
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling the mechanism of the one-pot synthesis of exchange coupled Co-based nano-heterostructures with a high energy product†
Beatrice
Muzzi
 abc,
Martin
Albino
c,
Claudia
Innocenti
bc,
Michele
Petrecca
*bc,
Brunetto
Cortigiani
c,
César de Julián
Fernández
d,
Giovanni
Bertoni
de,
Rodrigo
Fernandez-Pacheco
fgh,
Alfonso
Ibarra
fgh,
Clara
Marquina
if,
M. Ricardo
Ibarra
ifgh and
Claudio
Sangregorio
*bc
abc,
Martin
Albino
c,
Claudia
Innocenti
bc,
Michele
Petrecca
*bc,
Brunetto
Cortigiani
c,
César de Julián
Fernández
d,
Giovanni
Bertoni
de,
Rodrigo
Fernandez-Pacheco
fgh,
Alfonso
Ibarra
fgh,
Clara
Marquina
if,
M. Ricardo
Ibarra
ifgh and
Claudio
Sangregorio
*bc
aDept. of Biotechnology, Chemistry and Pharmacy, University of Siena 1240, I-53100 Siena, Italy
bICCOM – CNR, I-50019 Sesto Fiorentino (FI), Italy. E-mail: petrecca.michele@gmail.com; csangregorio@iccom.cnr.it
cDept. of Chemistry “U. Schiff”, University of Florence and INSTM, I-50019 Sesto Fiorentino (FI), Italy
dIMEM – CNR, I-43124 Parma, Italy
eCNR – Istituto Nanoscienze, I-41125 Modena, Italy
fDpto. de Física de la Materia Condensada, Universidad de Zaragoza, 50009-Zaragoza, Spain
gInstituto de Nanociencia de Aragón (INA), Universidad de Zaragoza, 50018-Zaragoza, Spain
hLaboratorio de Microscopias Avanzadas (LMA), Universidad de Zaragoza, 50018-Zaragoza, Spain
iInstituto de Ciencia de Materiales de Aragón (ICMA), Consejo Superior de Investigaciones Científicas (CSIC)-Universidad de Zaragoza, 50009-Zaragoza, Spain
First published on 5th June 2020
Abstract
The development of reproducible protocols to synthesize hard/soft nano-heterostructures (NHSs) with tailored magnetic properties is a crucial step to define their potential application in a variety of technological areas. Thermal decomposition has proved to be an effective tool to prepare such systems, but it has been scarcely used so far for the synthesis of Co-based metal/ferrite NHSs, despite their intriguing physical properties. We found a new approach to prepare this kind of nanomaterial based on a simple one-pot thermal decomposition reaction of metal-oleate precursors in the high boiling solvent docosane. The obtained NHSs are characterized by the coexistence of Co metal and Co doped magnetite and are highly stable in an air atmosphere, thanks to the passivation of the metal with a very thin oxide layer. The investigation of the influence of the metal precursor composition (a mixed iron–cobalt oleate), of the ligands (oleic acid and sodium oleate) and of the reaction time on the chemical and structural characteristics of the final product, allowed us to rationalize the reaction pathway and to determine the role of each parameter. In particular, the use of sodium oleate is crucial to obtain a metal phase in the NHSs. In such a way, the one-pot approach proposed here allows the fine control of the synthesis, leading to the formation of stable, high performant, metal/ferrite NHSs with tailored magnetic properties. For instance, the room temperature maximum energy product was increased up to 19 kJ m−3 by tuning the Co content in the metal precursor.
1. Introduction
The capability of shaping matter to create complex hybrid nanoarchitectures has represented a major step forwards in nanoscience to develop innovative and multifunctional materials. Combining components at the nanoscale endowed with different physical properties, either strongly interacting or not, can indeed largely expand and enhance the material functionalities, and, in some cases, enable the emergence of new properties. The resulting multifunctional nanomaterials are then promising candidates to address many of the challenges of the next future in a wealth of research areas, ranging from biomedicine to electronics. In this regard, magnetic hybrid nanostructures are particularly promising. Coupling at the nanoscale materials with intrinsically different magnetic properties, indeed, allows the observation of novel magnetic phenomena, the most prominent being exchange bias and exchange spring coupling. The former originates from the exchange interactions between differently ordered magnetic phases, usually an antiferromagnet and a ferro- or ferrimagnetic one,1 while the second is obtained by combining at the nanoscale a high anisotropy hard magnetic phase with a high magnetization soft magnetic phase.2These phenomena have been proposed in the recent past as efficient strategies to enhance the performances of permanent magnets (PM).3–5 PMs are key elements of many technological devices used in everyday life,6 such as hard disks, automotive devices, wind turbines, electronic components, and hybrid electric vehicles. Their performance is quantified by the so-called maximum energy product, BHmax, which describes the maximum amount of magnetic energy storable in a magnet.7 By far, the most powerful magnets comprise rare earth (RE) elements. However, the criticality about the sustainability of the RE chain supply that emerged in the last decade8 has brought about renewed interest towards novel RE-free materials which can provide a viable alternative to RE, at least in all those applications where large performances are not strictly required.9
Among the various strategies investigated to date, considerable interest has been directed towards exchange coupled hard/soft nano-heterostructures (NHSs). The M/CoxFe3−xO4 nanosystem, where M is Fe, Co or a Co–Fe alloy, is indeed one of the best materials to investigate the feasibility of this approach, as it comprises a soft metal with the highest saturation magnetization, MS, (CoFe2 has MS = 245 A m2 kg−1),10 and a hard ferrite (CoxFe3−xO4 has magnetic anisotropy up to 106 J m−3 and a magnetization of 88 A m2 kg−1 for x = 0.6);11–13 moreover, it offers the opportunity to convert one phase into the other by a simple oxidation/reduction process. Nevertheless, although simple in principle, the practical realization of high quality Co-based NHSs is a rather complex task. On the one hand, in fact, the synthesis of stable metal cobalt or cobalt–iron cores by microemulsion14 or thermal decomposition of Co2(CO)8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15 and metal acetylacetonate is already well established.16 However, on the other hand, the following reduction/oxidation step required to obtain exchange coupled NHSs often leads to low crystallinity materials with not well-defined and partially disordered interfaces. Consequently, the resulting products are characterized by low saturation magnetization, small remanence and/or weak magnetic coupling.17–20 On the other hand, thermal decomposition of metal–organic precursors has proved to be effective for producing Fe/Fe3O4 core–shell NPs with well-defined architectures, high quality interfaces and good magnetic properties.21 However, to our knowledge, the technique has been scarcely extended to the synthesis of Co-analogous heterostructures, Co/CoxFe3−xO4. This is rather surprising since Co-based NHSs are extremely appealing for several applications, particularly when materials combining large hysteresis loops and high magnetization are required.11 The replacement of Fe(II) ions in octahedral sites with the high anisotropy Co(II) ones, indeed, largely increases the magnetic anisotropy of the spinel ferrites without compromising the MS.13 For this reason, cobalt ferrite has been proposed for application in electronics, biomedicine, catalysis and as building blocks for the realization of rare earth free permanent magnets.11,22–26
15 and metal acetylacetonate is already well established.16 However, on the other hand, the following reduction/oxidation step required to obtain exchange coupled NHSs often leads to low crystallinity materials with not well-defined and partially disordered interfaces. Consequently, the resulting products are characterized by low saturation magnetization, small remanence and/or weak magnetic coupling.17–20 On the other hand, thermal decomposition of metal–organic precursors has proved to be effective for producing Fe/Fe3O4 core–shell NPs with well-defined architectures, high quality interfaces and good magnetic properties.21 However, to our knowledge, the technique has been scarcely extended to the synthesis of Co-analogous heterostructures, Co/CoxFe3−xO4. This is rather surprising since Co-based NHSs are extremely appealing for several applications, particularly when materials combining large hysteresis loops and high magnetization are required.11 The replacement of Fe(II) ions in octahedral sites with the high anisotropy Co(II) ones, indeed, largely increases the magnetic anisotropy of the spinel ferrites without compromising the MS.13 For this reason, cobalt ferrite has been proposed for application in electronics, biomedicine, catalysis and as building blocks for the realization of rare earth free permanent magnets.11,22–26
Despite the large experimental work carried out in the last 20 years, the synthesis of magnetic NPs by thermal decomposition is still far from being fully rationalized. Indeed, a very large number of parameters concur to determine the nature of the final product,27,28 making the unambiguous identification of their role a complex task. As a result, the synthesis by this technique of Co-based NHSs with pre-determined architecture and composition is still challenging.
Here, we report an experimental investigation on the use of one pot thermal decomposition of mixed iron–cobalt oleate in a high boiling organic solvent, (docosane) to prepare Co/spinel ferrite NHSs with high crystallinity and a controlled composition. In particular, the influence of ligands, metal precursors and reaction time was carefully investigated. The study allowed us to propose a reaction mechanism where sodium oleate plays a crucial role in the formation of a metal cobalt phase. In this way, we were able to identify the key parameters controlling the reaction pathway, paving the way towards the rational design of Co-based NHSs with controlled magnetic properties.
2. Experimental
2.1 Chemicals and materials
Iron(III) chloride hexahydrate (FeCl3·6H2O, 98% from Sigma-Aldrich), cobalt(II) chloride hexahydrate (CoCl2·6H2O, 98% from Honeywell), sodium oleate (NaOL, >97%, TCI Chemicals), oleic acid (OA, 90%, Aldrich), docosane (97%, Aldrich), ethanol (99.8%, Fluka), hexane (99%, Honeywell) and 2-propanol (98%, Honeywell) were used without any further purification.2.2 Synthesis of the iron–cobalt oleate (x-FeCo(OL)) precursor
Bimetal oleate precursors, x-FeCo(OL), with a Fe:Co molar ratio of 2:1 (1-FeCo(OL)), 1:1 (2-FeCo(OL)) and 1:2 (3-FeCo(OL)) were synthesized by dissolving the desired stoichiometric amount of FeCl3·6H2O and CoCl2·6H2O in 20 mL of hexane, 10 mL of ethanol and 10 mL of distilled water. At room temperature, a stoichiometric amount of NaOL was added and the mixture was heated at 75 °C, under vigorous stirring, for 5 hours. The brown colored organic phase was separated from the aqueous phase and a brown-dark waxy product was obtained after solvent evaporation. The stoichiometry of the final product was confirmed by Inductively Coupled Plasma analysis. The thermal stability of the ligand was studied by thermogravimetric analysis, (TGA) using Q-600 (DTA-TG) apparatus with open aluminum pans under a nitrogen atmosphere.
2.3 Synthesis of Co/CoyFe3−yO4 NPs
The Co/CoyFe3−yO4 NPs were synthesized by thermal decomposition of the bimetallic precursor. 1.5 g (ca. 2 mmol of metal ions) of x-FeCo(OL), 0.57 g (2 mmol) of OA and 0.57 g (1.8 mmol) of NaOL were dissolved in 10 mL of docosane at 80 °C. The mixture was heated from 80 to 370 °C, at 2.9 °C min−1, under vigorous stirring and nitrogen flux. The suspension was kept at 370 °C for 40 minutes and then allowed to cool to room temperature. The resulting black powder was separated by application of an external magnet, washed with 2-propanol, hexane and ethanol and finally dried under nitrogen flux.2.4 Characterization techniques
Transmission electron microscopy (TEM, CM12 Philips equipped with a LaB6 filament operating at 100 kV) was employed to determine morphology and size distribution of the NPs. The mean diameter and the NP size distribution for each sample were obtained by statistical analysis over more than 100 NPs, using Image Pro-Plus software. Powder X-Ray diffraction (XRD) data were recorded using a Bruker New D8 ADVANCE ECO diffractometer equipped with a Cu Kα (1.5406 Å) radiation source and operating in θ–θ Bragg–Brentano geometry at 40 kV and 40 mA. The measurements were carried out in the range of 25–70°, with a step size of 0.03° and a collection time of 1 s. Elemental analysis was performed in triplicate using a PerkinElmer Optima 2000 PerkinElmer Inductively Coupled Plasma Optical Emission Spectrophotometer (ICP-OES) Dual Vision. The amount of organic ligand on the surface of NHSs was determined by CHN analysis with an Elemental Analyzer CHN-S Flash E1112 Thermofinnigan. X-ray Photoelectron Spectroscopy (XPS) investigation was carried out using a non-monochromatic X-ray source (V.S.W. Scientific Instrument Limited model TA10, Mg Kα radiation, 1253.6 eV), operating at 12 kV and 12 mA, equipped with a hemispherical analyser (V.S.W. Scientific Instrument Limited, model HA100). Powders were deposited on a KBr disk and pressed to obtain a single pellet. XPS spectra were analyzed with CasaXPS software: the inelastic background was subtracted by means of the Shirley method29 and the deconvolution of the XPS spectra was carried out using the product of Gaussian and Lorentzian line shapes for each component in the spectra. Calibration was performed using the O 1s peak at 530.0 eV. Scanning transmission electron microscopy (STEM) images were acquired on a probe-corrected Titan (Thermo Fischer Scientific), with a working voltage of 300 kV. The microscope was equipped with a high annular dark field (HAADF) detector for imaging (Fischione) and an energy filter Gatan Tridiem 866 ERS (Gatan, Inc.) for Electron Energy Loss Spectroscopy (EELS) mapping. The EELS elemental maps were obtained by fitting the (x, y, E) spectrum images by the model based technique,30 to extract the intensity of the O-K edge (at ∼532 eV), the Fe-L2,3 edge (at ∼710 eV) and Co-L2,3 edge (at ∼780 eV). The spectrum images were acquired with probe convergence semi-angle α = 24.8 mrad and spectrometer collection angle semi-angle β = 51.3 mrad. See the ESI† for details on the quantification procedure. In order to obtain the crystalline structure of the different phases present in the system, an image-corrected Titan3 (Thermo Fischer Scientific) ultra-high resolution electron transmission microscope (UHRTEM) was operated at 300 kV. Magnetic properties were investigated using a Quantum Design MPMS SQUID magnetometer on randomly oriented pressed powder pellets. The field was always applied parallel to the pellet plane. The Zero Field Cooled/Field Cooled (ZFC/FC) procedure was performed applying a 5 mT probe field.3. Results and discussion
Metal/ferrite NHSs were prepared by thermal decomposition of 2:1 mixed iron–cobalt oleate (1-FeCo(OL)) in the high boiling solvent docosane (Teb = 368.7 °C) in the presence of sodium oleate and oleic acid. Fig. 1, left panel, shows a STEM image of sample NHS-1 and the corresponding size distribution. The sample consists of polydisperse NPs coated with ca. 4% w/w of oleate surfactant, as evaluated by CHN Analysis, with an irregular shape and an average diameter, estimated from the largest dimension measured on each NP, of ca. 90 ± 12 nm.
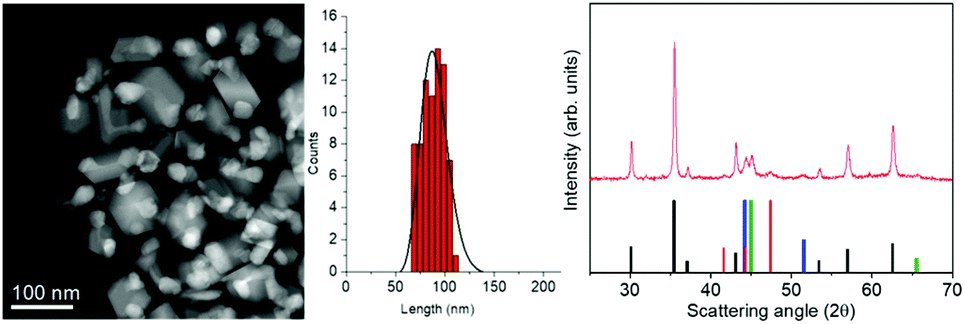 | ||
Fig. 1 (Left) A representative STEM image of NHS-1 NPs and the corresponding size distribution as evaluated from TEM analysis (see also Fig. S1 in the ESI†); (right) experimental XRD pattern (red line) and reference patterns of:  CoFe2O4, (JCPDS PDF #22-1086); CoFe2O4, (JCPDS PDF #22-1086);  hcp Co(0) (JCPDS PDF #89-7373); hcp Co(0) (JCPDS PDF #89-7373);  fcc Co(0), (JCPDS PDF #15-0806), and fcc Co(0), (JCPDS PDF #15-0806), and  Co–Fe alloy, (JCPDS PDF #65-4131). Co–Fe alloy, (JCPDS PDF #65-4131). | ||
The XRD pattern (Fig. 1, right panel) indicates that different crystallographic phases occur in the nanopowder. Indeed, the peaks characteristic of the partially inverse spinel structure of cobalt doped ferrite (black reference), hexagonal and face-centered-cubic metal cobalt (red and blue references) can be recognized. The peak at 2θ = 45.1 degrees can be indexed as arising from the diffraction plane (110) of the cobalt–iron alloy (green reference). The Rietveld analysis of the experimental pattern, detailed in Table 1, showed for the Fd![[3 with combining macron]](https://www.rsc.org/images/entities/b_char_0033_0304.gif) m structure of CoyFe3−yO4 NPs an average crystal size of 41(1) nm, with a lattice parameter a = 0.8397(1) nm. The metal cobalt crystallized in the cubic (space group Fmm) and hexagonal (space group P63/mmc) structures, with an average crystallite size of 17(1) nm and 5(1) nm, respectively, while the Co1−xFex alloy grains (cubic structure, Imm) have an average size of 12(1) nm and a = 0.2847(1) nm, which is consistent with a Co:Fe ratio of ca. 1:1. The weight percentages of the three crystallographic structures are 77(2)% for the ferrite, 13(2)% for the metal Co (mostly fcc with traces of hcp) and 10(2)% for the alloy.
m structure of CoyFe3−yO4 NPs an average crystal size of 41(1) nm, with a lattice parameter a = 0.8397(1) nm. The metal cobalt crystallized in the cubic (space group Fmm) and hexagonal (space group P63/mmc) structures, with an average crystallite size of 17(1) nm and 5(1) nm, respectively, while the Co1−xFex alloy grains (cubic structure, Imm) have an average size of 12(1) nm and a = 0.2847(1) nm, which is consistent with a Co:Fe ratio of ca. 1:1. The weight percentages of the three crystallographic structures are 77(2)% for the ferrite, 13(2)% for the metal Co (mostly fcc with traces of hcp) and 10(2)% for the alloy.
| NHS-1 | XRD | XPS | ||
|---|---|---|---|---|
| Lattice parameters (nm) | Crystal size (D) (nm) | Phase percentage (%) | Phase percentage (%) | |
| Co (fcc) | a = 0.3546(1) | 17(1) | 11(2) | 19(5) |
| Co (hcp) | a = 0.2507(1) | 5(1) | 2(1) | |
| c = 0.4085(1) | ||||
| Co–Fe alloy | a = 0.2847(1) | 12(1) | 10(2) | |
| CoyFe3−yO4 | a = 0.8397(1) | 41(1) | 77(2) | 81(5) |
Further information about the chemical composition of the magnetic phases was obtained by XPS measurements. Since the technique is surface sensitive, the NHS-1 powder was pressed on top of a pre-consolidated KBr pellet and then subjected to a series of ion beam etching and spectrum acquisition cycles (removal of ca. 5 nm thickness per cycle) until a constant spectrum profile was obtained (see Fig. S2, ESI†). Once this condition is reached, the collected signal becomes representative of the whole sample, independent of the element distance from the NP surface. Fig. 2 reports the XPS spectra in the Co and Fe threshold energy regions. The deconvolution of the Co 2p3/2 peak (Fig. 2, left panel) revealed the presence of three components, which can be attributed to Co(0) metal (red peak at 778.2 eV)31 and to Co(II) in octahedral (Oh) and tetrahedral (Td) sites of the non-stoichiometric CoyFe3−yO4 (blue and green peaks at 780 and 782.4 eV, respectively)32 and, at higher binding energy, a satellite peak (light blue one at 784.7 eV). The Fe 2p3/2 peak (Fig. 2, right panel) was successfully deconvoluted by five components: the lowest binding energy component at 706.7 eV (purple peak) was attributed to Fe(0),31 while the three components at 708.9 eV (green), 710.7 eV (blue) and 712.7 eV (red) were ascribed to octahedral Fe(II)/Fe(III) and tetrahedral Fe(III), respectively.31 The satellite peaks related to Fe(II) and Fe(III) were resolved as a single peak at 715.3 eV (light blue).33,34
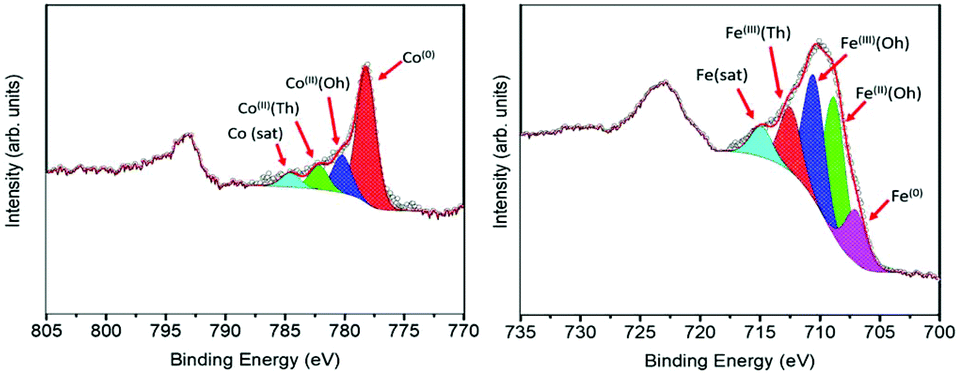 | ||
| Fig. 2 XPS spectra of NHS-1 in the region of cobalt (left) and iron (right). Spectra were recorded after four Ar ion beam etching cycles (removing ca. 5 nm thickness per cycle). The Co 2p3/2 peak was fitted with four components: Co(0) metal (red peak), Co(II) in octahedral (Oh) and tetrahedral (Td) sites of the CoyFe3−yO4 (blue and green, respectively) and a satellite peak (cyan). The Fe 2p3/2 peak was deconvoluted by five components: Fe(0) metal (purple), octahedral Fe(II) (green) and Fe(III), (blue), tetrahedral Fe(III) (red), and a satellite peak (cyan). | ||
The quantitative analysis of these signals, summarized in Table 1, indicated the formation of 19(5)% of metal phase (11% of Co(0) and 8% of Fe(0)) and 81(5)% of a spinel ferrite doped with a very low content of cobalt, according to the results obtained by Rietveld analysis of the XRD pattern.
With the aim to understand the structure of the synthesized sample, the element distribution was examined through the STEM-EELS technique. The STEM image of a single NP (Fig. 3, left panel) showed the presence of well-defined areas with higher brightness, probably corresponding to Co(0) and the Co–Fe alloy, and regions with lower brightness, corresponding to the metal oxide. The STEM-EELS mapping confirmed this hypothesis, depicting an overlapped distribution of oxygen and iron (Fig. 3, right panel) in the darker areas, while cobalt was inhomogeneously distributed, with a much higher percentage in the bright areas of the STEM image. From EELS measurements, it was harder to discriminate the presence of metallic Fe(0) due to the high amount of Fe from both the spinel ferrite phase and the oxide shell.
 | ||
| Fig. 3 (Left) STEM image of NHS-1 NPs; (right) EELS mapping of NHS-1 and distributions inside one NP of oxygen (O-K edge, red), iron (Fe-L2,3 edge, green) and cobalt (Co-L2,3 edge, blue). | ||
STEM images and EELS data therefore indicated that the NPs can be described as heterodimers comprising iron and cobalt in the metal state and a Co-poor spinel ferrite. Moreover, they suggested that the metal is covered by a very thin metal oxide layer (ca. 3 nm), whose presence justifies the long-time stability of the NPs. This was confirmed by an in-depth STEM-EELS analysis of the metal Co surface (Fig. 4 and Fig. S4 in the ESI†).
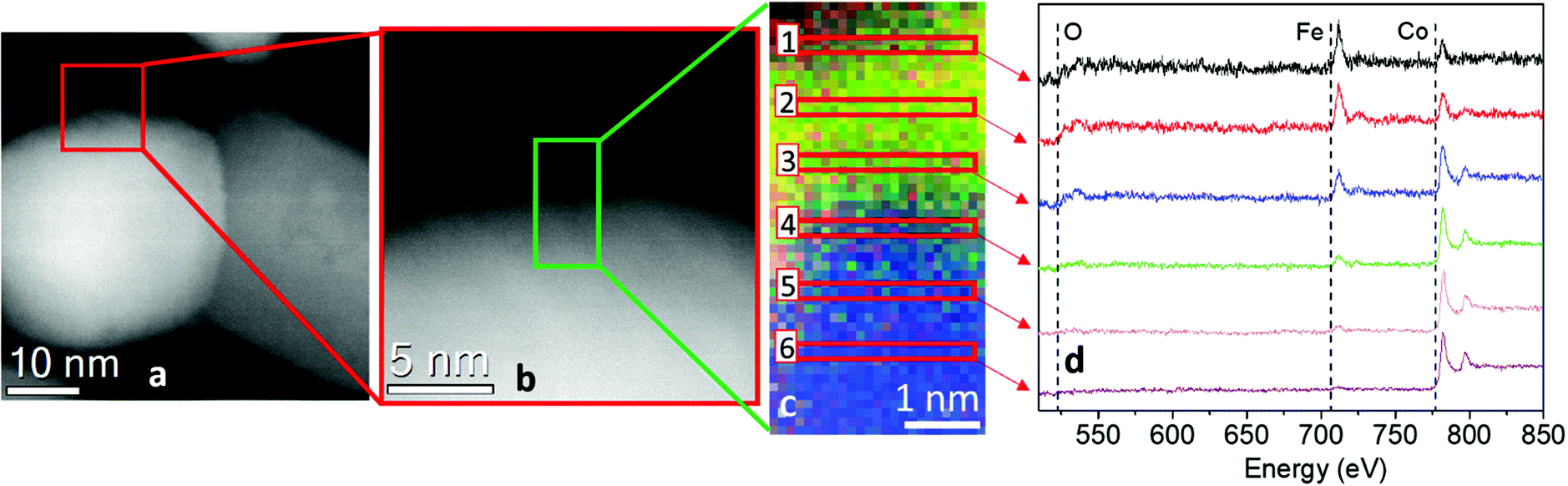 | ||
| Fig. 4 (a) STEM image of NHS-1 NP and (b) magnification of the shell region (5 nm thickness) from the red square in (a). (c) EELS color mapping of the selected green region (ca. 3 × 6 nm) in b panel; this area was chosen as representative of the shell at the metal surface, where the blue and green spots refer to cobalt and iron, respectively. (d) Normalized EELS spectra related to the regions in the EELS color map. The peaks at 708 eV and 779 eV correspond to the L2,3 edge of iron and of cobalt, respectively. In the outer surface (region 1 in c), it is possible to distinguish both iron and cobalt edges (intensity 60(6)% and 40(4)%, respectively). Moving towards the inner side (regions 2–6), the iron signal decreases until it disappears (region 6). | ||
The sub-nm resolved EELS mapping performed on a 5 nm thick layer of the NHS-1 NP surface highlighted the chemical composition of the shell. In the most external part of the shell (region 1), ca. 1 nm of thickness, the percentage of iron and cobalt was 60(6)% and 40(4)%, respectively. Moving deeper inside the NP, the amount of iron decreased progressively until it was no more detected close to the metal core (region 6). Thus, we can conclude that the oxide surrounding the metal is a quasi-stoichiometric cobalt ferrite. The other phase has a composition close to that of magnetite, with a very low amount (y < of 0.04) of cobalt.
In order to better understand the crystalline structure of the NHS, HRTEM images were acquired and their local Fast Fourier Transformation (FFT) was analyzed. In Fig. 5I, the image of a NP of NHS-1 is shown as representative of the whole powder sample. Two parts with spherical and cubic morphology can be distinguished. The corresponding magnifications (a and b, respectively), together with the one related to the outer shell of the spherical part (c), are shown in Fig. 5II. The FFT analysis, performed on a and b, revealed the presence of the fcc-structure of metal cobalt (Fig. 5III-a′) and of the cubic spinel structure of CoyFe3−yO4 (Fig. 5III-b′). The FFT performed on the selected region of the outer metal shell (d in Fig. 5II-c) showed the coexistence of spots related to both the fcc cobalt and the spinel ferrite (Fig. 5III-d′), confirming the presence of an interface between the two phases. Altogether this information indicated that NHS-1 comprises two components, a faceted spinel ferrite and a metal core, the latter being surrounded by a highly crystalline, thin ferrite layer.
 | ||
Fig. 5 (I) HRTEM image of a NHS-1 NP, where 3 regions (a–c) are selected: a and b correspond to metal and ferrite cores, respectively, while the region c to the shell around the metal core; (II) enlargement of the selected regions in I; (III) FFT analysis of the regions a, b, and d (yellow square in c) showing the presence of different crystallographic structures: the labelled spots are related to crystallographic planes that can be indexed as: (a′) fcc metal phase (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), in zone axis [011]Co-fcc, (b′) cubic spinel structure (Fdm), [011]spinel and (d′) metal (white) and cubic spinel oxide (green) phases, [01−1]spinel and [−112]Co-fcc; the presence of both phases confirms a sharp interface. m), in zone axis [011]Co-fcc, (b′) cubic spinel structure (Fdm), [011]spinel and (d′) metal (white) and cubic spinel oxide (green) phases, [01−1]spinel and [−112]Co-fcc; the presence of both phases confirms a sharp interface. | ||
The magnetic properties of NHS-1 were first investigated by the measurement of the hysteresis loops at 5 K and 300 K (Fig. 6, left panel). In both loops, a single step magnetization reversal process was observed, as generally expected for strongly exchange coupled magnetic phases. The MS values at 300 K and 5 K, extrapolated from the high field data, were 114 A m2 kg−1 and 116 A m2 kg−1, respectively. The remanence, MR, increased from 43 A m2 kg−1 up to 66 A m2 kg−1 and the coercive field from 0.04 T to 0.16 T when decreasing temperature from 300 K to 5 K.
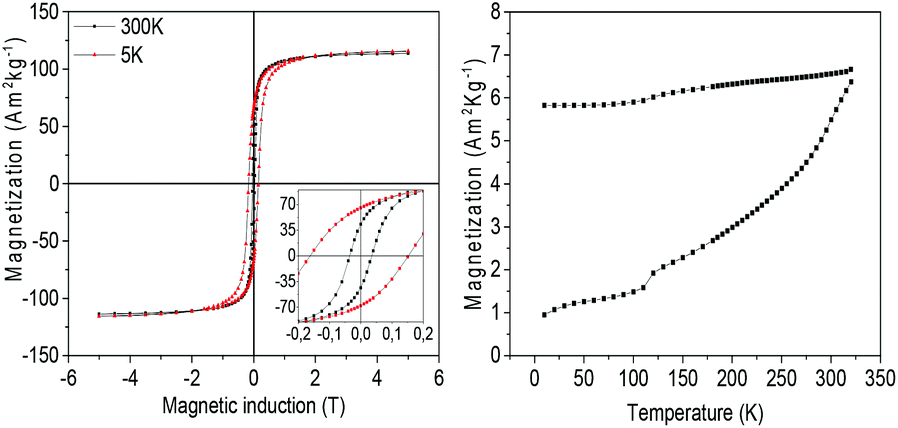 | ||
| Fig. 6 (Left) Hysteresis loops of NHS-1 measured at 300 K (black) and 5 K (red); the magnification of the low field region is shown in the inset; (right) temperature dependence of the magnetization recorded after ZFC and FC procedures, applying a constant field of 5 mT. | ||
The temperature dependence of the magnetization (Fig. 6, right panel) measured after ZFC and FC procedures displayed magnetic irreversibility up to the largest investigated temperature (340 K). Interestingly, no traces of any magnetic ordering transition appear in the ZFC and FC magnetization curves, confirming the absence of antiferromagnetic species such as iron or cobalt monoxides. A kink is however observed at ca. 120 K, which can be attributed to the Verwey transition.35 This observation confirmed that the oxide phase is mainly magnetite with a very low percentage of Co(II) in the lattice. Indeed, as reported by Abellan et al., the Verwey transition in CoyFe3−yO4 disappears when y is larger than 0.04.36 In our NHSs, the presence of metal phases strongly increases MS (30% more at room temperature) and the coercive field (90% more) with respect to those of magnetite NPs (for example, 42 nm magnetite NPs exhibit MS = 89 A m2 kg−1 and μ0HC = 0.021 T, at room temperature).37 It is interesting to note that when the hysteresis loop was measured at low temperature (5 K) after a 5 T FC procedure, no evidence of exchange bias occurred. Such observation allows us to exclude the presence of any surface spin coupling effect arising from ferro-(ferri-)/antiferro-magnetic interfaces or magnetically disordered shells in our NPs.
Despite the relatively large remanence value, the NPs in NHS-1 are still too soft to be used as building blocks for PM. In fact, the BHmax, evaluated after rescaling the magnetization for the volume estimated from the phase densities obtained from XPS and using the demagnetization factor (N∥ = 0.1)38 to take into account the internal field of the pellet, was 8 kJ m−3. Although the proposed synthesis method proved to be an efficient route to obtain a metal/ferrite exchange coupled nanostructure, designing a system with enhanced BHmax, exploitable for practical applications, requires the increase of the magnetic anisotropy of the spinel ferrite component. This can be accomplished by increasing the amount of Co(II) ions in the Oh sites of the spinel lattice. To this aim, the synthesis was repeated starting from two Co-richer mixed metal oleate complexes, 2-FeCo(OL) and 3-FeCo(OL), with a Fe:Co ratio of 1:1 and 1:2, respectively. The two precursors were prepared following the same synthetic strategy reported in the Experimental section for 1-FeCo(OL).
In Fig. 7 the STEM images and size distributions of the two obtained samples, NHS-2 (left) and NHS-3 (right), are shown. For both samples, it is possible to distinguish heterodimeric NPs with a mean size of about 40 ± 6 nm and 70 ± 10 nm for NHS-2 and NHS-3, respectively, and narrower size distribution compared to NHS-1 (±12 nm).
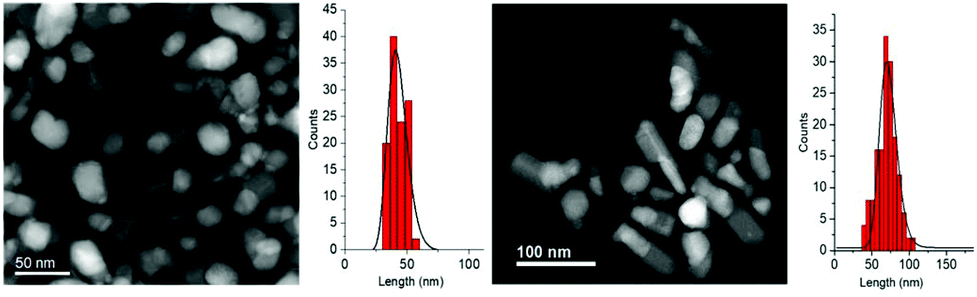 | ||
| Fig. 7 STEM images and particle size distributions of NHS-2 (left) and NHS-3 (right). The size distributions are evaluated from TEM images (see Fig. S1, ESI†) considering the length of the elongation axis. | ||
STEM images suggested that increasing the amount of cobalt in the bimetallic precursor leads to an evolution of the NPs’ shape to a more elongated heterodimeric architecture. XRD patterns of both samples are shown in Fig. 8, and, similarly to that of NHS-1, they exhibit diffraction peaks corresponding to the inverse spinel structure of cobalt ferrite, and to metal cobalt and the cobalt–iron alloy.
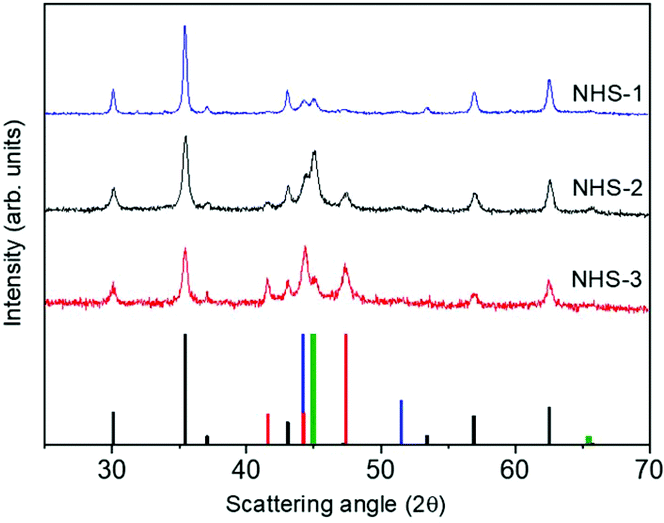 | ||
Fig. 8 XRD patterns of NHS-2 (black line) and NHS-3 (red line) NPs compared with NHS-1 (blue line). Reference bars are related to:  CoFe2O4, (JCPDS PDF #22-1086); CoFe2O4, (JCPDS PDF #22-1086);  hcp Co(0) (JCPDS PDF #89-7373); hcp Co(0) (JCPDS PDF #89-7373);  fcc Co(0), (JCPDS PDF #15-0806), and fcc Co(0), (JCPDS PDF #15-0806), and  Co–Fe alloy, (JCPDS PDF #65-4131). Co–Fe alloy, (JCPDS PDF #65-4131). | ||
The data obtained from the Rietveld refinement of the experimental XRD patterns are reported in Table 2. It is observed that increasing the concentration of Co in the mixed metal precursor leads to a decrease of the amount of spinel ferrite with respect to the metal phases. The weight percentages (% w/w) of the oxide and metal species were also estimated by fitting the XPS data (see Fig. S3, ESI†), obtained for both samples, following the procedure described for NHS-1. The results (Table 2) indicate the formation of 61% and 56% of Co-doped ferrite of average composition Co0.3Fe0.7Fe2.0O4 and Co0.4Fe0.6Fe2.0O4, for NHS-2 and NHS-3, respectively. The remarkable agreement between XRD and XPS data underlines the validity of the approach used to extract quantitative information of the overall composition of the NPs from a surface sensitive technique as XPS.
| XRD | XPS | |||
|---|---|---|---|---|
| Lattice parameters (nm) | Crystal size (D) (nm) | Phase percentage % (w/w) | Phase percentage % (w/w) | |
| NHS-2 | ||||
| Co (fcc) | a = 0.3541(1) | 16(1) | 10(2) | 39(5) |
| Co (hcp) | a = 0.2510(1) | 15(1) | 4(2) | |
| c = 0.4079(1) | ||||
| Co–Fe alloy | a = 0.2846(1) | 13(1) | 28(3) | |
| CoyFe3−yO4 | a = 0.8389(1) | 22(1) | 58(2) | 61(5) |
| NHS-3 | ||||
| Co (fcc) | a = 0.3541(1) | 21(1) | 19(2) | 44(5) |
| Co (hcp) | a = 0.2506(1) | 19(1) | 13(2) | |
| c = 0.4104(1) | ||||
| Co–Fe alloy | a = 0.2850(1) | 16(1) | 10(3) | |
| CoyFe3−yO4 | a = 0.8389(1) | 22(1) | 58(2) | 56(5) |
Therefore, increasing the amount of cobalt in the bimetallic precursor FeCo(OL) leads to a larger amount of metal species and to a Co-richer spinel ferrite. This, in turn, is expected to modify the magnetic properties of the resulting NHSs, such as the disappearance of the Verwey transition in the ZFCFC measurements due to the larger amount of Co in the spinel lattice (Fig. S5, ESI†). In Table 3, the magnetic parameters extracted from the hysteresis loops recorded at low and room temperature (Fig. S6, ESI†) are reported. The data obtained at room temperature showed an increase with respect to NHS-1 both in MS (9% for NHS-2 and 14% for NHS-3) and in MR (23% and 39%, respectively). Considering the bulk magnetization values of the Co–Fe alloy (MS = 245 A m2 kg−1),10fcc and hcp Co, (160 A m2 kg−1),39 magnetite (92 A m2 kg−1)7 and cobalt ferrite (80 A m2 kg−1),12 and the weight percentage obtained from XRD measurements, average MS = 114, 136, and 122 A m2 kg−1 are estimated for NHS-1, NHS-2 and NHS-3, respectively. These values are in good agreement with the experimental ones (Table 3). The difference observed for NHS-2 and NHS-3 (ca. 7%) can be ascribed to variation in the alloy composition.
| Sample | 5 K | 300 K | |||||
|---|---|---|---|---|---|---|---|
| M S (A m2 kg−1) | M R (R%) (A m2 kg−1) | μ 0 H C (T) | M S (A m2 kg−1) | M R (R%) (A m2 kg−1) | μ 0 H C (T) | BHmax (kJ m−3) | |
| NHS-1 | 115 | 66 (57%) | 0.16 | 114 | 43 (38%) | 0.04 | 8 |
| NHS-2 | 131 | 80 (61%) | 0.20 | 127 | 53 (42%) | 0.05 | 14 |
| NHS-3 | 132 | 79 (60%) | 0.20 | 130 | 60 (46%) | 0.07 | 19 |
The increase of coercivity of NHS-2 and NHS-3 with respect to that of NHS-1 is related to the magnetic anisotropy enhancement due to the larger amount of Co(II) in the CoyFe3−yO4 lattice and of hcp Co(0). The effective magnetic anisotropy of the hybrid NPs, indeed, is proportional to the HCMS product and increases along the series, doubling for NHS-3 the value of NHS-1, the one with the lowest Co(II) amount. The hysteresis loops display a single step magnetization reversal process, in the same way as NHS-1. This result is unexpected when theoretical predictions for spring magnets are considerded.3 The latter suggest that in a bilayer 2D system, when the size of the soft component exceeds 2–3 times the exchange length of the hard phase, the magnetic phases are decoupled.40,41 However, the application of this theory to NHSs has not been validated yet. Quesada et al.26 investigated the magnetic properties of CoFe2O4–CoFe composites of NPs with core@shell morphology, where the size of both phases is above the exchange length and below the single domain size. The authors demonstrated that even a weak coupling between the soft and hard phases can give rise to single step loops with reduced coercivity with respect to that of the hard phase. Similarly, in the present systems, the size of all the components is below their respective single domain critical values (110 nm for hcp Co, 120 nm for cobalt ferrite, 70 nm for fcc Co, and ca. 50 nm for Co–Fe alloy),7,42 and above the exchange coupling length (3–5 nm). Therefore, we can hypothesize that the exchange coupling between metal phases in the core is strong, while the coupling between the core and its oxide shell is weak, giving rise anyway to a collective reversal process.
Changing the cobalt concentration in FeCo(OL) precursors, we were thus able to tune the percentage of the metal phase and the hardness of the ferrite. Accordingly, BHmax increased from 8 (NHS-1) up to 19 (NHS-3) kJ m−3, demonstrating the effectiveness of the proposed approach in increasing the energy product. It is worth noting that this BHmax value is almost two times larger than those previously found for other CoFe2O4/CoFe2 nanosystems26,43 and among the highest reported for non-oriented rare-earth free nanostructures.11,44 Moreover, it is comparable to the energy product of some strontium ferrite-based commercial magnets.
It is also important to stress that thanks to the passivation of the metal phase with the highly crystalline spinel oxide, which allows metal preservations, these heterodimeric systems are highly stable in the air atmosphere over a long time. After one year of exposure to air, no variation of the structural and magnetic properties was indeed observed (Fig. S8 and S9, ESI†). This feature, which is important from the point of view of application, is not trivial as Co and FeCo based nanostructures are commonly reported to be unstable upon oxidation when exposed to air for a long time.14,16,26,45
The chemical and structural characterization unambiguously demonstrated that the one step synthesis presented here produces NHSs comprising exchange coupled metal and spinel ferrite phases, resulting in a high remanence nanosystem. The system was obtained starting from a mixed iron cobalt precursor decomposed at high temperature in docosane, in the presence of oleic acid and sodium oleate. This points out an important difference with respect to standard synthetic approaches based on thermal decomposition of mixed iron cobalt oleate, which lead to monoxides species, or less commonly, to ferrite or complex hybrids.46–50 Here, we succeeded in the reduction of part of the metal precursor to the metal state, and we did not observe the presence of the antiferromagnetic monoxide. We attributed these to the high amount of NaOL used in the synthesis. It is indeed well documented that NaOL plays a key role in the thermal decomposition reaction. For example, it has been reported that the presence of NaOL can induce preferential crystal growth along predetermined directions and allow the morphology control of iron oxide NPs.51–53 The latter was justified by the influence of the cation on the interaction between the carboxylic group and the various crystallographic faces of the oxide, resulting in the control of the surface energy of the NPs. In our case, we believe that the high amount of NaOL used (the NaOL:FeCo(OL) ratio is 1:1) can determine, under the synthetic conditions adopted (temperature and reactant molar ratio), an increase of the reducing strength of the reaction medium, allowing the reduction of transition metal cations Fe(III) and Co(II) down to the metal state.
The fundamental role of NaOL as a reducing agent was highlighted by performing four syntheses using a variable ratio between OA and NaOL, while keeping the precursor to surfactant ratio constant (see ESI, Table S1,† for details of the synthesis). XRD patterns of the obtained samples are reported in Fig. 9 and clearly show how the formation of the metal phase critically depends on the NaOL concentration. Indeed, as expected from the literature data, the synthesis with 1-FeCo(OL) and oleic acid alone led to the formation of mixed iron–cobalt monoxide (red line). Interestingly, a pure monoxide phase was also observed when the synthesis was carried out without any kind of surfactant, i.e. using 1-FeCo(OL) alone (Fig. 9, black line). Increasing the NaOL to OA molar ratio to 1:9, it was possible to observe the growth of the spinel ferrite phase (blue line) and, for larger values (1:1, green line), the full disappearance of the rock salt oxide with the formation of metal phases. The synthesis carried out with only the precursor and NaOL (purple line) led to the crystallization of the ferrite and metal phases. These results showed how the presence of NaOL is fundamental for obtaining a metal phase and providing, at the same time, a simple and effective method to control the spinel ferrite to metal ratio.
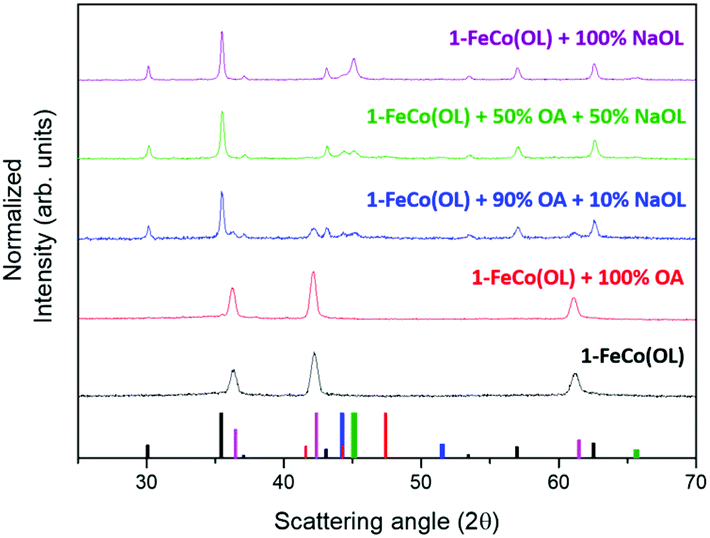 | ||
Fig. 9 XRD patterns of samples obtained using a different NaOL:OA ratio in the synthetic process: 100% OA (red line), 90% OA and 10% NaOL (blue line), 50% OA and 50% NaOL (green line) and 100% NaOL (purple line). The black bottom pattern corresponds to the powder obtained by using 1-FeCo(OL) only. Reference bars are related to:  CoFe2O4, (JCPDS PDF #22-1086); CoFe2O4, (JCPDS PDF #22-1086);  CoO (JCPDS PDF #71-1178); CoO (JCPDS PDF #71-1178);  hcp Co(0) (JCPDS PDF #89-7373); hcp Co(0) (JCPDS PDF #89-7373);  fcc Co(0), (JCPDS PDF #15-0806), and fcc Co(0), (JCPDS PDF #15-0806), and  Co–Fe alloy, (JCPDS PDF #65-4131). Co–Fe alloy, (JCPDS PDF #65-4131). | ||
It is worth noting that also the use of a mixed metal oleate precursor has an influence on the composition of the final product. Indeed, repeating the synthesis using only one monometallic precursor (Fe(OL) or Co(OL)) in the presence of OA and NaOL (molar ratio 1:1), the XRD pattern of the obtained NPs displayed the presence of a pure wüstite phase (FeO) when starting from Fe(OL); conversely, when pure Co(OL) was used, a mixed system comprising CoO and metal cobalt Co(0) was obtained (Fig. S7, ESI†). Therefore, we can conclude that both the presence of cobalt in the precursor and that of NaOL in the synthetic medium are essential for obtaining the metal species.
To better understand the formation mechanism of the metal/ferrite NHSs, we investigated the evolution of the crystallographic phases with the reaction time. To this aim, four different syntheses were carried out following the procedure described in the Experimental method section, changing only the dwelling time at reflux. The XRD patterns (Fig. 10) showed the formation, after 10 minutes at 370 °C, of three species: a ferrite (denoted as * in the figure) with lattice parameter a = 0.840 nm, cobalt–iron monoxide ( , Co1−xFexO) with a very low content of iron (a = 0.427(1) nm) and fcc Co(0) (
, Co1−xFexO) with a very low content of iron (a = 0.427(1) nm) and fcc Co(0) ( ). The Co1−xFexO disappeared after 25 minutes of reaction, the only phase observed being metal fcc cobalt and a spinel ferrite. On increasing the time at 370 °C up to 45 minutes a decrease of the lattice parameter value of ferrite (a = 0.837 nm) suggested the increase of cobalt content in the spinel lattice, while the crystal structure of Co(0) evolved from cubic to hexagonal (hcp,
). The Co1−xFexO disappeared after 25 minutes of reaction, the only phase observed being metal fcc cobalt and a spinel ferrite. On increasing the time at 370 °C up to 45 minutes a decrease of the lattice parameter value of ferrite (a = 0.837 nm) suggested the increase of cobalt content in the spinel lattice, while the crystal structure of Co(0) evolved from cubic to hexagonal (hcp,  ) and a small amount of cobalt–iron alloy formed (
) and a small amount of cobalt–iron alloy formed ( ). When the reaction time was further increased up to 300 minutes, only reduced species appeared, according to the XRD pattern, the predominant one being the Co–Fe alloy, with a minor amount of hcp Co.
). When the reaction time was further increased up to 300 minutes, only reduced species appeared, according to the XRD pattern, the predominant one being the Co–Fe alloy, with a minor amount of hcp Co.
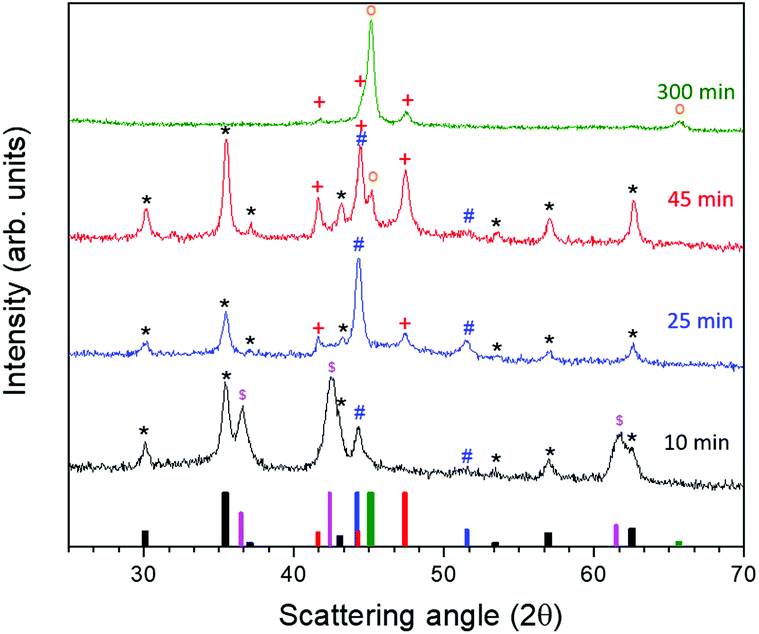 | ||
Fig. 10 XRD pattern of nanopowders prepared by increasing the reaction time: 10′ (black), 25′ (blue), 45′ (red) and 300′ (green). Reference bars are related to:  CoFe2O4, (JCPDS PDF #22-1086); CoFe2O4, (JCPDS PDF #22-1086);  CoO (JCPDS PDF #71-1178); CoO (JCPDS PDF #71-1178);  hcp Co(0) (JCPDS PDF #89-7373); hcp Co(0) (JCPDS PDF #89-7373);  fcc Co(0), (JCPDS PDF #15-0806), and fcc Co(0), (JCPDS PDF #15-0806), and  Co–Fe alloy, (JCPDS PDF #65-4131). The black asterisks (*) indicate the peaks attributed to the cubic spinel structure, the magenta dollars ( Co–Fe alloy, (JCPDS PDF #65-4131). The black asterisks (*) indicate the peaks attributed to the cubic spinel structure, the magenta dollars ( ) those of the monoxide phase, the blue pounds ( ) those of the monoxide phase, the blue pounds ( ) of fcc Co, the red crosses ( ) of fcc Co, the red crosses ( ) of hcp Co and the orange circles ( ) of hcp Co and the orange circles ( ) of the cobalt–iron alloy. ) of the cobalt–iron alloy. | ||
The obtained results can be explained by considering the extreme conditions of the reaction media in terms of temperature and redox strength, which allow the total transformation of the monoxide species in metals and spinel ferrite during the first 25 minutes. We can in fact hypothesize a mechanism of reaction as that reported in Scheme 1. The first two reactions that occur in the media are the release of Co(II) and Fe(III) by the oleate (black arrows). The decomposition of oleic acid and sodium oleate, in large amount and under an inert atmosphere, leads to a high concentration of reducing species as hydrogen (H2) and carbon monoxide (CO), which allow the partial reduction of Fe(III) to Fe(II) and the combination of cobalt and iron divalent cations to form a mixed rock-salt monoxide (Co1−xFexO) and, with other Fe(III) ions still available in solution, the ferrites (Fe3O4 or CoyFe3−yO4) (red arrows). The further step is the reduction of Co(II) down to the metal state. This process can be activated by the mixed mono-oxide. It is indeed well known and largely reported in the literature54–56 that cobalt monoxide plays a role as a activator in the oxidation of CO to carbon dioxide (CO2). According to this scheme, after 10 minutes of reaction Co-rich Co1−xFexO, Co(II)yFe(II)1−yFe(III)2O4 and fcc Co(0) NPs are found in the reaction medium (for TEM images and magnetic measurements see Fig. S11a in the ESI†). After 25 minutes (Fig. S11b, ESI†) the reduction of the cobalt in the monoxide is complete and fcc Co(0) starts to transform in hcp, which is the most thermodynamically stable Co phase at the reaction temperature (green arrows). The reduction process of Fe(II) to Fe(0) is slower than the one involving the cobalt, and the formation of the cobalt–iron alloy was observed only after 45 minutes (blue arrows). This is probably due to the lower reduction potential of Fe(II) under the experimental conditions adopted, which makes its reduction possible only when the initial Co(II) concentration has significantly reduced. The reduction process goes further on, until, after 300 minutes most of the metal precursors have transformed in the cobalt–iron alloy, with a minor amount of hcp Co (orange arrows).
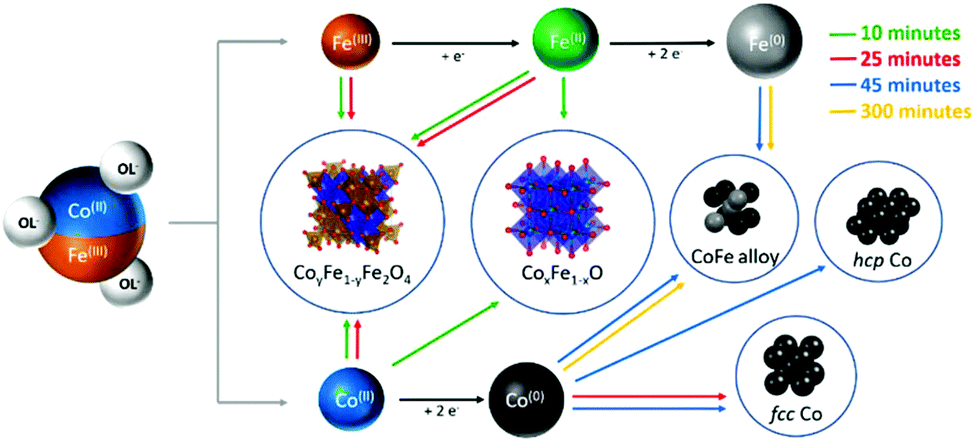 | ||
| Scheme 1 Evolution of the composition of the final NHSs as a function of the dwelling time: the decomposition of the mixed Co–Fe precursor and the resulting reducing conditions lead to the simultaneous presence of metal atoms at different oxidation states, whose relative amount depends on the reaction time. Colored arrows indicate the phases found at different reaction times (green, red, blue and yellow correspond to 10, 25, 45 and 300 minutes, respectively). | ||
Our experimental results unambiguously demonstrate that the presence of NaOL is essential to make the proposed reaction scheme effective. In the literature, it is reported that the partial substitution of OA with NaOL is an effective route to reduce the amount of H2O and CO2 in the reaction medium, released during the condensation via heterocyclic cleavage of OA (ketonic decarboxylation).57,58 In our synthesis, OA and NaOL are present in the same molar ratio; thus, we cannot exclude that the ketonic decarboxylation reaction partially occurred and then water was released. The water in the mixture can interact with the Na(I) cation of NaOL and increase the amount of OL− available in the reaction medium. Moreover, the carboxylate group of this oleate has a stronger affinity for the surface of the metal monoxide53 than OA. In the thermal decomposition synthesis, metal ions from smaller NPs are usually re-dissolved to reprecipitate forming bigger particles.59–61 In our case, the large amount of OL− available can modify the dissolution/precipitation process of the metal ions from the initial Co–Fe monoxide NPs, stabilizing the re-dissolved metal ions as organo-metallic complexes. These complexes in solution are Co(OL)2 and Fe(OL)2, which partially undergo the decarboxylation reaction leading to the formation of H2, CO2, radical alkenes and “free” metal ions.58 The latter are more prone to reduction by the reducing species occurring in the reaction medium than metal ions enclosed in the oxide. The proposed mechanism supports the presence of NaOL as stabilizing and reducing agents, as essential for obtaining metal/ferrite NHSs by this one-pot thermal decomposition synthesis. Finally, we would like to note that the heterodimeric morphology of the final product supports the proposed mechanism.
4. Conclusions
Summarizing, we reported here a one-pot thermal decomposition synthesis for obtaining metal/ferrite NHSs with very high saturation magnetization and large remanence and coercivity. The thorough investigation of the mechanism of the proposed synthesis approach as a function of the amount of stabilizing agents, of the nature of the metal precursors and of the decomposition time, allowed us to identify the key role played by NaOL at different steps of the reaction, down to the formation of the metal/ferrite NHSs. NaOL, indeed, does not enter the reaction pathway as a stabilizing agent only, but it is fundamental for the reduction of the mixed metal precursor down to the metal state, too. Moreover, the increase of the cobalt content in the Co–Fe oleate precursor caused the evolution of the NP shape, leading to a more elongated heterodimeric architecture, and to a modification of the relative amount of metal and spinel ferrite phases. Overall, these results suggest that the control of the relative amount of metal oleates (FeCoOL and NaOL) is a simple and efficient tool for tuning the hardness of the final NHSs. As an example, here we showed how, playing with these parameters, it is possible to more than double the BHmax at room temperature, from 8 (NHS-1) up to 19 (NHS-3) kJ m−3. This value, which is comparable to that of some commercial Strontium ferrite based magnets, can be further and consistently improved by increasing the amount of Co(II) ions in the spinel lattice. Finally, it is worth stressing that unlike most of the metal/spinel ferrite NHSs prepared so far, the presence of a very thin, highly crystalline, quasi-stoichiometric cobalt ferrite shell surrounding the metal makes the NHSs highly stable in the air atmosphere for a very long time, which is crucial for further technological applications. No modification of the magnetic properties of our NHSs was in fact observed over more than one year.Author contribution
MP and CS conceived the experiment. BM synthesized the NHSs. XPS measurements were performed by BC and BM. BM, MA, CI, MP and CS recorded and analysed the XRD patterns and magnetic data. STEM, EELS and HRTEM measurements and data analysis were performed by BM, CJF, GB, RFP, AI, CM and RMI. MP, BM and CS wrote the manuscript, which was critically revised by all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Francesca Ridi for TGA measurements and Dr Laura Chelazzi and Dr Samuele Ciattini, at CRIST laboratory (Univ. of Florence), for XRD data analysis. This work was supported by EU-H2020 AMPHIBIAN Project (no. 720853) and by European Union's Horizon 2020 research and innovation programme under grant agreement no. 823717-ESTEEM3.References
- W. H. Meiklejohn and C. P. Bean, Phys. Rev., 1956, 102, 1413–1414 CrossRef
.
- H. Zeng, J. Li, J. P. Liu, Z. L. Wang and S. Sun, Nature, 2002, 420, 395–398 CrossRef CAS PubMed
- E. F. Kneller and R. Hawig, IEEE Trans. Magn., 1991, 27, 3588–3560 CAS
-
F. Jimenez-Villacorta and L. H. Lewis, in Nanomagnetism, ed. J. M. Gonzalez Estevez, One Central Press, 2014, pp. 160–189 Search PubMed
- J. Sort, J. Nogués, S. Suriñach, J. S. Muñoz, M. D. Baró, E. Chappel, F. Dupont and G. Chouteau, Appl. Phys. Lett., 2001, 79, 1142–1144 CrossRef CAS
- O. Gutfleisch, M. a. Willard, E. Brück, C. H. Chen, S. G. Sankar and J. P. Liu, Adv. Mater., 2011, 23, 821–842 CrossRef CAS PubMed
-
J. M. D. Coey, Magnetism and Magnetic Materials, 2010 Search PubMed
-
A. H. King, R. G. Eggert and K. A. Gschneidner, in Handbook on the Physics and Chemistry of Rare Earths, Elsevier B.V., 1st edn, 2016, vol. 50, pp. 19–46 Search PubMed
- N. T. Nassar, X. Du and T. E. Graedel, J. Ind. Ecol., 2015, 19, 1044–1054 CrossRef
- P. Weiss and R. Forrer, Ann. Phys., 1929, 10, 279–372 Search PubMed
- A. López-Ortega, E. Lottini, C. D. J. Fernández and C. Sangregorio, Chem. Mater., 2015, 27, 4048–4056 CrossRef
- E. Fantechi, C. Innocenti, M. Albino, E. Lottini and C. Sangregorio, J. Magn. Magn. Mater., 2015, 380, 365–371 CrossRef CAS
- E. Fantechi, G. Campo, D. Carta, A. Corrias, C. de Julián Fernández, D. Gatteschi, C. Innocenti, F. Pineider, F. Rugi and C. Sangregorio, J. Phys. Chem. C, 2012, 116, 8261–8270 CrossRef CAS
- C. Petit and M. P. Pileni, J. Phys. Chem. B, 1999, 103, 1805–1810 CrossRef CAS
- V. F. Puntes, K. M. Krishnan and P. Alivisatos, Appl. Phys. Lett., 2001, 78, 2187–2189 CrossRef CAS
- G. S. Chaubey, C. Barcena, N. Poudyal, C. Rong, J. Gao, S. Sun and J. P. Liu, J. Am. Chem. Soc., 2007, 129, 7214–7215 CrossRef CAS PubMed
- B. Aslibeiki, Ceram. Int., 2016, 42, 6413–6421 CrossRef CAS
- L. J. Zhao and Q. Jiang, Mater. Lett., 2010, 64, 677–679 CrossRef CAS
- A. Quesada, F. Rubio-Marcos, J. F. Marco, F. J. Mompean, M. García-Hernández and J. F. Fernández, Appl. Phys. Lett., 2014, 105, 202405 CrossRef
- B. Nakhjavan, M. N. Tahir, M. Panthöfer, H. Gao, T. Gasi, V. Ksenofontov, R. Branscheid, S. Weber, U. Kolb, L. M. Schreiber and W. Tremel, Chem. Commun., 2011, 47, 8898 RSC
- Z. Zhou, Y. Sun, J. Shen, J. Wei, C. Yu, B. Kong, W. Liu, H. Yang, S. Yang and W. Wang, Biomaterials, 2014, 35, 7470–7478 CrossRef CAS PubMed
- L. Wu, P.-O. Jubert, D. Berman, W. Imaino, A. Nelson, H. Zhu, S. Zhang and S. Sun, Nano Lett., 2014, 14, 3395–3399 CrossRef CAS PubMed
- J. H. Jung, S. Kim, H. Kim, J. Park and J. H. Oh, Small, 2015, 11, 4976–4984 CrossRef CAS PubMed
- E. Fantechi, C. Innocenti, M. Zanardelli, M. Fittipaldi, E. Falvo, M. Carbo, V. Shullani, L. Di Cesare Mannelli, C. Ghelardini, A. M. Ferretti, A. Ponti, C. Sangregorio and P. Ceci, ACS Nano, 2014, 8, 4705–4719 CrossRef CAS PubMed
- K. F. Ortega, S. Anke, S. Salamon, F. Özcan, J. Heese, C. Andronescu, J. Landers, H. Wende, W. Schuhmann, M. Muhler, T. Lunkenbein and M. Behrens, Chem. – Eur. J., 2017, 23, 12443–12449 CrossRef CAS PubMed
- A. Quesada, C. Granados-Miralles, A. López-Ortega, S. Erokhin, E. Lottini, J. Pedrosa, A. Bollero, A. M. Aragón, F. Rubio-Marcos, M. Stingaciu, G. Bertoni, C. de Julián Fernández, C. Sangregorio, J. F. Fernández, D. Berkov and M. Christensen, Adv. Electron. Mater., 2016, 2, 1500365 CrossRef
- G. Cotin, C. Kiefer, F. Perton, D. Ihiawakrim, C. Blanco-Andujar, S. Moldovan, C. Lefevre, O. Ersen, B. Pichon, D. Mertz and S. Bégin-Colin, Nanomaterials, 2018, 8, 881 CrossRef PubMed
- W. Baaziz, B. P. Pichon, J.-M. Grenèche and S. Begin-Colin, CrystEngComm, 2018, 20, 7206–7220 RSC
- D. A. Shirley, Phys. Rev. B: Solid State, 1972, 5, 4709–4714 CrossRef
- G. Bertoni and J. Verbeeck, Ultramicroscopy, 2008, 108, 782–790 CrossRef CAS PubMed
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS
- W. P. Wang, H. Yang, T. Xian and J. L. Jiang, Mater. Trans., 2012, 53, 1586–1589 CrossRef CAS
- D. Wilson and M. A. Langell, Appl. Surf. Sci., 2014, 303, 6–13 CrossRef CAS
- T. Aghavnian, J. B. Moussy, D. Stanescu, R. Belkhou, N. Jedrecy, P. Ohresser, H. Magnan, M.-A. Arrio, Ph. Sainctavit and A. Barbier, J. Electron Spectrosc. Relat. Phenom., 2016, 202, 16–21 CrossRef
- E. J. W. Verwey, Nature, 1939, 144, 327–328 CrossRef CAS
- J. Abellan and M. Ortuno, Phys. Status Solidi, 1986, 96, 581–586 CrossRef CAS
- T. Orlando, S. Mannucci, E. Fantechi, G. Conti, S. Tambalo, A. Busato, C. Innocenti, L. Ghin, R. Bassi, P. Arosio, F. Orsini, C. Sangregorio, M. Corti, M. F. Casula, P. Marzola, A. Lascialfari and A. Sbarbati, Contrast Media Mol. Imaging, 2016, 11, 139–145 CrossRef CAS PubMed
- B. K. Pugh, D. P. Kramer and C. H. Chen, IEEE
Trans. Magn., 2011, 47, 4100–4103 Search PubMed
- Y. Bao and K. M. Krishnan, J. Magn. Magn. Mater., 2005, 293, 15–19 CrossRef CAS
- G. Asti, M. Ghidini, R. Pellicelli, C. Pernechele, M. Solzi, F. Albertini, F. Casoli, S. Fabbrici and L. Pareti, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 094406 CrossRef
- M. Ghidini, G. Asti, R. Pellicelli, C. Pernechele and M. Solzi, J. Magn. Magn. Mater., 2007, 316, 159–165 CrossRef CAS
- F. E. Luborsky and T. O. Paine, J. Appl. Phys., 1960, 31, S68–S70 CrossRef
- G. C. P. Leite, E. F. Chagas, R. Pereira, R. J. Prado, A. J. Terezo, M. Alzamora and E. Baggio-Saitovitch, J. Magn. Magn. Mater., 2012, 324, 2711–2716 CrossRef CAS
- Y. Kumar, A. Sharma, M. A. Ahmed, S. S. Mali, C. K. Hong and P. M. Shirage, New J. Chem., 2018, 42, 15793–15802 RSC
- C. Dobbrow and A. M. Schmidt, Beilstein J. Nanotechnol., 2012, 3, 75–81 CrossRef PubMed
- C. J. Chen, R. K. Chiang, S. Kamali and S. L. Wang, Nanoscale, 2015, 7, 14332–14343 RSC
- C. Moya, G. Salas, M. del P. Morales, X. Batlle and A. Labarta, J. Mater. Chem. C, 2015, 3, 4522–4529 RSC
- W. Baaziz, B. P. Pichon, Y. Liu, J. M. Grenèche, C. Ulhaq-Bouillet, E. Terrier, N. Bergeard, V. Halté, C. Boeglin, F. Choueikani, M. Toumi, T. Mhiri and S. Begin-Colin, Chem. Mater., 2014, 26, 5063–5073 CrossRef CAS
- N. Bao, L. Shen, Y. Wang, P. Padhan and A. Gupta, J. Am. Chem. Soc., 2007, 129, 12374–12375 CrossRef CAS PubMed
- E. Lottini, A. López-Ortega, G. Bertoni, S. Turner, M. Meledina, G. Van Tendeloo, C. de Julián Fernández and C. Sangregorio, Chem. Mater., 2016, 28, 4214–4222 CrossRef CAS
- M. V. Kovalenko, M. I. Bodnarchuk, R. T. Lechner, G. Hesser, F. Schäffler and W. Heiss, J. Am. Chem. Soc., 2007, 129, 6352–6353 CrossRef CAS PubMed
- L. Wang and L. Gao, J. Phys. Chem. C, 2009, 15914–15920 CrossRef CAS
- Z. Zhou, X. Zhu, D. Wu, Q. Chen, D. Huang, C. Sun, J. Xin, K. Ni and J. Gao, Chem. Mater., 2015, 27, 3505–3515 CrossRef CAS
- M. Kang, Appl. Catal., A, 2003, 251, 143–156 CrossRef CAS
- C.-B. Wang, C.-W. Tang, H.-C. Tsai and S.-H. Chien, Catal. Lett., 2006, 107, 223–230 CrossRef CAS
- P. Thormählen, M. Skoglundh, E. Fridell and B. Andersson, J. Catal., 1999, 188, 300–310 CrossRef
- J. Muro-Cruces, A. G. Roca, A. López-Ortega, E. Fantechi, D. Del-Pozo-Bueno, S. Estradé, F. Peiró, B. Sepúlveda, F. Pineider, C. Sangregorio and J. Nogues, ACS Nano, 2019, 13, 7716–7728 CrossRef CAS PubMed
- S. J. Kemp, R. M. Ferguson, A. P. Khandhar and K. M. Krishnan, RSC Adv., 2016, 6, 77452–77464 RSC
-
N. Pinna and M. Kamaoui, Advanced Wet-Chemical Synthetic Approaches to Inorganic Nanostructures, 2008 Search PubMed
-
T. Sugimoto, Monodispersed Particles, 2001 Search PubMed
- J. Park, J. Joo, S. G. Kwon, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 4630–4660 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: TEM images; XPS spectra; EELS elemental and quantification analysis; magnetic measurements; all experimental details about the synthesis performed to study the synthetic protocol; XRD patterns. See DOI: 10.1039/d0nr01361g |
| This journal is © The Royal Society of Chemistry 2020 |