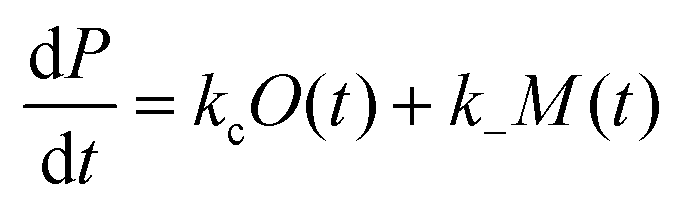DOI:
10.1039/D0NR01481H
(Paper)
Nanoscale, 2020,
12, 18663-18672
Amelioration of aggregate cytotoxicity by catalytic conversion of protein oligomers into amyloid fibrils†
Received
20th February 2020
, Accepted 29th May 2020
First published on 14th August 2020
Abstract
The aggregation of peptides and proteins into amyloid fibrils is a molecular self-assembly phenomenon associated with both biological function and malfunction, notably in the context of neurodegenerative diseases. Oligomeric species formed early in the aggregation process are generally associated with cytotoxicity. Extrinsic molecules such as peptides have been found to influence amyloid formation kinetics and regulate this cellular process. Here, we use single-molecule FRET and bulk assays combined with global kinetic analysis to study quantitatively the effect of an 8-residue peptide (LQVNIGNR) on fibril formation by the yeast prion protein Ure2. This peptide, which is derived from a segment of the Ure2 prion domain, forms vesicular assemblies that accelerate fibril formation of Ure2 by promoting conformational conversion of oligomeric intermediates into fibrillar species in a catalytic manner. This reduces oligomer longevity and consequently ameliorates cytotoxicity. The LQVNIGNR peptide was found to accelerate fibril formation of unrelated proteins including Tau and α-Synuclein, suggesting a general ability to catalyse fibrillation. This study provides a general strategy for investigating the microscopic mechanism of extrinsic factors on amyloid aggregation. This approach can readily be applied to other amyloid systems and demonstrates that acceleration of oligomer conversion is a promising strategy to reduce amyloid toxicity.
Introduction
The aggregation of polypeptide chains into β-sheet rich amyloid fibrils is not only associated with a series of neurodegenerative disorders including Alzheimer's disease and Parkinson's disease, but is also related to a range of cellular functions in living organisms.1–3 Microscopic reactions underlying the assembly process of amyloidogenic proteins are complex, typically including multiple events such as primary nucleation and elongation, as well as secondary pathways such as fragmentation and monomer-dependent secondary nucleation.2,4,5 The oligomeric species populated early in the process of amyloid fibril formation have attracted great interest in recent years because such species have been observed to be significantly more cytotoxic than fibrils.2,6,7 However, the low-populated, transient and heterogeneous nature of the oligomers makes it challenging to study them by traditional biochemical and biophysical methods. During the past decades, the development of single molecule fluorescence techniques offers a powerful approach for exploring the formation and properties of oligomers, because of their ability to probe the nature of individual molecular species.8–12
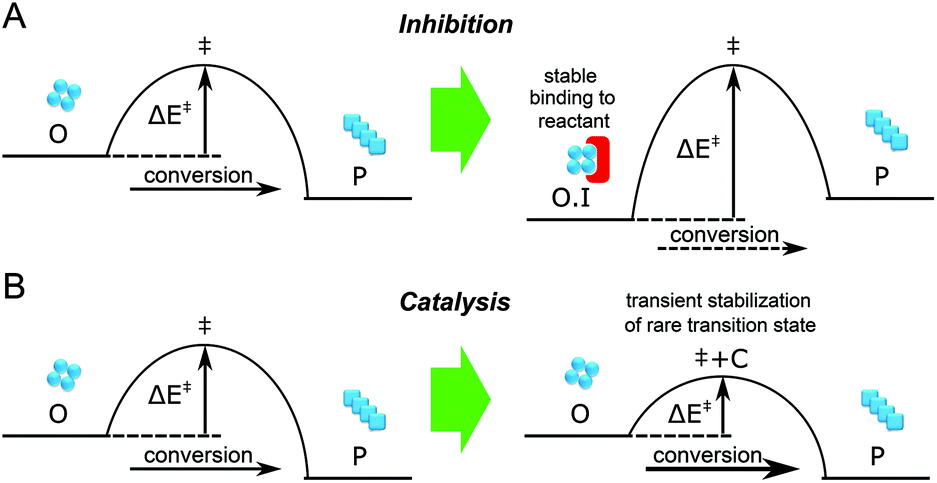
Due to the generic toxicity of amyloid oligomers,13 significant efforts have been devoted to finding inhibitors of the aggregation process. Extrinsic factors such as small molecules, peptides and antibodies have been shown to inhibit the aggregation of many amyloidogenic proteins, including the Aβ peptide, α-Synuclein and islet amyloid polypeptide (IAPP) associated with Alzheimer's disease, Parkinson's disease, and type II diabetes, respectively.14 Of these factors, short peptides are simple and effective in this regard, especially those derived from self-recognition regions of amyloidogenic proteins.14–20 However, inhibitors must be present at saturating concentration to achieve stable binding to the target (Fig. 1A); this may be challenging to attain in vivo. Moreover, the detailed mechanisms of how these molecules interfere with amyloid aggregation are still under investigation, and it is not guaranteed that a factor that inhibits the overall aggregation process will also reduce the concentration of toxic oligomeric intermediates of this process. By contrast, some amphiphilic peptides have been reported to accelerate the formation of amyloid fibrils.21 Unlike inhibition, it is possible for acceleration to be catalytic (Fig. 1B), with the accelerating agent binding only transiently to the target before being regenerated, and thus the catalyst is likely to be effective at significantly lower concentrations. In this paper we show that acceleration of the appropriate microscopic steps in the aggregation reaction can offer an orthogonal approach to lower the concentration of toxic oligomeric intermediates, that has the advantage of requiring in principle far lower doses of drug than an inhibition-based approach. We furthermore show how the concerted use of single molecule spectroscopy and kinetic analysis can allow the modulation of individual molecular level processes in the formation and depletion of oligomers to be defined in quantitative detail.
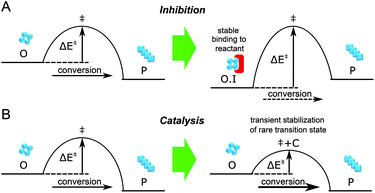 |
| | Fig. 1 Differing requirements for catalysis vs. inhibition. (A) Inhibition of oligomers requires stable binding of oligomers such that most of them become chemically inactive and unable to exert toxic effects. This may require a high concentration of inhibitor (e.g. antibodies). (B) On the other hand, catalysis of oligomer conversion to fibrils requires only enough catalyst to meaningfully increase the flux through the transition state. Since the transition state is high-energy and therefore rare for slow oligomer conversion, in principle only substoichiometric concentrations of catalyst may be required (e.g. vesicular assemblies of the kind studied in the present work). O, oligomer; P, product; I, inhibitor, C, catalyst. | |
As a model system we focus on the yeast prion protein Ure2, which is the protein determinant of the prion state [URE3] in Saccharomyces cerevisiae.22 Native Ure2 consists of a flexible N-terminal prion domain and a globular C-terminal domain; the N-terminal domain is responsible for amyloid formation. In our previous work, we discovered that the amyloid aggregation of Ure2 begins with the formation of oligomers that are functionally distinct from Ure2 fibrils, and that subsequently undergo a conformational conversion into rapidly growing fibrillar species. We also discovered that these nonfibrillar oligomers are vastly more numerous than those fibrillar species short enough to be considered oligomeric. We developed a detailed kinetic model for this phenomenon, which now makes possible the study of the microscopic reaction steps of Ure2 aggregation in the presence of acceleratory or inhibitory factors.23 In the present study, using single-molecule FRET (smFRET) and kinetic modelling, we elucidate the way in which short peptides affect the fibril formation of Ure2. In particular, we find that the LQVNIGNR peptide promotes Ure2 aggregation by forming vesicular assemblies that result in an increase in the oligomer conformational conversion rate, kc. The accelerated oligomer conversion to non-toxic fibrillar species reduces the duration of the oligomer-generating lag phase in the aggregation process and thus decreases the cellular toxicity. These findings enhance our understanding of the interaction between short peptides and amyloid proteins, and also indicate that the acceleration of oligomer conversion could be beneficial to cell viability.
Results
Screening the effects of peptides derived from the prion domain sequence of Ure2
Short peptides can influence fibril formation by amyloid proteins, especially peptides related to self-recognition regions of such proteins.14 These regions generally include amyloidogenic core sequences, for example residues 20–29 of hIAPP and residues 17–21 of Aβ, which have been found to inhibit fibril formation of the parent protein.15,24 In the case of Ure2, we performed an amyloidogenicity prediction using the FoldAmyloid algorithm (Fig. S1†). The results indicate that residues 16–20 (LRQVN) has the highest amyloidogenicity, in agreement with our previous study showing that the segment QVNI (residues 18–21) represents a potential amyloid-initiation region.25 Additionally, a β-sheet-forming region involving residues 8–12 has also been suggested to be crucial for fibril formation of Ure2.26 Therefore, we designed several peptides derived from these regions with different lengths and hydrophobicities (Table S1†) as potential candidates to modulate the fibril formation of Ure2.
We tested each of the peptides individually by incubating them with full-length Ure2 to observe their effects on fibril formation (Fig. S2†). Most of the designed peptides have no effect on the aggregation behaviour, except for the peptide QVSNL, which slightly prolongs the lag phase of the reaction (Fig. S2A†), indicating a weak inhibitory effect. The peptide LQVNIGNR was, however, found to dramatically accelerate the fibril formation of Ure2 (Fig. 2A and S2B†), indicating that this peptide is an accelerator of Ure2 self-assembly rather than an inhibitor. This finding is in contrast to the previously observed inhibition effect of fibril core sequences on amyloid proteins for Aβ peptide, PrP, α-Synuclein, Tau, IAPP, poly-Q and insulin.15–20,27,28 The mechanism for these inhibition effects has been proposed to be a result of the ability of these peptides to bind stably to the similar sequence in the full-length protein and hinder its self-assembly into highly-ordered β-sheet structures. The observation that the LQVNIGNR peptide has the reverse effect when it interacts with the full-length sequence makes it an interesting system through which to probe the origin of such effects.
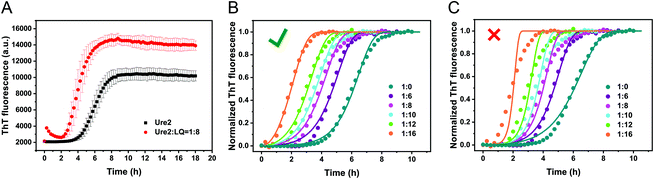 |
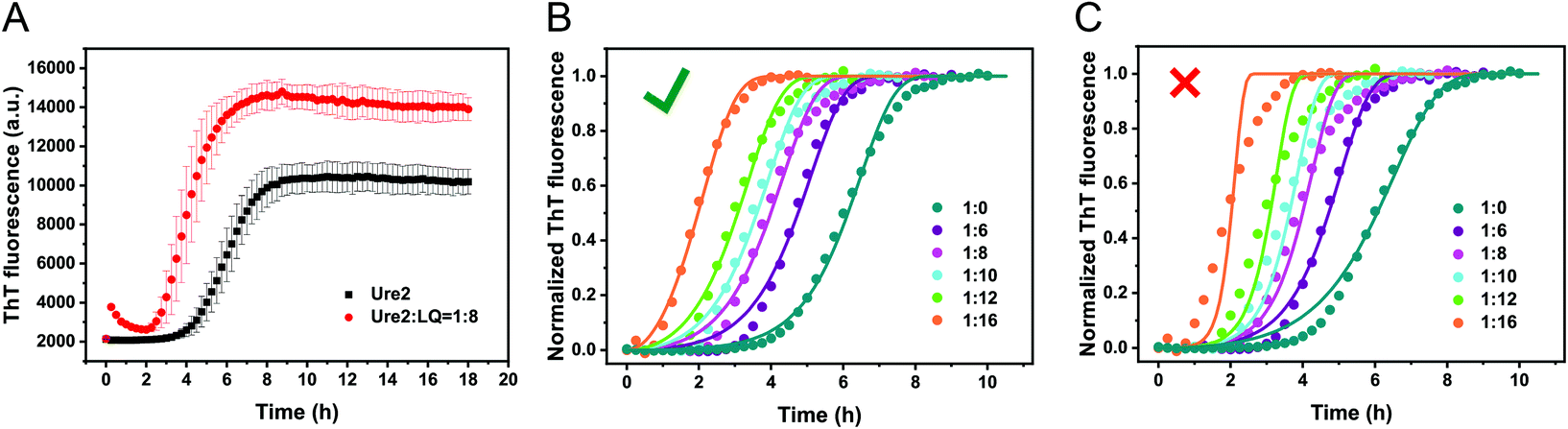
| | Fig. 2 Kinetics of Ure2 fibril formation in the presence of LQVNIGNR. (A) ThT curves for 5 μM Ure2 in the absence (black) or presence (red) of 40 μM LQVNIGNR. The data shown are the average of three replicates and the error bars represent the SD. (B) and (C) The normalized ThT curves of 5 μM Ure2 with different concentrations of LQVNIGNR were globally fitted to a kinetic model for breakable biofilaments which is described in detail in the Methods. The concentrations of LQVNIGNR are 0 μM (dark green), 30 μM (purple), 40 μM (pink), 50 μM (cyan), 60 μM (light green), and 80 μM (orange). Three repetitions were averaged and analyzed for each concentration. (B) Global fitting of the model to the data using a different knk+ but the same k+k− for each concentration of LQVNIGNR is able to faithfully reproduce the kinetics. (C) Global fitting with the same knk+ value but different k+k− values for each concentration of LQVNIGNR yields misfits. This indicates that LQVNIGNR affects only kn and not k+ or k−. | |
The kinetics of fibril formation by Ure2 in the presence of the LQVNIGNR peptide
We used kinetic analysis4,29 to probe the mechanism of the acceleration effect of LQVNIGNR on amyloid fibril formation of Ure2. Since Ure2 is a highly stable dimer in solution and the dimeric unit of Ure2 seldom dissociates during fibril formation,23 we therefore refer to the dimer concentration throughout this study. Our previous work has demonstrated that the aggregation of Ure2 is dominated by fragmentation of mature fibrils.23,30 Global fitting of the kinetics of fibril formation measured by ThT fluorescence over a range of initial Ure2 concentrations yielded the two combined kinetic parameters, knk+ and k+k−, where kn, k+ and k− represent the primary nucleation, fibril elongation and fragmentation rate constants, respectively.23 To determine which microscopic reaction steps are influenced by the peptide, we measured the kinetics of Ure2 aggregation using ThT assays in the presence of different concentrations of LQVNIGNR (Fig. 2B and C). We fitted these kinetic curves globally to a fragmentation-dominant model with variable knk+ but fixed k+k−, or with fixed knk+ but variable k+k− (see Methods); the former but not the latter showed a good fit to the experimental data (Fig. 2B and C). Allowing both knk+ and k+k− to vary did not improve the quality of the fit compared to that obtained using fixed k+k−. The results show that both k+ and k− were unaffected by the presence of LQVNIGNR, whereas the primary nucleation rate kn increased with increasing concentration of peptide (Table S2†).
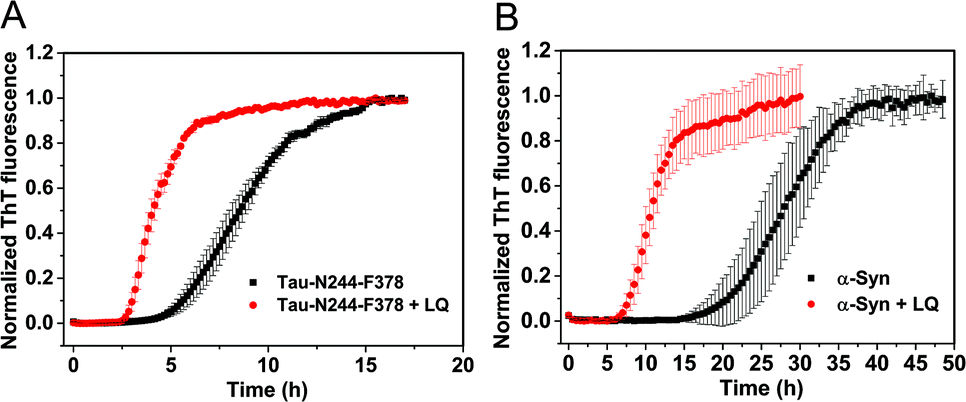
We next investigated the mechanism of the acceleration effect of LQVNIGNR on Ure2 primary nucleation during fibril formation. We noted that this peptide was shown previously to be capable of forming fibrillar aggregates and that preformed fibrils can promote the aggregate formation of Ure2.25 Therefore, we investigated the possibility that the aggregation of LQVNIGNR might promote Ure2 aggregate formation under our experimental conditions (Fig. 3). Since the ribbon-like fibrils formed by LQVNIGNR (Fig. 3A) did not stain with ThT (Fig. S3†), we carried out a turbidity assay to monitor the self-assembly of LQVNIGNR as well as its effect on Ure2 aggregation (Fig. 3B). The results showed that the lag time of Ure2 or LQVNIGNR aggregation when incubated individually was about 4 h, indicating that the peptide does not assemble into fibrillar species more rapidly than Ure2. When incubated together, the aggregation was accelerated and the lag phase decreased to about 2 h, consistent with the ThT assay (Fig. 3C). Furthermore, addition of 10% fibril seeds (0.5 μM) of LQVNIGNR did not promote formation of Ure2 fibrils, in contrast to the large effects of the addition of Ure2 fibril seeds (Fig. 3C). In a previous study,25 seeding of WT Ure2 by LQVINGNR was observable when using a 200-fold higher concentration (100 μM) of seeds. However, at the seed concentration used here, no change in kinetics was observed (Fig. 3C). Additionally, we used TEM to observe the species formed by LQVNIGNR or by the LQVINGNR/Ure2 mixture after 1 h incubation. The TEM images of LQVNIGNR showed no evidence for fibril formation, but showed some vesicle-like clusters, while the LQVNIGNR/Ure2 mixture had already formed visible protofibrils (Fig. 3A). Taken together, the results indicate that the observed acceleration effect of LQVNIGNR peptide on Ure2 fibril formation was not caused by a seeding effect of fibrillar aggregates of LQVNIGNR. To further investigate whether the LQVINGNR vesicles form sequence-specific interactions with Ure2, or whether the vesicles have a more general ability to accelerate fibril formation, we tested the effect of the LQVINGNR peptide on aggregation of the fibril core segment of Tau protein (N244-F378) and α-Synuclein. The results show that LQVINGNR vesicles are likewise able to accelerate the aggregation of these amyloid proteins (Fig. 4), implying that LQVINGNR vesicles have a general function as a catalytic surface to promote fibril formation, and do not operate by a specific interaction or seeding effect.
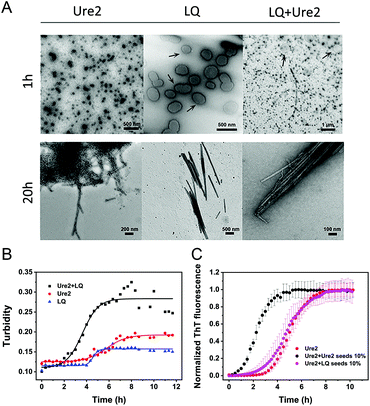 |
| | Fig. 3 The ThT and turbidity measurements of the seeding of Ure2 aggregates by LQVNIGNR. (A) TEM images of aggregates formed by Ure2, LQVNIGNR, and the mixture of Ure2 and LQVNIGVR taken during the lag phase (after 1 h incubation) and at the plateau (after 20 h incubation). Black arrows indicate vesicular assemblies. (B) Turbidity assay of aggregation kinetics of 5 μM Ure2, 40 μM LQVNIGNR (“LQ”), and 5 μM Ure2 plus 40 μM LQVNIGNR (“Ure2 + LQ”). The data shown are the average of three replicates. The data are fitted to sigmoidal curves as a guide to the eye. (C) Kinetic curves derived from ThT measurements for Ure2 alone, or in the presence of seed fibrils of Ure2 or of LQVNIGNR (“LQ”). | |
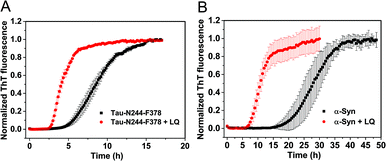 |
| | Fig. 4 The fibril formation of other amyloid proteins in the absence or presence of LQVNIGNR peptide. (A) The fibril core segment of Tau (N244-F378). The concentrations of Tau-N244-F378 and LQVNIGNR peptide (“LQ”) were 10 μM and 40 μM. (B) α-Synuclein protein (α-Syn). The concentrations of α-Synuclein and LQVNIGNR peptide (“LQ”) were 200 μM and 40 μM. | |
Combined smFRET and kinetic analysis of the acceleration effect of LQVNIGNR
Since the primary nucleation of Ure2 was found to be influenced by the LQVNIGNR peptide, and we have previously developed methods to extricate the rates of individual molecular steps in Ure2 fibril formation (Fig. 5A),23 we therefore applied single-molecule FRET to detect oligomeric intermediate species formed during the process of Ure2 fibril formation. In the previous study of Ure2 oligomerization, confocal smFRET experiments were performed by detecting the fluorescence-labeled molecules that diffuse across the focal volume of the excitation laser beam.23 Here we flowed the samples into a microfluidic channel when performing the single molecule fluorescence detection,31 which greatly improved the detection efficiency of low populations of oligomeric species and shortened the data acquisition time from 2 h to 10 min. We used the variant Ure2-S53C for smFRET experiments, as fluorescent labelling at this position has been shown to have no significant influence on fibril formation of Ure2.23 An equimolar mixture of AF488- and AF647-labeled Ure2-S53C was incubated to form fibrils. Aliquots were taken at different time points during aggregation and diluted for fast-flow smFRET detection (see Methods). The species containing both AF488- and AF647-labeled Ure2-S53C generated FRET signals when passing across the diffraction-limited focus. Since the dimeric unit of Ure2 seldom dissociates or exchanges during fibril formation,23 the FRET signal can only be caused by association of Ure2 dimers into oligomers. The numbers of FRET events were converted into concentrations of oligomers (see Methods) and plotted as a function of time (Fig. 5B, left panel). Meanwhile, fibril formation of unlabeled Ure2 in bulk solution was monitored by ThT fluorescence under the same incubation conditions as the smFRET experiments (Fig. 5B, right panel). The smFRET data show that the concentration of oligomers rose to its highest level within 4 h (Fig. 5B, blue data points), which occurred during the lag phase of fibril formation monitored by ThT fluorescence. The concentration of oligomers then decreased as Ure2 was sequestered into mature fibrils that precipitate and could not be detected by the confocal smFRET technique. When 40 μM LQVNIGNR was added, the maximum oligomer concentration was reached within 2 h and the level of oligomer accumulation was about 30% lower (Fig. 5B, red data points), a finding consistent with the shortened lag time indicated by the ThT assay.
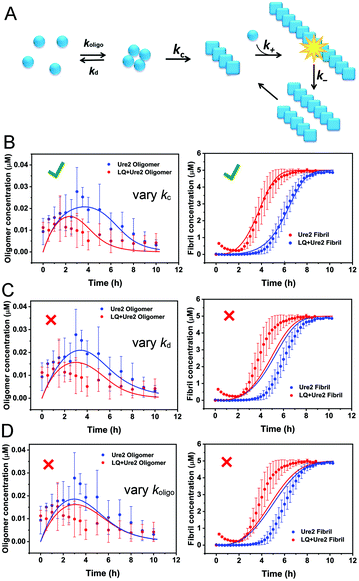 |
| | Fig. 5 Global fitting of Ure2 oligomer and fibril concentration data to a multi-step nucleation model reveals that LQVNIGNR accelerates the conformational conversion of oligomers to fibrils. (A) Scheme of the kinetic model that was used to describe the multi-step nucleation process of Ure2 fibril formation. (B)–(D) Global fitting of Ure2 oligomer and fibril concentration in the presence and the absence of the LQVNIGNR peptide. Left panels, concentration of AF488/AF647-labeled Ure2 S53C oligomers throughout the aggregation reaction monitored by smFRET. Right panels, the bulk aggregation kinetics of 5 μM unlabeled Ure2 S53C monitored by ThT assay. The smFRET and bulk ThT data in the absence (blue) or presence (red) of 40 μM LQVNIGNR were globally fitted to a multistep theoretical model (see Methods). The fitting parameter k+k− was set to be unchanged in the presence or absence of LQVNIGNR according to the fitting results of Fig. 2. (B) Allowing solely kc to have a different value in the absence and in the presence of LQVNIGNR yields reasonable fits. (C) Allowing solely kd to change and (D) allowing solely koligo to change upon addition of LQVNIGNR yields poor fits. This indicates that LQVNIGNR increases the effective rate of primary nucleation by increasing the rate of conversion kc. The data shown are the average of three independent measurements and the error bars represent the SD. | |
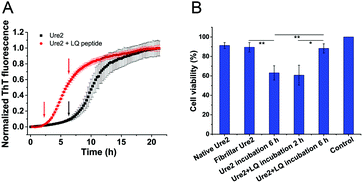
We recently developed a kinetic model for Ure2 oligomer-mediated fibril formation, based on data obtained by smFRET and ThT assays, that explicitly accounts for the intermediates in the primary nucleation step and provides additional insights into the nucleation process of amyloid proteins.23 In this model, non-fibrillar oligomers are formed through an initial assembly process, occurring with rate constant koligo, and subsequently convert into growth-competent fibrillar species with rate constant kc or dissociate with rate constant kd. These species can then elongate by addition of native Ure2 with rate constant k+, and fragment with rate constant k−. The vast majority of detectable oligomeric species were found to be non-fibrillar; thus, measured Ure2 oligomer concentrations correspond to those of the non-fibrillar intermediate. In our previous study,23 we derived an expression for the bulk primary nucleation rate kn in terms of the oligomer-mediated fibril formation model rate constants:
where
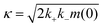
and
α =
koligom(0)
nc. In this study, since fibril elongation and fragmentation were shown to be unaffected by LQVNIGNR, the increase in primary nucleation must be due either to a decrease in oligomer dissociation, or to an increase in oligomer formation or conversion.
We globally fitted the combined smFRET and bulk ThT data using this model to reveal further details of the microscopic steps that account for the acceleration of kn (Fig. 5B–D). The bulk ThT data discussed above showed that k+ and k− are unaffected by the presence of LQVNIGNR (Fig. 2B and C), so these two parameters were set to be the same in the presence and absence of the peptide, while koligo, kd and kc were allowed to vary. The results showed that considering either only kd or only koligo as a variable gave poor fits to the data (Fig. 5C and D), while allowing only kc to vary gave a good fit to both the smFRET and the ThT data (Fig. 5B). Allowing kc and either koligo or kd to vary gave only a marginal improvement in fitting quality and a marginal difference in the values of koligo or kd compared with those obtained when only kc is variable, indicating that koligo and kd are not dramatically changed in the presence of the LQVNIGNR peptide. The results (Table S3†) revealed that the principal effect of LQVNIGNR was a large increase (by an order of magnitude) in kc, suggesting that LQVNIGNR interacts with Ure2 oligomers to promote their conversion from prefibrillar oligomers to β-sheet containing fibrillar species, resulting in a shortened lag phase and reduced accumulation of oligomers.
LQVNIGNR forms vesicular assemblies which influence Ure2 oligomer conformational conversion
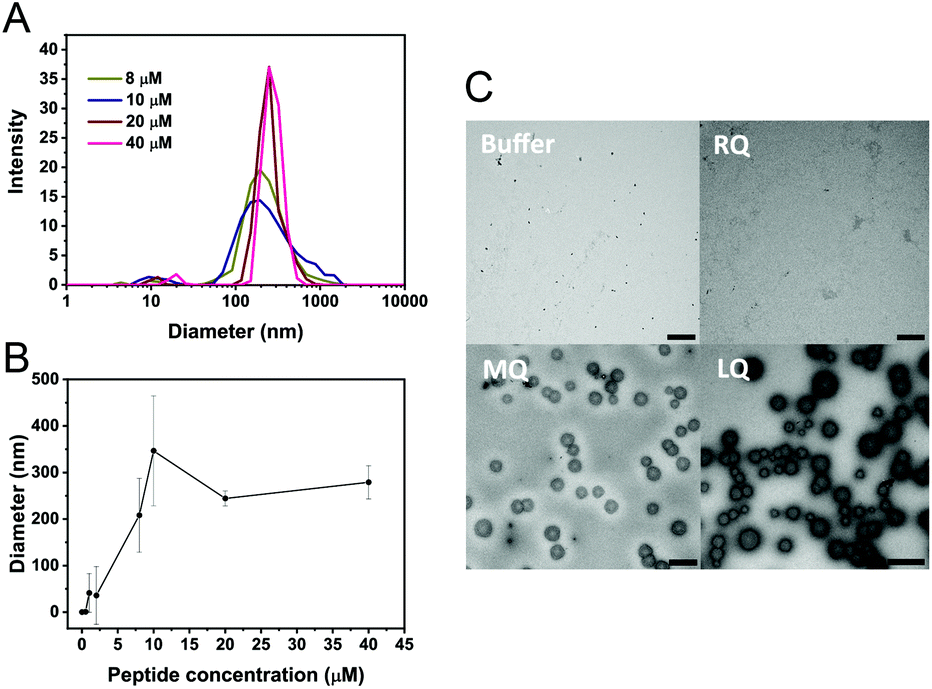
Vesicle-like clusters were observed in TEM images of both LQVNIGNR and LQVNIGNR + Ure2 samples after incubation for 1 h (Fig. 3A). To further characterize the formation of vesicles, we performed dynamic light scattering (DLS) experiments to determine the sizes of the species in solution (Fig. 6A and B). The DLS results showed an average size of around 300 to 500 nm, which is consistent with the TEM data (Fig. 6C). When the concentration of LQVNIGNR reached 20 μM, the size of the species became uniform (Fig. 6A and B). Thus, the critical concentration for the formation of homogeneous vesicles is about 20 μM, as indicated by the DLS results (Fig. 6B). It has been reported that amyloidogenic proteins, such as PrP and a fragment of hIAPP (residues 20–29) can assemble into micelle-like structures that are typically 30 nm in diameter.32,33 Our results showed that the species formed by the Ure2 fragment LQVINGNR have a larger size of hundreds of nanometers (Fig. 6), indicating they are vesicle-like clusters. When LQVNIGNR concentrations were lower than the critical concentration, no significant acceleration effect on the fibril formation of Ure2 could be observed (Fig. S4†), indicating that the acceleration effect is related to the existence of vesicular assemblies of LQVNIGNR. We also used TEM to compare species in the samples of LQVNIGNR, MQVNIGNR and RQVNIGNR, at a concentration of 40 μM (Fig. 6C). The TEM images showed that both LQVNIGNR and MQVNIGNR samples contained vesicular assemblies, although in the latter sample these could only be seen in a minority of frames and in much smaller numbers, while such species were not evident at all in RQVNIGNR samples. As LQVNIGNR is the only one of these three peptides that has a significant acceleration effect on Ure2 aggregation (Fig. S2B and C†), this result further supports the conclusion that the acceleration effect is related to the ability of the peptide to form vesicles.
 |
| | Fig. 6 Characterization of vesicles formed by LQVNIGNR. (A) The size distributions of vesicles formed by LQVNIGNR peptide of different concentrations measured by DLS. (B) The average size of vesicles formed by LQVNIGNR peptide of different concentrations measured by DLS. The data shown are the average of three independent measurements and the error bars represent the SD. (C) TEM images of LQVNIGNR (“LQ”), MQVNIGNR (“MQ”), and RQVNIGNR (“RQ”) 40 μM peptide samples; the scale bars are 1 μm. | |
Fast oligomer conversion leads to reduced cell toxicity
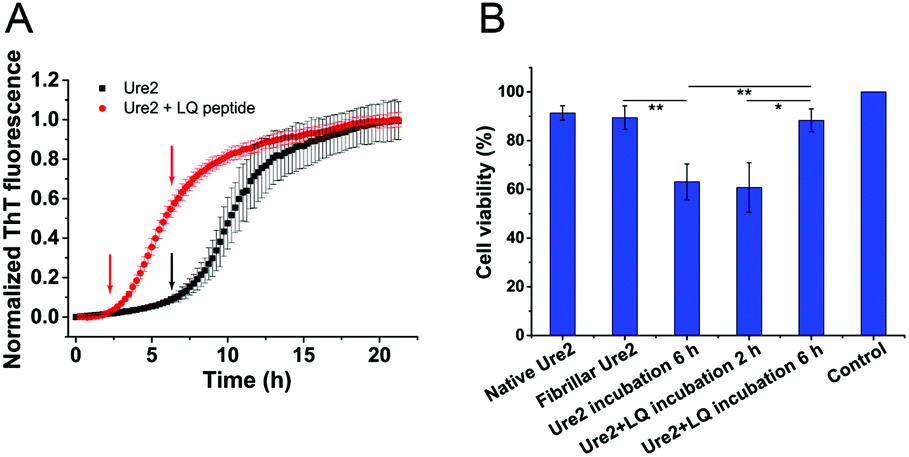
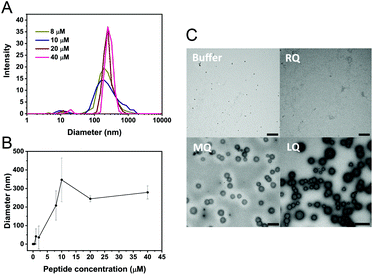
Growing evidence indicates that amyloid oligomers are the most cytotoxic species formed during the aggregation of amyloidogenic proteins.2,6,7 Since our analysis above shows that the oligomers persist for less time in the presence of LQVNIGNR, the toxic intermediates are expected to accumulate less in cells. Thus we performed MTT assays using the human neuroblastoma cell-line SH-SY5Y to detect the cytotoxicity of the Ure2 samples in the presence or absence of LQVNIGNR during amyloid formation. The MTT results showed that the samples taken from near the end of the lag phase of Ure2 aggregation in the presence of LQVNIGNR (2 h) or without LQVNIGNR (6 h) have similar levels of cytotoxicity due to the generation of oligomers. In contrast, the cell viability at 6 h in the presence of LQVNIGNR is significantly higher than without LQVNIGNR (Fig. 7), which means the duration of the cytotoxicity caused by the presence of oligomers is reduced. This result supports our proposal that the accelerated conversion of oligomers to less toxic fibrillar species leads to lower cytotoxicity and may be beneficial for cell growth.
 |
| | Fig. 7 MTT cell viability assays. (A) ThT measurements of Ure2 fibril formation in the absence (black) and presence of LQVNIGNR (red). (B) Cell viability measured by MTT assay. Human SH-SY5Y cells were treated with aliquots of the solutions (final concentration 0.5 μM) taken from the time points indicated in ThT curves. As additional controls, 0.5 μM native Ure2 or Ure2 fibrils were also used to treat the cells but showed no significant toxicity. The data shown are the average of three independent measurements and the error bars represent the SD. *p < 0.05, **p < 0.01. | |
Discussion
Previous studies have demonstrated that specific interfaces between water and other media such as air, detergent, polystyrene and lipids are able to increase significantly the rate of amyloid formation.34–39 Vesicles or small micelles formed by amphiphilic molecules such as LQVNIGNR can provide such an interface in solution and influence the aggregation kinetics of amyloidogenic proteins40–44 by forming hydrogen bonds, hydrophobic or electrostatic interactions with amyloid proteins.45–47 Such effects of interfaces are thought to be related mainly to the promotion of primary nucleation. For instance, the rate of primary nucleation of α-Synuclein can be enhanced by three orders of magnitude or more in the presence of lipid bilayers provided by small unilamellar vesicles.34 Our results not only reaffirm the importance of interfaces in promoting primary nucleation but also demonstrate that one of the ways interfaces may achieve this is by increasing the rate of conformational conversion of oligomers to fibrils. We propose that the amphiphilic LQVNIGNR peptide may accelerate Ure2 fibril formation by self-assembling into vesicles, which then provide a surface with which Ure2 oligomers can interact through non-specific interactions, which in turn catalyses the conversion of Ure2 oligomers into elongation-competent fibrillar species (Fig. 8). The fact that vesicles formed from LQVNIGNR peptide are also able to accelerate aggregation of unrelated amyloidogenic proteins (Fig. 4), suggests that it is the ability of the peptide to form a catalytic surface that is key, rather than the peptide sequence per se.
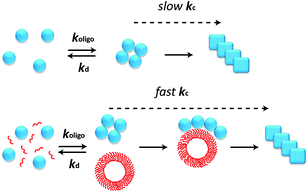 |
| | Fig. 8 A proposed model of the acceleration mechanism of Ure2 fibril formation by LQVNIGNR. LQVNIGNR forms vesicles that promote the oligomer conformational conversion step, resulting in acceleration of the aggregation process to form less toxic fibrillar species. | |
There have been examples showing that acceleration of amyloid fibril formation can decrease the population of toxic oligomeric species and reduce cytotoxicity.48–51 Our results show that the increased rate of conversion of Ure2 oligomers to elongation-competent fibrillar species has a negligible effect on the oligomer half-life (the median survival time for an individual oligomer), as the rate of disappearance of oligomers remains dominated by the much faster dissociation process. However, the accelerated formation of new Ure2 fibrils by oligomer conversion results in a shorter lag phase during which native Ure2 has yet to be significantly depleted. This shortens the time period during which significant concentrations of new oligomers may be assembled and in so doing indirectly reduces the concentration of oligomers seen during the aggregation process. In systems where oligomer formation is catalysed by existing fibrils, accelerating oligomer conversion is expected to have the same effect although for subtly different reasons. Rather than reducing the lag phase, this will increase the effective rate of secondary growth processes, reducing the period of time during which both native and fibrillar protein species co-exist in significant amounts, and so reducing the period of time during which oligomers are formed.
These results therefore suggest an alternative strategy for reducing the cellular toxicity caused by amyloid aggregation under conservation-of-mass conditions: rather than taking the more common approach of inhibiting amyloid formation to reduce toxicity, the desired effect may instead be achieved by accelerating amyloid formation and so reducing the period of time during which toxic oligomeric intermediates may form. Clearly, this approach is likely to be most effective when the acceleration of amyloid formation is not caused by an acceleration in the rate of oligomer formation itself. Although it has not been explicitly verified precisely how interfaces (such as those formed by lipids and surfaces) accelerate primary nucleation of amyloid fibrils, it seems highly likely that an increase in oligomer formation is usually responsible, given that heterogeneous nucleation is initiated by interfaces. The great advantage of the LQVNIGNR peptide studied here is therefore that it can promote aggregation without promoting oligomer formation. This new strategy also offers a significant advantage over traditional inhibition-based approaches. The acceleration of a reaction step can be achieved with a catalytic agent that binds transiently to the reactant and is released upon formation of product. For a slow reaction step such as conversion, relatively low quantities of catalyst may be required, since only a fraction of oligomers need interact with the catalyst at any one time to dramatically boost the conversion rate. By contrast, an inhibitor must remain bound to a significant proportion of the oligomers (or monomers) to noticeably reduce the concentration of free oligomers; effective inhibition therefore always requires maintenance of saturating concentrations of inhibitory agent. Where a proposed drug for the suppression of toxic amyloid oligomers in e.g. neurodegenerative diseases is not very bioavailable, is expensive to manufacture, is inherently toxic, or does not readily cross the blood–brain barrier, this advantage may be decisive.
In the present study, the critical assembly concentration (CAC) of the LQ vesicle formation is about 20 μM. However, taking into account the relative sizes of the peptide and the intact Ure2 protein, the CAC is only 5% of the Ure2 mass concentration. Nonetheless, a promising line of future enquiry might be to develop catalytic vesicles from peptides with far lower CACs. The potential ways to improve the CAC include increasing the length of the non-polar hydrophobic residue tails, introducing other synthetic hydrocarbon tails, and decreasing the electrostatic repulsion between charged head groups while keeping the amphiphilic balance of the peptide.52,53 The optimization of the sequence could be realized with the assistance of molecular simulation. Further, finding non-vesicular catalysts that have similar effects to the vesicular catalyst could also lower the concentration required and present an additional approach. These improvements will facilitate application of this catalytic strategy in drug development.
Conclusions
In this study, we have found that the amphiphilic peptide LQVNIGNR derived from the Ure2 prion domain sequence forms vesicular assemblies that can accelerate the aggregation of native Ure2 as well as unrelated amyloidogenic proteins into insoluble fibrils. Oligomer concentration and fibril mass concentration of Ure2 were measured over time by smFRET and by ThT assay, respectively. Global fitting of these data to a multistep kinetic model revealed that this acceleration in fibril formation by LQVNIGNR is achieved by a 10-fold increase in the conversion rate of pre-fibrillar oligomers to fibrillar species (Fig. 8). The faster transition from oligomers to fibrils reduces the amount of toxic intermediates accumulated in cells. These findings demonstrate the power of combining single molecule detection with kinetic analysis to investigate factors that influence the microscopic reactions in amyloid formation, a method which could be applied in the future to determine the mechanisms by which other accelerators or inhibitors influence amyloid formation.
Experimental
Single-molecule FRET measurement of Ure2 oligomerization under fast flow conditions
Single molecule measurements were carried out using a home-built confocal microscope as described previously,23,54 except that the sample was flowed into a microfluidic device instead of being dropped onto the coverslip. An equimolar concentration (5 μM) of AF647-labeled Ure2 and AF488-labeled Ure2 were mixed in the presence or absence of 40 μM LQVNIGNR peptide and incubated in a Fluostar Omega plate reader (BMGLabtech) at 30 °C under 200 rpm orbital shaking. The concentrations of Ure2 protein we used throughout this study refer to dimer concentrations because the lack of dissociation of the Ure2 dimer during amyloid formation has been demonstrated previously.23 During the fibril formation process, a 2 μL aliquot was taken from the well at each time point and diluted 104-fold for smFRET data collection. A 150 μL volume of diluted sample was sucked into a tip that plugged the inlet of the microfluidic channel and the sample was pumped with a flow velocity of 200 μL−1 h−1. Under fast flow conditions, fewer photons could be detected compared with static conditions because of the shorter residence time of molecules in the laser focus. Therefore, the laser power was increased to 1.25 mW to obtain a better signal-to-noise ratio. Accordingly, the bin time for data collection was decreased to 50 μs to adapt to the shortened average residence time of fluorescent molecules in the focus, which is 27 μs under the flow velocity of 200 μL−1 h−1 as measured by the fluorescence correlation spectroscopy described in the ESI (Fig. S5 and Table S4†); 10 photons per bin for the donor channel and the acceptor channel was set as the threshold to select the oligomer events. Large particles that occupy multiple time bins were assumed to be fibrillar species and were excluded from further analysis of the oligomers.55 The oligomer burst rate was converted to oligomer concentration using donor-labeled soluble Ure2 as the concentration standard. At least three independent experiments were performed, and the results were averaged.
Bulk kinetic assay and analysis of Ure2 fibril formation
A 150 μL volume of 5 μM Ure2 protein mixed with different concentrations of peptide (0, 30, 40, 50, 60, 80 μM) supplemented with 5 μM ThT was pipetted into a 96-well clear bottomed plate (Costar). The reaction was carried out in a Fluostar Omega plate reader (BMGLabtech) under the same conditions as used in the single-molecule experiments. The ThT fluorescence was recorded every 15 min with excitation at 450 nm and emission at 485 nm. At least three replicates were performed to obtain the averaged values and standard deviations. The normalized ThT data for Ure2 aggregation in the presence of LQVNIGNR peptide were fitted to an analytical solution of the kinetics of breakable filament assembly.5,29 The evolution of the fibril mass concentration is given in terms of the rate constants as a double exponential form as shown:| | 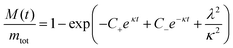 | (1) |
where M(t) is the concentration of protein in fibrillar form at time t, known as the mass concentration; mtot is the total concentration of Ure2; and the constants, C+ and C−, are fixed by the initial conditions
| | 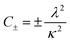 | (2) |
where
λ and
k are two combined kinetic parameters related to the primary nucleation rate
kn, fibril elongation rate
k+ and fibril fragmentation rate
k−.
| | 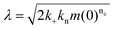 | (3) |
| | 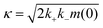 | (4) |
By fitting either or both of these combined parameters locally at each peptide concentration instead of globally across all peptide concentrations, we were able to identify which mechanistic step is affected by the LQVNIGNR peptide. All fitting was carried out using the online fitting platform AmyloFit.56
Combined bulk ThT and smFRET data analysis
We previously reported a simplified coarse-grained kinetic model suitable for describing Ure2 fibril formation via oligomeric intermediates,23 that explicitly includes the oligomer concentration O(t) as well as the native dimer concentration m(t), the fibril concentration P(t) and the fibril mass concentration M(t). The rate equations for this model are written as a master equation:| | 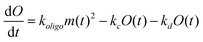 | (5) |
| | 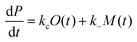 | (6) |
| | 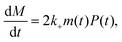 | (7) |
and obey the conservation-of-mass relation m(t) + M(t) = m(0). This model features the new rate constants koligo (for formation of new oligomers by dimer association), kc (conversion of oligomers to new fibrils), and kd (oligomer dissociation). We also previously developed a customized offline extension to the fitting platform AmyloFit to numerically fit kinetic data for M(t) and O(t) simultaneously to these equations. We carried out the same procedure in the current paper, using kinetic data for M(t) and O(t) collected both with and without a single concentration of LQVNIGNR peptide. Having determined the effect of LQVNIGNR on the bulk rate parameters from fitting ThT data, we could use this information to constrain our fits and determine its effect on the reaction steps involving oligomers directly. We did so by globally fitting as many parameters as possible across both datasets, to determine the minimum set of parameters that need to be varied between datasets to yield good fits. We identified the reaction mechanisms corresponding to these parameters as the ones being primarily affected by LQVNIGNR peptide.
Other experimental details including sample preparation, TEM, DLS, and MTT assay are described in the ESI.†
Author contributions
J. Y., S. W. and S. P. designed the experiments; J. Y., Q. Y., Z. L. and S. W. performed the experiments; A. J. D. and T. P. J. K. performed the theoretical modelling; J. Y., A. J. D., C. M. D., T. P. J. K., S. W. and S. P. analyzed the data and wrote the paper.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Chris Taylor (University of Cambridge) for providing the mask for construction of the single-channel microfluidics. We thank Prof. David Klenerman (University of Cambridge) for helpful discussions about single-molecule experiments under fast flow conditions. The Perrett group acknowledges support from the National Key R&D Program of China (2017YFA0504000), the National Natural Science Foundation of China (31920103011, 21673278), the National Laboratory of Biomacromolecules, and the CAS Centre of Excellence in Biomacromolecules. A. J. D. was supported by the Schiff Foundation. Our electron microscopy work was performed at the Center for Biological Imaging (Institute of Biophysics, Chinese Academy of Sciences).
Notes and references
- D. M. Fowler, A. V. Koulov, W. E. Balch and J. W. Kelly, Trends Biochem. Sci., 2007, 32, 217–224 CrossRef CAS PubMed.
- T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef CAS PubMed.
- S. I. A. Cohen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, J. Mol. Biol., 2012, 421, 160–171 CrossRef CAS PubMed.
- S. I. Cohen, M. Vendruscolo, M. E. Welland, C. M. Dobson, E. M. Terentjev and T. P. J. Knowles, J. Chem. Phys., 2011, 135, 065105 CrossRef PubMed.
- J. P. Cleary, D. M. Walsh, J. J. Hofmeister, G. M. Shankar, M. A. Kuskowski, D. J. Selkoe and K. H. Ashe, Nat. Neurosci., 2005, 8, 79–84 CrossRef CAS PubMed.
- B. Winner, R. Jappelli, S. K. Maji, P. A. Desplats, L. Boyer, S. Aigner, C. Hetzer, T. Loher, M. Vilar, S. Campioni, C. Tzitzilonis, A. Soragni, S. Jessberger, H. Mira, A. Consiglio, E. Pham, E. Masliah, F. H. Gage and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4194–4199 CrossRef CAS PubMed.
- N. Cremades, S. I. Cohen, E. Deas, A. Y. Abramov, A. Y. Chen, A. Orte, M. Sandal, R. W. Clarke, P. Dunne, F. A. Aprile, C. W. Bertoncini, N. W. Wood, T. P. J. Knowles, C. M. Dobson and D. Klenerman, Cell, 2012, 149, 1048–1059 CrossRef CAS PubMed.
- M. Iljina, G. A. Garcia, M. H. Horrocks, L. Tosatto, M. L. Choi, K. A. Ganzinger, A. Y. Abramov, S. Gandhi, N. W. Wood, N. Cremades, C. M. Dobson, T. P. J. Knowles and D. Klenerman, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 15 CrossRef PubMed.
- P. Narayan, A. Orte, R. W. Clarke, B. Bolognesi, S. Hook, K. A. Ganzinger, S. Meehan, M. R. Wilson, C. M. Dobson and D. Klenerman, Nat. Struct. Mol. Biol., 2012, 19, 79–83 CrossRef CAS PubMed.
- S. L. Shammas, G. A. Garcia, S. Kumar, M. Kjaergaard, M. H. Horrocks, N. Shivji, E. Mandelkow, T. P. J. Knowles, E. Mandelkow and D. Klenerman, Nat. Commun., 2015, 6, 7025 CrossRef CAS PubMed.
- M. Iljina, L. Hong, M. H. Horrocks, M. H. Ludtmann, M. L. Choi, C. D. Hughes, F. S. Ruggeri, T. Guilliams, A. K. Buell, J.-E. E. Lee, S. Gandhi, S. F. Lee, C. E. Bryant, M. Vendruscolo, T. P. J. Knowles, C. M. Dobson, E. De Genst and D. Klenerman, BMC Biol., 2017, 15, 57 CrossRef PubMed.
- M. Bucciantini, E. Giannoni, F. Chiti, F. Baroni, L. Formigli, J. Zurdo, N. Taddei, G. Ramponi, C. M. Dobson and M. Stefani, Nature, 2002, 416, 507–511 CrossRef CAS PubMed.
- B. Cheng, H. Gong, H. Xiao, R. B. Petersen, L. Zheng and K. Huang, Biochim. Biophys. Acta, 2013, 1830, 4860–4871 CrossRef CAS PubMed.
- C. Soto, M. S. Kindy, M. Baumann and B. Frangione, Biochem. Biophys. Res. Commun., 1996, 226, 672–680 CrossRef CAS PubMed.
- J. Chabry, B. Caughey and B. Chesebro, J. Biol. Chem., 1998, 273, 13203–13207 CrossRef CAS PubMed.
- Y. Nagai, T. Tucker, H. Ren, D. J. Kenan, B. S. Henderson, J. D. Keene, W. J. Strittmatter and J. R. Burke, J. Biol. Chem., 2000, 275, 10437–10442 CrossRef CAS PubMed.
- L. A. Scrocchi, Y. Chen, S. Waschuk, F. Wang, S. Cheung, A. A. Darabie, J. McLaurin and P. E. Fraser, J. Mol. Biol., 2002, 318, 697–706 CrossRef CAS PubMed.
- O. M. El-Agnaf, K. E. Paleologou, B. Greer, A. M. Abogrein, J. E. King, S. A. Salem, N. J. Fullwood, F. E. Benson, R. Hewitt, K. J. Ford, F. L. Martin, P. Harriott, M. R. Cookson and D. Allsop, FASEB J., 2004, 18, 1315–1317 CrossRef CAS PubMed.
- T. J. Gibson and R. M. Murphy, Protein Sci., 2006, 15, 1133–1141 CrossRef CAS PubMed.
- N. V. Artemova, V. A. Stein-Margolina, Z. M. Bumagina and Y. B. Gurvits, Biotechnol. Prog., 2011, 27, 846–854 CrossRef CAS PubMed.
- R. B. Wickner, Science, 1994, 264, 566–569 CrossRef CAS PubMed.
- J. Yang, A. J. Dear, T. C. T. Michaels, C. M. Dobson, T. P. J. Knowles, S. Wu and S. Perrett, J. Am. Chem. Soc., 2018, 140, 2493–2503 CrossRef CAS PubMed.
- X. Zhang, B. Cheng, H. Gong, C. Li, H. Chen, L. Zheng and K. Huang, FEBS Lett., 2011, 585, 71–77 CrossRef CAS PubMed.
- L. Fei and S. Perrett, J. Biol. Chem., 2009, 284, 11134–11141 CrossRef CAS PubMed.
- S. Ngo, V. Chiang and Z. Guo, J. Struct. Biol., 2012, 180, 374–381 CrossRef CAS PubMed.
- L. O. Tjernberg, J. Näslund, F. Lindqvist, J. Johansson, A. R. Karlström, J. Thyberg, L. Terenius and C. Nordstedt, J. Biol. Chem., 1996, 271, 8545–8548 CrossRef CAS PubMed.
- P. M. Seidler, D. R. Boyer, J. A. Rodriguez, M. R. Sawaya, D. Cascio, K. Murray, T. Gonen and D. S. Eisenberg, Nat. Chem., 2018, 10, 170–176 CrossRef CAS PubMed.
- T. P. J. Knowles, C. A. Waudby, G. L. Devlin, S. I. Cohen, A. Aguzzi, M. Vendruscolo, E. M. Terentjev, M. E. Welland and C. M. Dobson, Science, 2009, 326, 1533–1537 CrossRef PubMed.
- Y. Q. Wang, A. K. Buell, X. Y. Wang, M. E. Welland, C. M. Dobson, T. P. J. Knowles and S. Perrett, J. Biol. Chem., 2011, 286, 12101–12107 CrossRef CAS PubMed.
- M. H. Horrocks, H. Li, J.-U. U. Shim, R. T. Ranasinghe, R. W. Clarke, W. T. Huck, C. Abell and D. Klenerman, Anal. Chem., 2012, 84, 179–185 CrossRef CAS PubMed.
- E. Hingant, P. Fontes, M. T. Alvarez-Martinez, J.-D. D. Arnaud, J.-P. P. Liautard and L. Pujo-Menjouet, PLoS Comput. Biol., 2014, 10 Search PubMed.
- E. Rhoades and A. Gafni, Biophys. J., 2003, 84, 3480–3487 CrossRef CAS PubMed.
- C. Galvagnion, A. K. Buell, G. Meisl, T. C. Michaels, M. Vendruscolo, T. P. J. Knowles and C. M. Dobson, Nat. Chem. Biol., 2015, 11, 229–234 CrossRef CAS PubMed.
- J. W. P. Brown, G. Meisl, T. P. J. Knowles, A. K. Buell, C. M. Dobson and C. Galvagnion, Chem. Commun., 2018, 54, 7854–7857 RSC.
- S. Campioni, G. Carret, S. Jordens, L. Nicoud, R. Mezzenga and R. Riek, J. Am. Chem. Soc., 2014, 136, 2866–2875 CrossRef CAS PubMed.
- R. Vacha, S. Linse and M. Lund, J. Am. Chem. Soc., 2014, 136, 11776–11782 CrossRef CAS PubMed.
- L. Giehm, C. L. P. Oliveira, G. Christiansen, J. S. Pedersen and D. E. Otzen, J. Mol. Biol., 2010, 401, 115–133 CrossRef CAS PubMed.
- M. Rabe, A. Soragni, N. P. Reynolds, D. Verdes, E. Liverani, R. Riek and S. Seeger, ACS Chem. Neurosci., 2013, 4, 408–417 CrossRef CAS PubMed.
- Z. Jiang, M. de Messieres and J. C. Lee, J. Am. Chem. Soc., 2013, 135, 15970–15973 CrossRef CAS PubMed.
- C. M. Pfefferkorn, Z. Jiang and J. C. Lee, Biochim. Biophys. Acta, 2012, 1818, 162–171 CrossRef CAS PubMed.
- T. L. Williams and L. C. Serpell, FEBS J., 2011, 278, 3905–3917 CrossRef CAS PubMed.
- Y. Han, X. Huang, M. Cao and Y. Wang, J. Phys. Chem. B, 2008, 112, 15195–15201 CrossRef CAS PubMed.
- J. R. Brender, S. Salamekh and A. Ramamoorthy, Acc. Chem. Res., 2012, 45, 454–462 CrossRef CAS PubMed.
- S. Bag, S. Chaudhury, D. Pramanik, S. DasGupta and S. Dasgupta, Proteins, 2016, 84, 1213–1223 CrossRef CAS PubMed.
- K. E. Kumar and P. N. Prabhu, Phys. Chem. Chem. Phys., 2014, 16, 24076–24088 RSC.
- Z. Jiang and J. C. Lee, J. Mol. Biol., 2014, 426, 4074–4086 CrossRef CAS PubMed.
- T. Mohamed, S. S. Gujral and P. P. N. Rao, ACS Chem. Neurosci., 2018, 9, 773–782 CrossRef CAS PubMed.
- P. K. Singh, V. Kotia, D. Ghosh, G. M. Mohite, A. Kumar and S. K. Maji, ACS Chem. Neurosci., 2013, 4, 393–407 CrossRef CAS PubMed.
- J. Bieschke, M. Herbst, T. Wiglenda, R. P. Friedrich, A. Boeddrich, F. Schiele, D. Kleckers, J. M. Lopez del Amo, B. A. Gruning, Q. Wang, M. R. Schmidt, R. Lurz, R. Anwyl, S. Schnoegl, M. Fandrich, R. F. Frank, B. Reif, S. Gunther, D. M. Walsh and E. E. Wanker, Nat. Chem. Biol., 2011, 8, 93–101 CrossRef PubMed.
- C. M. Cremers, D. Knoefler, S. Gates, N. Martin, J. U. Dahl, J. Lempart, L. Xie, M. R. Chapman, V. Galvan, D. R. Southworth and U. Jakob, Mol. Cell, 2016, 63, 768–780 CrossRef CAS PubMed.
- X. Zhao, F. Pan, H. Xu, M. Yaseen, H. Shan, C. A. Hauser, S. Zhang and J. R. Lu, Chem. Soc. Rev., 2010, 39, 3480–3498 RSC.
- A. Dasgupta and D. Das, Langmuir, 2019, 35, 10704–10724 CrossRef CAS PubMed.
- F. Lou, J. Yang, S. Wu and S. Perrett, Chem. Commun., 2017, 53, 7986–7989 RSC.
- M. H. Horrocks, L. Tosatto, A. J. Dear, G. A. Garcia, M. Iljina, N. Cremades, M. Dalla Serra, T. P. J. Knowles, C. M. Dobson and D. Klenerman, Anal. Chem., 2015, 87, 8818–8826 CrossRef CAS PubMed.
- G. Meisl, J. B. Kirkegaard, P. Arosio, T. C. Michaels, M. Vendruscolo, C. M. Dobson, S. Linse and T. P. Knowles, Nat. Protoc., 2016, 11, 252–272 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr01481h |
| ‡ Current address: Department of Cell Biology, Yale School of Medicine, New Haven, CT 06511, USA |
| § Deceased 8 September 2019 |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  c,
Qiong-Qiong
Yao
c,
Qiong-Qiong
Yao



 and α = koligom(0)nc. In this study, since fibril elongation and fragmentation were shown to be unaffected by LQVNIGNR, the increase in primary nucleation must be due either to a decrease in oligomer dissociation, or to an increase in oligomer formation or conversion.
and α = koligom(0)nc. In this study, since fibril elongation and fragmentation were shown to be unaffected by LQVNIGNR, the increase in primary nucleation must be due either to a decrease in oligomer dissociation, or to an increase in oligomer formation or conversion.