject a thin liquid film on the metal target15 as well as atomic or cluster species16–18 and primary particles.19–22 Aggregation of monomers has been modelled to occur on a sub-microsecond time scale.15,21 Subsequently, the material gets in contact to the liquid phase, the latter is also evaporating due the strong excitation. On a sub-millisecond time scale a vapour bubble of the surrounding liquid forms with a size of hundreds of micrometers to millimetres, depending on pulse energy and number of pulses.23,24 The bubble collapse fosters nanoparticle agglomeration and formation of secondary particles.25 Further particle ripening can extend to much longer time scales.26 The interior of the bubble is known to present quite unique conditions, in particular, if a laser-induced plasma is active. Size quenching has been seen to be active already inside the vapour,27,28 if electrolytes or surfactants are present.18Standard laboratory approaches to capture dynamics in this early time scale have been limited to high-speed videography29,30 or light (plasma) emission or scattering.31–35 Snapshot imaging with visible light has been used extensively to visualize the formation of a cavitation bubble after the intense heating of the target-liquid interface. High-speed cameras are combined with shadowgraphy to film the bubble on a microsecond time scale.29 Alternatively, pump-probe setups with lasers36,37 or flash lamps25,38 as probes can be used to reconstruct a film-like sequence of the different stages. Plasma emission delivers valuable information on cluster temperature or ionic species.17 Optical spectroscopy12 is virtually impossible to apply before the bubble has vanished due to the strong perturbation by refraction and opaque micro bubble clouds. Novel approaches via creation of a laser-induced flat transparent window allow the clear observation of the formation of nanoparticles inside the ablation bubble through laser scattering,35 but without chemical speciation or exact size determination. Thus, particle sensitivity and quantification of ablated mass yield is difficult to obtain in situ.
Therefore, synchrotron methods with short-wavelength radiation become extremely valuable, because they are less perturbed by the inhomogeneities during ablation, are sensitive to an atomic to nanometre length scale and also yield spectral information that can be rationalized for deriving the chemical state of the involved elements.
Here, we describe the use of synchrotron-based X-ray probes that are capable of a ‘single shot’ approach to acquire a data set of spatial or spectroscopic information in one laser shot. This does not necessarily mean that one single measurement suffices, but averaging over multiple events to cope with the low interaction cross-section may still be necessary. Yet, the signal will not be affected by drifts in sequential scanning approaches. Time-resolved small angle X-ray scattering (TR-SAXS) has been among the first techniques that were able to resolve emitted nanoparticles.19,39,40 Complemented by time-resolved powder X-ray scattering (TR-WAXS) a quite good picture of the nanoparticle size distribution in PLAL of noble metals has been established.22,40 X-ray radiography has been used to penetrate the bubble phase to confirm the homogeneous first bubble and emission of a part of the particle mass only after the second or third bubble20 in a symmetric collapse, but is insensitive to the particle size distribution.
Contrast variation in X-ray imaging, on the other hand, can produce also a dark-field contrast in addition to the well-known absorption and phase contrast. Dark-field contrast can be detected, if very faint deviations in the beam direction can be resolved. This has, for example, been achieved in Talbot interferometry.41,42 An approach more suitable for pulsed acquisition is the Hartmann (TR-XHI) approach, where a large X-ray field is separated into an array of small beamlets, whose positions and scattering-induced broadening may be resolved.43–45 It can be regarded as a multiplexed SAXS with information on the nanoscale. This allows to interpret the dark-field channel as signature of spatial particle size distribution.24,28,46
Finally, we demonstrate here that time resolved X-ray absorption fine structure spectroscopy (TR-XAFS) in dispersive mode can be employed to focus on the chemical speciation47 of zinc during the ablation process. When tuning the X-ray energy around characteristic absorption edges of the elements the increase in absorption above the edge can be identified with the specific element under investigation. The magnitude of absorption can be directly related to the total mass of the investigated element in the beam, while the shape of the absorption edge reflects the chemical state.
2 Experimental
2.1 Ablation chamber
The ablation process was performed in a setup, which ensured reproducible conditions for stroboscopic and continuous-wave probing for an extended number of individual laser pulses. A description can be found elsewhere.14,20,22 The main features are a 3D printed chamber body that encloses a 10 × 10 × 5 mm3 reaction volume. The volume is continuously flushed by water to remove the ablated material between successive laser pulses. The laser beam enters the volume through a plano-convex lens of 38 mm effective focal length that also seals the chamber. X-rays enter through side windows of polyimide (Kapton, DuPont). The zinc wire (supported by the back wall)48 is pulled through the chamber at a speed that replaces the irradiated spot within around 5 laser pulses in order to optimize the ablation efficiency.492.2 Laser
An Nd:YAG laser (Continuum Minilite I, 7 ns pulse length) with up to 40 mJ and 10 Hz maximum repetition rate was employed for all experiments except for SAXS. The pulse energy was set to 12 mJ, with a fluence of about 380 kJ m−2 at a 0.2 mm focus diameter. Synchronization was done by a master trigger that actuates the laser Q-switch as well as the detector (a single image for stroboscopic videography, an image sequence of the 2D camera in XHI, or the 1D camera in dispersive TR-XAFS, respectively). Time resolution is not limited by synchronization jitter, but rather by the exposure time (10 μs for videography, 20 μs for XHI, 11.3 μs for TR-XAFS).2.3 Materials
The ablation was performed in ultrapure water (MilliQ, Millipore), which is pumped from a reservoir (20 L) by a gear pump. A Zn wire with purity of 99.99% and 1 mm diameter was purchased from Chempur (Germany).2.4 Videography
Visible-light snapshots of the cavitation bubble are performed by synchronizing a Xenon flashlamp (Rheintacho, 10 μs) and a video camera (Basler ACE) to the laser pulse with variable delay in 2 μs steps. The image sequence is stroboscopic, i.e. every image relates to an individual laser pulse. The images are only corrected for dynamic range.SAXS was performed at the beamline cSAXS at the Swiss Light source (PSI Villigen, CH) using a narrow X-ray beam of 6 × 24 μm2 cross-section. A pixellated X-ray detector (Pilatus 2M) at 7.1 m distance from the ablation chamber recorded the scattering from the sample at a height of 4 mm above the target surface and out of the laser path to avoid detecting secondary fragmentation. The time resolution was set to 1 second between SAXS measurements, not resolving the fast bubble-related dynamics.22 An Nd:YAG picosecond laser (Edgewave PX400-2-GM) with 1 mJ at 1 kHz repetition rate and 12 ps pulse length was used (fluence 60 kJ m−2 at 1064 nm). Water was not exchanged for the time of irradiation. Scattering was accumulated for 1000 pulses. Analysis is done by radial averaging and standard corrections to obtain the one-dimensional scattering curves I(q), with q = 4π/λ·sin(2Θ/2) at an X-ray wavelength of λ = 1.107 Å as function of scattering angle 2Θ. Particle size fitting was performed by a reverse Monte Carlo approach using the software McSAS.50
X-ray Shack–Hartmann imaging was performed at the UFO instrument at the synchrotron KARA (KIT Karlsruhe, D). A wide, polychromatic X-ray beam from the bending magnet source was spectrally filtered by a beryllium window (0.25 mm). The median X-ray energy after passing the additional 5 mm of water was centred at 17.8 keV. The beam is shaped into a square array of 32 × 32 beamlets by a Shack–Hartmann mask (CARL, stack of concave lens arrays in Mylar foil)46 with 65 μm pitch. The beamlets were imaged onto a thin scintillator (Lu:aluminium garnet, Crytur, Czechia) after passing the chamber volume. The scintillator light was recorded by a fast frame CMOS (complementary metal oxide semiconductor) camera (DIMAX, PCO). An effective frame rate of 50 kHz was achieved by interleaving two subsequent films at 25 kHz with a shifted delay of 20 μs to obtain an effective image rate with a 20 μs delay step. Averaging was done over 105 laser pulses. The intensity, position and width of each beamlet were obtained by a Fourier analysis43,45,51,52 and decomposed into the 3 image modalities: absorption, differential phase and dark-field (scattering) contrast. The latter contains contributions from scattering from sub-micrometre-sized inhomogeneities, such as nanoparticles. The sensitivity here ranges from 25 to 100 nm in diameter as given by geometry.44 The lower limit in our case to 25 nm (50% signal height per unit mass), given by the strongly varying scattering cross section with size. The upper limit is given by the angular (spatial) resolution on beamlet detection, 100 nm in the present case. Further details on the method can be found elsewhere.28,43,45,46,53 The interleaving scheme is displayed in the ESI.†
X-ray absorption spectroscopy was performed in two experiments with 11.3 μs and 300 μs time resolution, respectively at the beamline ID24 (European Synchrotron Radiation Facility, Grenoble) in a dispersive setup.54 A polychromator focuses a part of the X-ray spectrum from the insertion device around the zinc K edge onto the sample position to a <4 μm spot size. The beam disperses in energy along one spatial axis behind the sample to be spatially resolved on a fast linear detector (Ge microstrip detector55 with 11.3 μs and Frelon (ESRF), scintillator coupled, with 300 μs time bins, respectively). After standard image corrections (dark current, flat-field response) an absorption spectrum can ideally be recorded in a single shot. We used averages over about 2000 pulses each (after rejecting spectra with strong signal perturbation). The energy resolution is 1.3 eV.54 The X-ray absorption near edge structure (XANES) is used to gain information about Zn speciation.
2.5 Reference spectra and calculations
XAFS spectra of reference substances were recorded at the beamline SUL-X at KARA. A zinc foil, ZnO and hydrozincite Zn5(CO3)2(OH)6 powders (Sigma Aldrich) as cellulose pellets were measured in transmission geometry and ZnCl2 (Sigma Aldrich) as solution in fluorescence geometry. The absorption spectrum of zinc vapour was taken from ref. 56. The X-ray energy was calibrated to the zinc foil spectrum as found at ref. 56 and differed less than 0.4 eV from established ref. 57 and 58.3 Results
Structure formation in PLAL covers several orders of magnitude in time and space from the ultrafast atomic-scale excitation or nanoscale phase explosion up to the millimetre-scaled shock wave and bubble formation. The latter provides distinct reaction conditions in temperature and pressure that define the morphology and chemical speciation of the products.6,24,59,60 Elucidation of the reaction pathways needs to resolve different observables of the emitted material, such as overall mass, size distribution or chemical state with high time resolution. Short-wavelength radiation can be of high value to the sensitivity on these properties, particularly because X-rays penetrate matter without strong refraction. The short wavelength is compatible with investigated length scales to resolve nanoscale structure. Finally synchrotron based X-ray radiation can be tuned to selectively address given atomic species via TR-XAFS. It remains a challenge to apply these methods with high time resolution.3.1 Macroscopic dynamics
The observation of the macroscopic dynamics is directly accessible in visible-light PLAL investigations. A large setback is the difficult access to the bubble interior35 as well as the low sensitivity on nanoparticle distribution. Dense clouds of nanoparticles are readily seen, however, a disambiguation from permanent-gas microbubbles is difficult.46 The perturbation-free imaging of the bubble can be readily achieved by X-rays in a conventional radiography setup, as the refractive index change at the water–gas interface amounts for only about 10−6. Imaging contrast in radiography primarily is an absorption contrast and to some extent a phase contrast by edge enhancement.20 Still, ablated nanoparticle material will be barely visible due to the low concentration. In typical ablation conditions for millijoule laser pulses focused to spots around 0.2 mm on a metal surface, the ablated material will roughly be in the range of 10−8 g, too low to cause significant absorption. Single visible snapshots of the bubble evolution during laser ablation are shown in the left column of Fig. 1, with the expansion phase at 15 μs, an extended bubble at 114 μs and the collapse phase at 234 μs. The expansion is blurred by the fast moving bubble and expelled material. Another observation is that the bubble surface is less smooth than observed earlier for PLAL on noble metals in the expanded phase. This might be due to the relatively strong ablation due to the low energy needed for evaporation of zinc as compared to other metals49 as well as ablation of pre-deposited nanoparticles in a multi-shot exposure. Part of the ablated mass may have sizes large enough to penetrate the bubble interface to cause ripples.21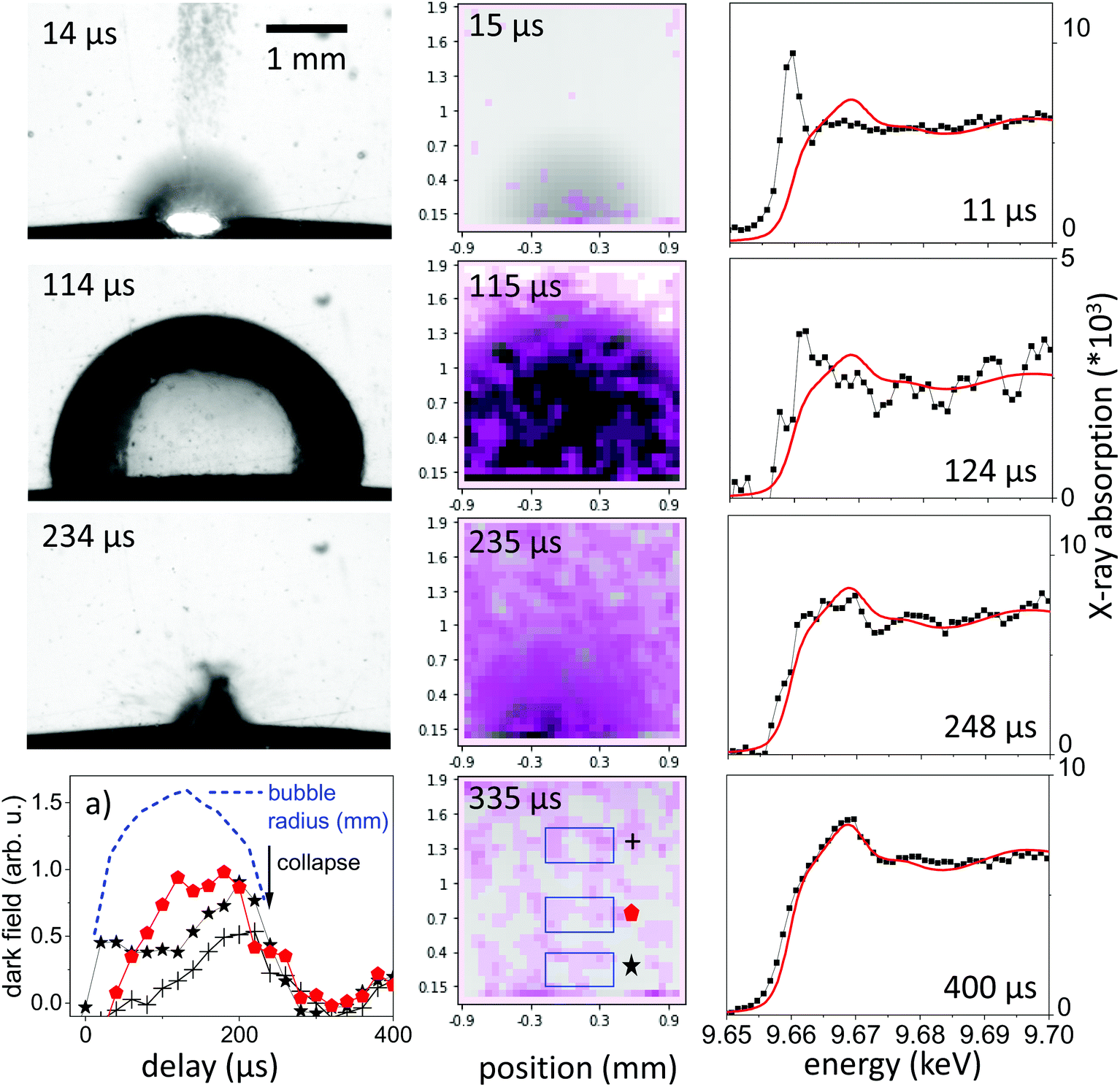 | ||
| Fig. 1 Visible stroboscopic images (left column), X-ray multicontrast images (center column, pink: dark-field contrast, gray: absorption contrast) and Zn K-edge X-ray absorption spectra (right column) as function of delay in microseconds after laser irradiation. The lower left plot (a) depicts the dark-field signal integrated over the 3 regions of interest sketched in the X-ray image with the bubble size marked as dashed line. The measured absorption spectra are normalized to absolute extinction (dots) and are compared to an individually scaled reference spectrum of a zinc foil (red line). | ||
Dark-field X-ray imaging, on the other hand, is sensitive on nanoparticle distribution by probing small-angle scattering around individual beamlets. In XHI the different contrasts absorption, phase and dark-field are separated for each image taken. In practice, the snapshots shown in Fig. 1 are visualizing the scattering distribution during the nanosecond ablation. The scattering is overlaid in violet colour upon an absorption image, where the vapour bubble is seen as dark grey feature following higher transmission through vapour than through water. The spatial resolution is given by the number of beamlets. The scattering distribution, which we link to a cloud of nanoparticles of sizes around 25–100 nm in diameter28,44 is first confined to the location of the expanding bubble. The confinement persists partially after the bubble starts to collapse after its largest extension. At collapse and later on the scattering distribution is spread over a larger area in the field of view, indicating a release of nanoparticles into the liquid. This is consistent with the earlier observation that the bubble represents a confinement for the particles and its collapse tends to concentrate the material again close to the ablated spot.20,25 Due to the strong averaging over a sequence of pulses the spatial distribution may be blurred, given that individual bubble sizes can vary with corrugation of the substrate.49,61
The integral of the scattering over an area close to the target in Fig. 1a indicates this concentration effect for the expanding bubble as well as during collapse through maxima in scattering (at 20 μs and 270 μs). The bubble size as function of delay is extracted from the absorption contrast images and is shown as the blue dashed line in Fig. 1a. Confinement in this case doesn't seem to be as strong as described earlier for picosecond ablation on a flat target.22 This is reasonable, as the bubble motion happens here on a curved wire, where the symmetry of the collapsing bubble is not maintained.24 This reduces the collapse strength and bubble rebound. Eventually, the rebounding bubbles would even detach from the wire and transport material into the bulk liquid.30,46,48 While it is not yet documented, such symmetry breaking might also reduce the agglomeration upon bubble collapse.25,40 At larger distance from the target the dark-field intensity peaks at maximum bubble extension to be reduced upon collapse.
3.2 Particle morphology
It should be emphasized that dark-field imaging has its merits for the detection of the overall distribution of scattering species, but due to the strongly varying scattering cross-section of particles for hard X-rays that scales with the 6th power of the radius the detection is very selective.62 Additionally, no clear size differentiation is possible, while a coarse discrimination may be achieved through detector distance variation.46 Therefore, a sensitive size determination requires a SAXS experiment, which, per definition has to be done on one single point in space, while spatial information has to be obtained in a sequential scanning setup.We have investigated size evolution of the emerging nanoparticles earlier for gold19,22 or silver20,40 and could see the influence of electrolytes and small organic molecules on size stabilization already on nanoparticles existing within the vapour bubble.27,28 The presented SAXS data in Fig. 2 of ablated zinc particles in the batch process shows a very broad scattering distribution, reflecting the large size dispersion after ablation. In Fig. 2 two scattering curves are shown at 10 and 440 seconds after starting ablation, respectively. The SAXS pattern show a general slope between 2.6 and 3.3, which already indicates that the sample is composed either of porous structures or of a broad size distribution.63 The scattering from an arbitrary size distribution of spherical particles is calculated in a Monte Carlo approach and varied until a good match between simulation and measured curves is achieved. The broad size distribution from 5 to 200 nm particles is shifting for prolonged irradiation towards larger particles. This indicates that ripening is ongoing for a prolonged time. This ripening could encompass processes like fragmentation and agglomeration, as well as growth by oxidation. Such growth and oxide formation processes have recently been reviewed by Zhang et al. addressing the ripening (and seed-growth) behaviour during laser ablation in liquids, in particular of oxidation-sensitive material,64 and Amendola et al. comprehensively summarizing the laser synthesis of oxide particles.65 Transmission electron micrographs in Fig. 3 of the collected particles generally confirm the broad size distribution, displaying small particles and agglomerates thereof in the 10 nm range as well as large solid spheres. The inset shows the number-weighted distribution. The oxide lattice spacing can be found in some of the particles.
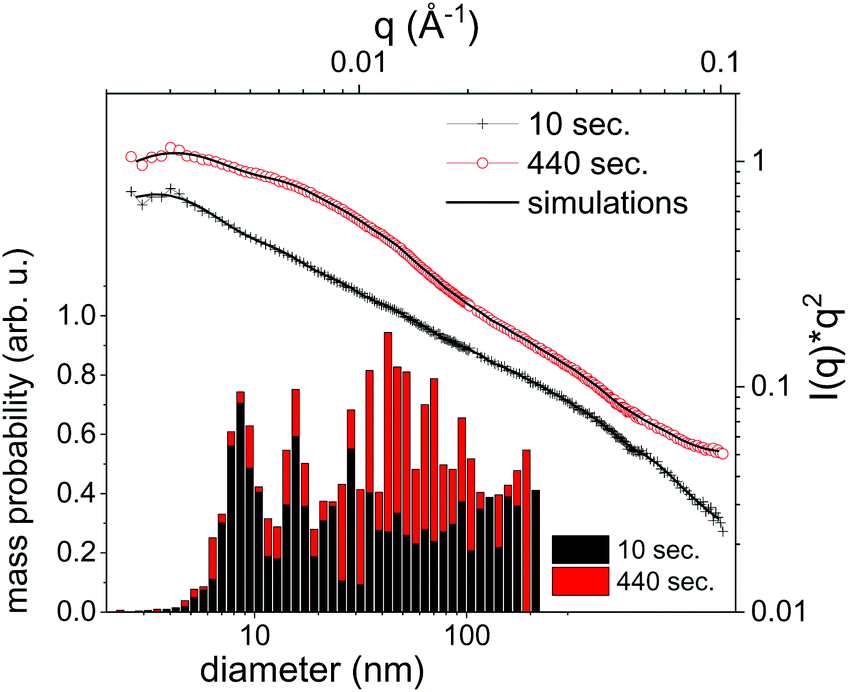 | ||
| Fig. 2 Selected SAXS measurements (symbols) after given time of batch processing of 10 and 440 seconds. The fit results from the particle size distribution are shown as lines. Lower left: Histogram of the mass-weighted particle size distribution as determined by the fits. | ||
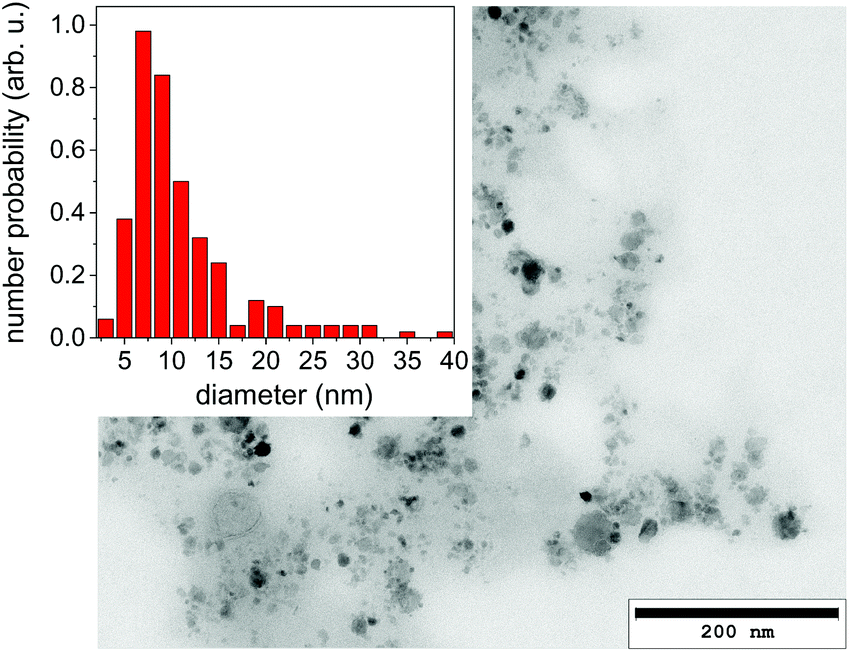 | ||
| Fig. 3 Transmission electron micrograph of a dried suspension of collected nanoparticles after ablation. The inset displays a size histogram. | ||
Nanoparticle size is a central information for the analysis of the PLAL process and SAXS is able to resolve even broad size distributions. Some caution should be spent on the uniqueness of this approach. Particularly, the Monte Carlo method produces one of many possible size distributions, that may equally fit the given measurement. This can be alleviated by repeating the iteration several times and using an average distribution from these several iterations. Still, artefacts may arise from using too many free parameters to describe the size distribution, such as the number of size bins. These can result in an apparently oscillatory distribution that is not based on real input. Sensitivity on small particles may additionally be masked by the presence of larger particles showing stronger scattering.66
Nevertheless, the basic information of SAXS is that particle size distribution in ablation of Zn is quite broad and the size distribution evolves after ablation. Oxidation and particle stabilization may play a role here. The SAXS signal can be used to quantify the total particle mass. However, it will only reproduce one fraction of the total mass as it selectively detects particles that fall into the resolved size range. In the present case particles in the size range of 5 to 200 nm are detected.
3.3 X-ray spectroscopy
X-ray spectroscopy, on the other hand, probes the absorption in the sample, which is linear in mass, irrespective of its condensation state. To probe the ablated zinc species, the energy of the impinging X-rays is tuned close to the K edge at 9.66 keV.Continuous series of absorption spectra were collected with a 11.3 μs time resolution and 300 μs for later delays. Spectra associated with selected time delays are shown in Fig. 1, right column, together with reference spectra of metallic zinc. Note that the spectra are normalized for absolute extinction, which is quite weak due to the low concentration of ablated material. At the same time, the absolute step height at the K edge can be converted in an effective thickness of zinc species that is crossed by the X-ray beam by comparison to tabulated values.67 The height of the absorption edge is not considered to be dependent on the valency of Zn, therefore a deduction of the total mass in the beam is appropriate.
Fig. 4a displays time sequences of the step height at the K edge as function of delay after laser irradiation for several distances to the target surface. At low distances of 0.1–0.3 mm the absorption clearly peaks at a very early delay. Subsequently, it drops again to go through a minimum at the largest bubble size and increases during bubble shrinkage. The bubble size is reflected by the total absorption through the chamber as dotted line. At bubble collapse the zinc absorption again drops to values that depend on the distance to target. Such behaviour has been described earlier22 as a consequence of the bubble retaining the ablated material of fixed amount. In our prior investigation only the nanoparticle fraction of the material had been quantified by SAXS. Here, characteristic X-ray absorption reflects the total mass distribution. The behaviour can be modelled by a function, where the X-ray beam intersects a variable length of the bubble, whose zinc content scales inversely with bubble volume. The observed contrast between maximum and minimum as function of time here is somewhat lower than calculated, pointing towards a higher material density close to the target surface for the expanded bubble. Another interesting point is that although the bubble collapse drags the ablated mass back towards the target, some material escapes the full collapse. This might be caused by a poorly uniform collapse, which limits the minimum bubble size or forms a non-uniform shape. It is nevertheless remarkable that the mass confinement region is less than 0.2–0.3 mm. At the larger distance (0.8 mm) the modulation is not strongly pronounced, while the later arrival of the expanding bubble at the given height is clearly visible. The absolute absorption step height fits reasonably well with the model of homogeneous bubble filling. The absolute scale also allows to infer the total ablated mass. By integrating the mass density over the whole bubble area we derive a mass of 2.2 × 10−7 g, corresponding to a volume of ablated zinc of ≤1 μm depth at a presumed crater area of 0.04 mm2. The derived ablation depth is much larger than the laser absorption length in zinc and probably caused by 2 effects: corrugation of the surface during the ablation with several pulses per unit area49 and a heat-affected zone that is considerable larger than the absorption length due to fast heat conduction by phonons and fast electrons.68
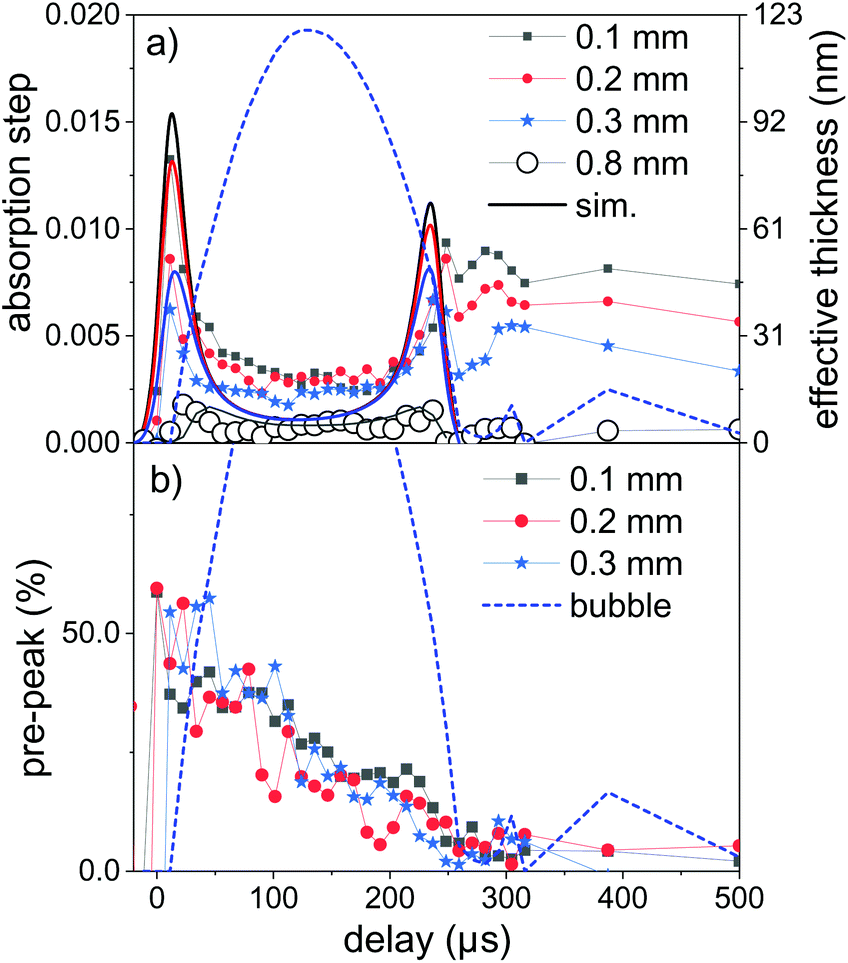 | ||
| Fig. 4 (a) Time dependence of the step height at the Zn K edge as a measure for the total intersected material by the X-ray beam at several distances above the target (symbols). The lines are model calculations assuming a homogeneous filling of the bubble with a fixed amount of material. (b) Time dependence of the ratio of the pre-peak to metallic Zn absorption as a measure for the relative atomic content at several distances. The dashed line in each sub-figure represents the X-ray transmission change due to bubble formation. | ||
While the total mass can be deduced from the absorption step height, the spectrum close to the edge can yield information about the ablated species. In Fig. 1 the measured near-edge spectra are compared to that of metallic zinc. Spectra at long delays after bubble collapse in particular show a very close similarity to metallic zinc. This is reasonable, considering earlier observations12–14 that oxidation in newly formed zinc nanoparticles takes place quantitatively on a 10 minutes time scale. The extended absorption fine structure (not shown in full) is less well defined, which points to some structural inhomogeneity.
Selected measured spectra of several stages as well as reference spectra are presented in Fig. 5. While the spectrum at 600 μs can be almost perfectly fitted by the metallic zinc reference, an astonishing difference is observed for the earliest spectra (here at 11.4 μs). This spectrum shows a very intense pre-peak at 9.662 keV, which is unlike any other condensed-phase reference in literature. The first peak of metallic zinc is found at 9.668 keV, the most common ZnO peaks at 9.669 keV. Some molecular species, such as Zn-acetate69 or solvated Zn2+,70 show peaks that are shifted to lower values down to 9.664 keV. The only found exception is that of zinc vapour, which shows an extra-ordinary pre-peak at the same energy, as displayed in the upper part of Fig. 5.56 This peak is, at the same time, exceptionally high. Similarly, prominent pre-peaks are also described for other transition metal elements, such as Na,71 Mn, Cr, Ca or Cu.72 This phenomenon is described as a resonance in the 1s → 4p transition, possibly including multiple electron excitation.
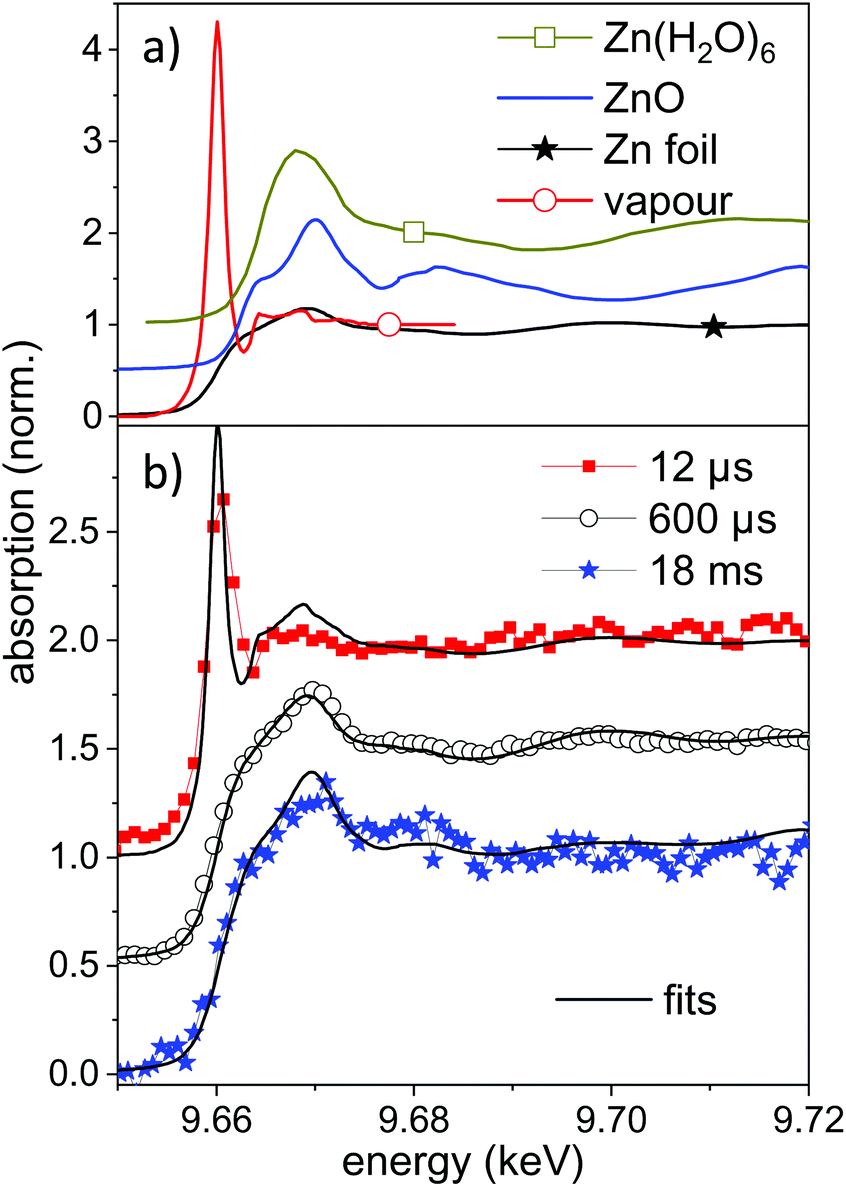 | ||
| Fig. 5 (a) Reference near-edge absorption spectra of several Zn containing samples. The spectra of Zn vapour and Zn(H2O)6 are digitized from the publication by Mihelič et al.56 and by Nicolis et al.,70 respectively. The latter are shifted up by 0.5 in absorption. (b) Near-edge absorption spectra for different delays during the ablation process compared to linear combinations of the reference spectra. The individual spectra are shifted by 0.5 for clarity. | ||
XANES references for other possible species, such as atom clusters or partially solvated atoms or clusters are not available. It could be that some of these molecular-like species show similar features as a transition between the atomic pre-peak and a weaker solvated-state peak at higher energy. Nevertheless a condensed phase structure seems to be unlikely. We conclude that for early time delays after irradiation with nanosecond laser pulses a large amount of zinc in an isolated, reactive state exists in the bubble. This is reasonable both because of the high prevailing temperature during the early expansion phase and the interaction with the plasma fueled by the long pulses. Results of atomic-scale simulations predict an emission of a mixture of small particles and atomic species.15,21,73 Most recently, Shih et al. have highlighted subtle differences between ultrashort pulse irradiation and sub-nanosecond irradiation.73 However, the simulations include only some nanoseconds time span and the excitation with picosecond to few-ns laser pulses.
In the present case the nanosecond laser will also irradiate the expanding ablation plume to intensify the emerging plasma to heat up the material further. A fit of the measured spectrum by a mixture of metallic and atomic zinc yields an atomic content of up to 40–50% of atomic species. An uncertainty comes from the degree of spectral broadening due to the limited time resolution. The atomic zinc content is deduced in first order by the intensity ratio of the two reference spectra at 9.662 keV, which is displayed in Fig. 5b. The life-time of the pre-peak is very long, extending over 100 μs (more spectra in the ESI†). In fact, a full disappearance only occurs at maximum bubble extension, which coincides with the lowest temperature inside the bubble. It should be noted that isolated atoms would be expected to be hydrated during the condensation phase, given that the number of water molecules in the vapour surpasses that of ablated atoms60 by far.
Indeed, the pre-peak position also tends to shift to a higher energy, which indicates that some chemical species are formed, including clusters within the first 100 μs. Therefore, the pre-peak intensity ratio as presented in Fig. 4b may be a mixture of ‘reactive species’ of isolated atoms and (hydrated) clusters. The transition from an atomic state to clustering would merit further attention, however, the full analysis of such spectra would require ab initio methods and is beyond the scope of the present report. Amans et al. have investigated the formation of ions, neutral atoms and clusters during ablation from aluminium oxide in details both theoretically and experimentally by plasma spectroscopy.32,60 In PLAL they find that the ratio of Al atoms over AlO clusters decays within a time span of several microseconds after UV laser (5 ns) impact.59
At the same time, the spectra in general show a less pronounced near-edge structure during the bubble phase than after bubble collapse. This additionally points to structural disorder during the violent bubble oscillation phase. At later delays of tens of milliseconds, the spectra slowly change to show a shift towards addition of ZnO features. A fit with both reference spectra allows to limit the oxidation at 18 ms to a range of 10–20%. An analysis of the oxidation has earlier shown that complete oxidation takes place on a 10 minutes time scale in an open vessel containing an ablated suspension.14 At the same time, hydrozincite by the dissolution of CO2 from air was formed. This compound is not identified here.
In Fig. 6 the percent contributions of reactive species and oxidized (ZnO) material are shown as function of delay and height above the target. The 3 reference spectra (vapour, metal, ZnO) were used to perform a fit of the reference spectra with variable relative contribution to the measured spectra. The metallic zinc contribution is not shown, but amounts for the remaining part to 100%. Noticeable oxidation is only seen on the millisecond time scale. The latest time delays >20 ms show a larger error due to the reduction of signal after partial flushing or the spatial region by the water flow.
 | ||
| Fig. 6 (a) Videography still of the vapour bubble at a delay of 114 μs. The dots indicate the positions, where the TR-XAFS spectra were acquired. (b) Time-resolved contributions of the reactive species and ZnO to the XANES part of the spectra as derived by a least squares fit. | ||
4 Discussion and conclusions
While the PLAL process shows a number of complicated facets, a deeper understanding can be gained by a combination of methods that can access the bubble phase and identify species from the atomic to the nanoscale. SAXS has earlier been demonstrated as a powerful method of choice. High time resolution can be achieved and a direct access is possible to the particle size distribution. However, classical SAXS with a micrometre-sized beam is experimentally very demanding (requiring a dedicated experimental station at a 3rd generation synchrotron). In a stroboscopic setup the spatial, as well as the time dimension has to be acquired sequentially. Therefore, dark-field X-ray imaging can to some extent complement the SAXS measurement by verifying the spatial distribution of the large-size fraction of the particles. Both techniques allowed here to conclude that particles are formed with a broad size distribution, extending to 100 nm and beyond. The dark-field images also confirm that the bubble acts as confinement for the particle mass. This has been identified earlier as a possible source for complications, as the collapse confines particles to a small volume, such that agglomeration can take place.X-ray scattering also misses a probably important fraction of ablated species, which is not condensed to particles or crystalline structures. Non-resonant scattering is in general not sensitive to chemical information. This can be complemented by XAFS spectroscopy, which has been explored for the first time in PLAL within this study. The near-edge structure confirms the prior knowledge that ablation produces species that end as metallic zinc after bubble collapse. This is not trivial, as we observe disorder in structure and reactive species within the bubble. Probably, bubble collapse and reheating of the particles sinter the emerging structures to clearly form metallic zinc. Oxidation is not predominant within the sub-millisecond time scale. Only some 10–20% of oxide is formed after about 20 ms.
The most surprising observation is that we find clear indication that very early after ablation (<100 μs) a large fraction of the ablated zinc is present as isolated, reactive species (atoms or atom clusters). This should be viewed in the context of the irradiation conditions with a nanosecond laser. The long laser duration implies that a forming plasma can still be heated by the trailing part of the pulse and increase the vapour temperature. The content of the reactive species drops during bubble expansion and cooling of the interior, but only disappears with bubble collapse. This observation indicates for the first time that reactions take place in the bubble cavity. It would be interesting to evaluate, whether this observation is an exception due to the low evaporation temperature of zinc or could also be observed for other metals, or is even relevant for ultra-short pulse PLAL. Copper, at least, is also expected to show a prominent pre-peak. On the other hand, recent plasma spectroscopy studies show the prospect of yielding information on this aspect.
In summary, a combination of synchrotron-based X-ray methods allow to deepen the knowledge of the ablation process, in particular in liquid. The methods are still difficult to implement, for instance to define procedures that allow for a non-sequential, ‘single-shot’ approach. Rapid developments in instrumentation, on the other hand, further support the case.
Author contributions
The research has been designed by SB and AP. Some experimental techniques (XHI) were developed by SR, MZ and TB. The experiments have been performed by SR, AZ, RS, AL, AM, OM, SP, MZ and AP with support by all authors. Data analysis has been done by SR, JG, MZ and AP. The manuscript has been written by SR, SB and AP. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by Deutsche Forschungsgemeinschaft under contract BA 3580/15-2 and PL325/8-2 and through research within “From Matter to Materials and Life” of the Helmholtz Association. We would like to acknowledge provision of beam time at ESRF(ID24), SLS (cSAXS) and KARA. Technical support at these beamlines from F. Perrin and X. Donath is gratefully acknowledged. HR-TEM by H. Störmer the Laboratory for Electron Microscopy (LEM, KIT) is acknowledged.References
- A. Fojtik and A. Henglein, Ber. Bunsenges. Phys. Chem., 1993, 97, 1493 CrossRef CAS.
- F. Mafuné, J. Kohno, Y. Takeda and T. Kondow, J. Phys. Chem. B, 2001, 105, 5114 CrossRef.
- G. Compagnini, A. A. Scalisi and O. Puglisi, Phys. Chem. Chem. Phys., 2002, 4, 2787–2791 RSC.
- S. Barcikowski, A. Menéndez-Manjón, B. Chichkov, M. Brikas and G. Račiukaitis, Appl. Phys. Lett., 2007, 91, 083113 CrossRef.
- D. Zhang, B. Gökce and S. Barcikowski, Chem. Rev., 2017, 117, 3990–4103 CrossRef CAS PubMed.
- S. Reichenberger, G. Marzun, M. Muhler and S. Barcikowski, ChemCatChem, 2019, 11, 4489–4518 CrossRef CAS.
- J.-Y. Lin, C. Xi, Z. Li, Y. Feng, D.-Y. Wu, C.-K. Dong, P. Yao, H. Liu and X.-W. Du, Chem. Commun., 2019, 55, 3121–3123 RSC.
- Ü. Özgür, Y. I. Alivov, C. Liu, A. Teke, M. A. Reshchikov, S. Doǧan, V. Avrutin, S.-J. Cho and H. Morkoç, J. Appl. Phys., 2005, 98, 041301 CrossRef.
- H. Usui, Y. Shimizu, T. Sasaki and N. Koshizaki, J. Phys. Chem. B, 2005, 109, 120 CrossRef CAS PubMed.
- J. M. Cho, J. K. Song and S. M. Park, Bull. Korean Chem. Soc., 2009, 30, 1616 CrossRef CAS.
- P. Wagener, A. Schwenke, B. N. Chichkov and S. Barcikowski, J. Phys. Chem. C, 2010, 114, 7618 CrossRef CAS.
- P. Camarda, L. Vaccaro, F. Messina and M. Cannas, Appl. Phys. Lett., 2015, 107, 013103 CrossRef.
- P. Camarda, F. Messina, L. Vaccaro, G. Buscarino, S. Agnello, F. M. Gelardi and M. Cannas, J. Appl. Phys., 2016, 120, 124312 CrossRef.
- S. Reich, J. Göttlicher, A. Letzel, B. Gökce, B. Barcikowski, T. dos Santos Rolo, T. Baumbach and A. Plech, Appl. Phys. A, 2018, 124, 71–77 CrossRef.
- C.-Y. Shih, M. V. Shugaev, C. Wu and L. V. Zhigilei, J. Phys. Chem. C, 2017, 121, 16549–16567 CrossRef CAS PubMed.
- H. Oguchi, T. Sakka and Y. H. Ogata, J. Appl. Phys., 2007, 102, 023306 CrossRef.
- J. Lam, D. Amans, F. Chaput, M. Diouf, G. Ledoux, N. Mary, K. Masenelli-Varlot, V. Motto-Ros and C. Dujardin, Phys. Chem. Chem. Phys., 2014, 16, 963–973 RSC.
- A. Matsumoto, A. Tamura, T. Honda, T. Hirota, K. Kobayashi, S. Katakura, N. Nishi, K.-I. Amano, K. Fukami and T. Sakka, J. Phys. Chem. C, 2015, 119, 26506 CrossRef CAS.
- S. Ibrahimkutty, P. Wagener, A. Menzel, A. Plech and S. Barcikowski, Appl. Phys. Lett., 2012, 101, 103104 CrossRef.
- S. Ibrahimkutty, P. Wagener, T. dos Santos Rolo, D. Karpov, A. Menzel, T. Baumbach, S. Barcikowski and A. Plech, Sci. Rep., 2015, 5, 16313 CrossRef CAS PubMed.
- C.-Y. Shih, R. Streubel, J. Heberle, A. Letzel, M. V. Shugaev, C. Wu, M. Schmidt, B. Gökce, S. Barcikowski and L. V. Zhigilei, Nanoscale, 2018, 10, 6900–6910 RSC.
- S. Reich, A. Letzel, A. Menzel, N. Kretzschmar, B. Gökce, S. Barcikowski and A. Plech, Nanoscale, 2019, 11, 6962–6969 RSC.
- S. Reich, P. Schönfeld, A. Letzel, S. Kohsakowski, M. Olbinado, B. Gökce, S. Barcikowski and A. Plech, ChemPhysChem, 2017, 18, 1084 CrossRef CAS PubMed.
- S. Barcikowski, A. Plech, K. S. Suslick and A. Vogel, MRS Bull., 2019, 44, 382–391 CrossRef.
- S. Reich, P. Schönfeld, P. Wagener, A. Letzel, S. Ibrahimkutty, B. Gökce, S. Barcikowski, A. Menzel, T. dos Santos Rolo and A. Plech, J. Colloid Interface Sci., 2017, 489, 106–113 CrossRef CAS PubMed.
- D. Zhang, J. Liu and C. Liang, Sci. China: Phys., Mech. Astron., 2017, 60, 074201 Search PubMed.
- A. Letzel, B. Gökce, P. Wagener, S. Ibrahimkutty, A. Menzel, A. Plech and S. Barcikowski, J. Phys. Chem. C, 2017, 121, 5356 CrossRef CAS.
- A. Letzel, S. Reich, T. dos Santos Rolo, A. Kanitz, J. Hoppius, A. Rack, M. Olbinado, A. Ostendorf, B. Gökce, A. Plech and S. Barcikowski, Langmuir, 2019, 35, 3038–3047 CrossRef CAS PubMed.
- K. Sasaki, T. Nakano, W. Soliman and N. Takada, Appl. Phys. Express, 2009, 2, 046501 CrossRef.
- A. De Giacomo, M. Dell'Aglio, A. Santagata, R. Gaudiuso, O. De Pascale, P. Wagener, G. C. Messina, G. Compagnini and S. Barcikowski, Phys. Chem. Chem. Phys., 2013, 15, 3083–3092 RSC.
- T. Sakka, S. Iwanaga, Y. H. Ogata, A. Matsunawa and T. Takemoto, J. Chem. Phys., 2000, 112, 8645 CrossRef CAS.
- J. Lam, V. Motto-Ros, D. Misiak, C. Dujardin, G. Ledoux and D. Amans, Spectrochim. Acta, B, 2014, 101, 86–92 CrossRef CAS.
- A. Tamura, A. Matsumoto, K. Fukami, N. Nishi and T. Sakka, J. Appl. Phys., 2015, 117, 173304 CrossRef.
- M. R. Gavrilović, M. Cvejić, V. Lazić and S. Jovićević, Phys. Chem. Chem. Phys., 2016, 18, 14629 RSC.
- K. Ando and T. Nakajima, Nanoscale, 2020, 12, 9640–9646 RSC.
- T. Tsuji, Y. Tsuboi, N. Kitamura and M. Tsuji, Appl. Surf. Sci., 2004, 229, 365–371 CrossRef CAS.
- J. Tomko, J. J. Naddeo, R. Jimenez, Y. Tan, M. Steiner, J. M. Fitz-Gerald, D. M. Bubb and S. M. O'Malley, Phys. Chem. Chem. Phys., 2015, 17, 16327 RSC.
- B. Thornton, T. Takahashi, T. Ura and T. Sakka, Appl. Phys. Express, 2012, 5, 102402 CrossRef.
- L. Lavisse, J.-L. Le Garrec, L. Hallo, J.-M. Jouvard, S. Charles, J. Perez, J. B. A. Mitchell, J. Decloux, M. Girault, V. Potin, H. Andrzejewski, M. C. M. de Lucas and S. Bourgeois, Appl. Phys. Lett., 2012, 100, 164103 CrossRef.
- P. Wagener, S. Ibrahimkutty, A. Menzel, A. Plech and S. Barcikowski, Phys. Chem. Chem. Phys., 2013, 15, 3068 RSC.
- A. Momose, S. Kawamoto, I. Koyama, Y. Hamaishi, K. Takai and Y. Suzuki, Jpn. J. Appl. Phys., 2003, 42, L866–L868 CrossRef CAS.
- F. Pfeiffer, M. Bech, O. Bunk, P. Kraft, E. F. Eikenberry, C. Brönnimann, C. Grünzweig and C. David, Nat. Mater., 2008, 7, 134–137 CrossRef CAS PubMed.
- H. H. Wen, E. E. Bennett, R. Kopace, A. F. Stein and V. Pai, Opt. Lett., 2010, 35, 1932–1934 CrossRef PubMed.
- F. A. Vittoria, M. Endrizzi, P. C. Diemoz, A. Zamir, U. H. Wagner, C. Rau, I. K. Robinson and A. Olivo, Sci. Rep., 2015, 5, 16318 CrossRef CAS PubMed.
- T. dos Santos Rolo, S. Reich, D. Karpov, S. Gasilov, T. Baumbach, D. Kunka and A. Plech, Appl. Sci., 2018, 8, 737 CrossRef.
- S. Reich, T. dos Santos Rolo, A. Letzel, T. Baumbach and A. Plech, Appl. Phys. Lett., 2018, 112, 151903 CrossRef.
- M. Fischer, J. Hormes, G. Marzun, P. Wagener, U. Hagemann and S. Barcikowski, Langmuir, 2016, 32, 8793–8802 CrossRef CAS PubMed.
- S. Kohsakowski, B. Gökce, R. Tanabe, P. Wagener, A. Plech, Y. Ito and S. Barcikowski, Phys. Chem. Chem. Phys., 2016, 18, 16585 RSC.
- S. Reich, A. Letzel, B. Gökce, A. Menzel, S. Barcikowski and A. Plech, ChemPhysChem, 2019, 20, 1036–1043 CrossRef CAS PubMed.
- B. R. Pauw, J. S. Pedersen, S. Tardif, M. Takata and B. B. Iversen, J. Appl. Crystallogr., 2013, 46, 365–371 CrossRef CAS PubMed.
- S. K. Lynch, V. Pai, J. Auxier, A. F. Stein, E. E. Bennet, C. K. Kemble, X. Xiao, W.-K. Lee, N. Y. Morgan and H. H. Wen, Appl. Opt., 2011, 50, 4310–4319 CrossRef PubMed.
- S. Reich and A. Plech, KITOpendata, repository, 2018 Search PubMed.
- M. Zakharova, S. Reich, A. Mikhaylov, V. Vlnieska, T. dos Santos Rolo, A. Plech and D. Kunka, Opt. Lett., 2019, 44, 2306–2309 CrossRef CAS PubMed.
- S. Pascarelli, O. Mathon, T. Mairs, I. Kantor, G. Agostini, C. Strohm, S. Pasternak, F. Perrin, G. Berruyer, P. Chappelet, C. Clavel and M. C. Dominguez, J. Synchrotron Radiat., 2016, 23, 353–368 CrossRef CAS PubMed.
- J. Headspith, J. Groves, P. N. Luke, M. Kogimtzis, G. Salvini, S. L. Thomas, R. C. Farrow, J. Evans, T. Rayment, J. S. Lee, W. D. Goward, M. Amman, O. Mathon and S. Diaz-Moreno, 2007 IEEE Nuclear Science Symposium Conference Record, U.S. ISSN Center, 2007, pp. N55–2.
- A. Mihelič, A. Kodre, I. Arčon, J. Padežnik Gomilšek and M. Borowski, Nucl. Instrum. Methods Phys. Res., Sect. B, 2002, 196, 194–197 CrossRef.
- J. A. Bearden and A. F. Burr, Rev. Mod. Phys., 1967, 39, 125–142 CrossRef CAS.
- S. Kraft, J. Stümpel, P. Becker and U. Kuetgens, Rev. Sci. Instrum., 1996, 67, 681–687 CrossRef CAS.
- J. Lam, D. Amans, F. Chaput, M. Diouf, G. Ledoux, N. Mary, K. Masenelli-Varlot, V. Motto-Ros and C. Dujardin, Phys. Chem. Chem. Phys., 2014, 16, 963–973 RSC.
- J. Lam, D. Amans, C. Dujardin, G. Ledoux and A.-R. Allouche, J. Phys. Chem. A, 2015, 119, 8944–8949 CrossRef CAS PubMed.
- A. Letzel, M. Santoro, J. Frohleiks, A. R. Ziefuß, S. Reich, A. Plech, E. Fazio, F. Neri, S. Barcikowski and B. Gökce, Appl. Surf. Sci., 2018, 473, 828–837 CrossRef.
- G. Beaucage, J. Appl. Crystallogr., 1995, 28, 717 CrossRef CAS.
- M. Sztucki, T. Narayanan and G. Beaucage, J. Appl. Phys., 2007, 101, 114304 CrossRef.
- D. Zhang, J. Liu and C. Liang, Sci. China, 2017, 60, 074201 Search PubMed.
- V. Amendola, D. Amans, Y. Ishikawa, N. Koshizaki, S. Scirè, G. Compagnini, S. Reichenberger and S. Barcikowski, Chem. – Eur. J., 2020 DOI:10.1002/chem.202000686.
- A. Letzel, B. Gökce, A. Menzel, A. Plech and S. Barcikowski, Appl. Surf. Sci., 2018, 435, 743–751 CrossRef CAS.
- B. Henke, E. Gullikson and J. Davis, At. Data Nucl. Data Tables, 1993, 54, 181–343 CrossRef CAS.
- Y. V. Petrov, V. A. Khokhlov, V. V. Zhakhovsky and N. A. Imogamov, Appl. Surf. Sci., 2019, 492, 285–297 CrossRef CAS.
- B. P. Grady, J. A. Floyd, W. B. Genetti, P. Vanhoorne and R. A. Register, Polymer, 1999, 40, 283–288 CrossRef CAS.
- I. Nicolis, P. Deschamps, E. Curis, O. Corriol, V. Acar, N. Zerrouk, J.-C. Chaumeil, F. Guyon and S. Bénazeth, J. Synchrotron Radiat., 2001, 8, 984–986 CrossRef CAS PubMed.
- C. Teodorescu, J. M. Esteva, M. Womes, A. El Afif, R. Karnatak, A. Flank and P. Lagarde, J. Electron Spectrosc. Relat. Phenom., 2000, 106, 233–245 CrossRef CAS.
- U. Arp, B. M. Lagutin, G. Materlik, I. D. Petrov, B. Sonntag and V. L. Sukhorukov, J. Phys. B: At., Mol. Opt. Phys., 1993, 26, 4381–4398 CrossRef CAS.
- C.-Y. Shih, M. V. Shugaev, C. Wu and L. V. Zhigilei, Phys. Chem. Chem. Phys., 2020, 22, 7077–7099 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: A large set of optical images, XHI images, XAFS spectra as function of delay, a scheme of the TR-XHI setup and selected XAS ference spectra. See DOI: 10.1039/D0NR01500H |
| ‡ Present address: Fraunhofer Institut for High-Speed Dynamics, Ernst-Mach-Institute, Freiburg. |
| This journal is © The Royal Society of Chemistry 2020 |